The bark of Nauclea latifolia contains tramadol at medicinal concentrations © imagebroker / Alamy
http://www.rsc.org/chemistryworld/2013/09/african-plant-natural-source-tramadol
In another example of nature beating chemists, the African plant Nauclea latifolia has been found to be a natural source of the synthetic opioid tramadol. First marketed in 1977, tramadol is frequently used to relive moderate to moderately-severe pain. While other synthetic drugs have later been found in nature, this is the first instance where the discovery involves clinically viable concentrations.
Colloquially known as the ‘African peach’ or ‘pin cushion tree’, N. latifolia is a flowering, sub-Saharan evergreen that grows widely across Central and West Africa and is used by local populations to treat a wide variety of ailments – including epilepsy, malaria, general pain and many infectious diseases………………………. READ ALL AT
http://www.rsc.org/chemistryworld/2013/09/african-plant-natural-source-tramadol

tramadol

tramadol hydrocloride
The chemical name for tramadol hydrochloride is (±)cis-2-[(dimethylamino)methyl]-1-(3methoxyphenyl) cyclohexanol hydrochloride
Tramadol (marketed as the hydrochloride salt by Janssen Pharmaceutica as Ultram in the United States, Ralivia by Biovail in Canada and many other companies throughout the world) is a centrally acting synthetic opioid analgesic used to treat moderate to moderately severe pain. The drug has a wide range of applications, including treatment of rheumatoid arthritis, restless legs syndrome, motor neurone disease and fibromyalgia.[citation needed] It was launched and marketed as Tramal by the German pharmaceutical company Grünenthal GmbH in 1977.
Tramadol is a weak μ-opioid receptor agonist, a serotonin releaser and a reuptake inhibitor of norepinephrine. Tramadol is metabolized to O-desmethyltramadol, a significantly more potent μ-opioid agonist. Tramadol and its major metabolite(s) are distinguished from other more potent opioid agonists by relative selectivity for μ-opioid receptors.
Chemistry
Characteristics
Structurally, tramadol closely resembles a stripped down version of codeine. Both codeine and tramadol share the 3-methyl ether group, and both compounds are metabolized along the same hepatic pathway and mechanism to the stronger opioid, phenol agonist analogs. For codeine, this is morphine, and for tramadol, it is the O-desmethyltramadol.
When administered through IV, patients notice very little clinical difference in subjective potency compared to morphine.
Comparison with related substances
Structurally, tapentadol is the closest chemical relative of tramadol in clinical use. Tapentadol is also an opioid, but unlike both tramadol and venlafaxine, tapentadol represents only one stereoisomer and is the weaker of the two, in terms of opioid effect. Both tramadol and venlafaxine are racemic mixtures. Structurally, tapentadol also differs from tramadol in being a phenol, and not an ether. Also, both tramadol and venlafaxine incorporate a cyclohexyl moiety, attached directly to the aromatic, while tapentadol lacks this feature.
Synthesis and stereoisomerism
-Tramadol.svg)
-Tramadol_gespiegelt.svg)
(1R,2R)-Tramadol (1S,2S)-Tramadol
-Tramadol.svg)
-Tramadol_gespiegelt.svg)
(1R,2S)-Tramadol (1S,2R)-Tramadol
The chemical synthesis of tramadol is described in the literature.[62] Tramadol [2-(dimethylaminomethyl)-1-(3-methoxyphenyl)cyclohexanol] has two stereogenic centers at the cyclohexane ring. Thus, 2-(dimethylaminomethyl)-1-(3-methoxyphenyl)cyclohexanol may exist in four different configurational forms:
- (1R,2R)-isomer
- (1S,2S)-isomer
- (1R,2S)-isomer
- (1S,2R)-isomer
The synthetic pathway leads to the racemate (1:1 mixture) of (1R,2R)-isomer and the (1S,2S)-isomer as the main products. Minor amounts of the racemic mixture of the (1R,2S)-isomer and the (1S,2R)-isomer are formed as well. The isolation of the (1R,2R)-isomer and the (1S,2S)-isomer from the diastereomeric minor racemate [(1R,2S)-isomer and (1S,2R)-isomer] is realized by the recrystallization of the hydrochlorides. The drug tramadol is a racemate of the hydrochlorides of the (1R,2R)-(+)- and the (1S,2S)-(–)-enantiomers. The resolution of the racemate [(1R,2R)-(+)-isomer / (1S,2S)-(–)-isomer] was described[63] employing (R)-(–)- or (S)-(+)-mandelic acid. This process does not find industrial application, since tramadol is used as a racemate, despite known different physiological effects[64] of the (1R,2R)- and (1S,2S)-isomers, because the racemate showed higher analgesic activity than either enantiomer in animals[65] and in humans.[66]
- 62…..Pharmaceutical Substances, Axel Kleemann, Jürgen Engel, Bernd Kutscher and Dieter Reichert, 4. ed. (2000) 2 volumes, Thieme-Verlag Stuttgart (Germany), p. 2085 bis 2086, ISBN 978-1-58890-031-9; since 2003 online with biannual actualizations.
- 63………Zynovy, Zinovy; Meckler, Harold (2000). “A Practical Procedure for the Resolution of (+)- and (−)-Tramadol”. Organic Process Research & Development 4 (4): 291–294. doi:10.1021/op000281v.
- 64……..Burke D, Henderson DJ (April 2002). “Chirality: a blueprint for the future”. British Journal of Anaesthesia 88 (4): 563–76. doi:10.1093/bja/88.4.563. PMID 12066734.
- 65…Raffa, R. B.; Friderichs, E.; Reimann, W.; Shank, R. P.; Codd, E. E.; Vaught, J. L.; Jacoby, H. I.; Selve, N. (1993). “Complementary and synergistic antinociceptive interaction between the enantiomers of tramadol”. The Journal of Pharmacology and Experimental Therapeutics 267 (1): 331–340. PMID 8229760.
- 66 ..Grond, S.; Meuser, T.; Zech, D.; Hennig, U.; Lehmann, K. A. (1995). “Analgesic efficacy and safety of tramadol enantiomers in comparison with the racemate: a randomised, double-blind study with gynaecological patients using intravenous patient-controlled analgesia”. Pain 62 (3): 313–320. doi:10.1016/0304-3959(94)00274-I. PMID 8657431.
-
tramadol hydrochloride, which is (RR, SS)-2-dimethylaminomethyl-1-(3-methoxyphenyl)cyclohexanol hydrochloride (trans), from a mixture of its (RS, SR) (cis) and trans bases, and to an improved process for the preparation of tramadol (base) monohydrate, sometimes used as an intermediate in the preparation of tramadol hydrochloride.
-
Tramadol is a well-established drug disclosed in US patent specification no. 3 652 589, which is used in the form of its hydrochloride salt as a non-narcotic analgesic drug. Tramadol is the pharmacologically active trans isomer of 2-dimethylaminomethyl-1-(3-methoxyphenyl)cyclohexanol, as opposed to the corresponding cis isomer, namely, (RS, SR)-2-dimethylaminomethyl-1-(3-methoxyphenyl)cyclohexanol.
-
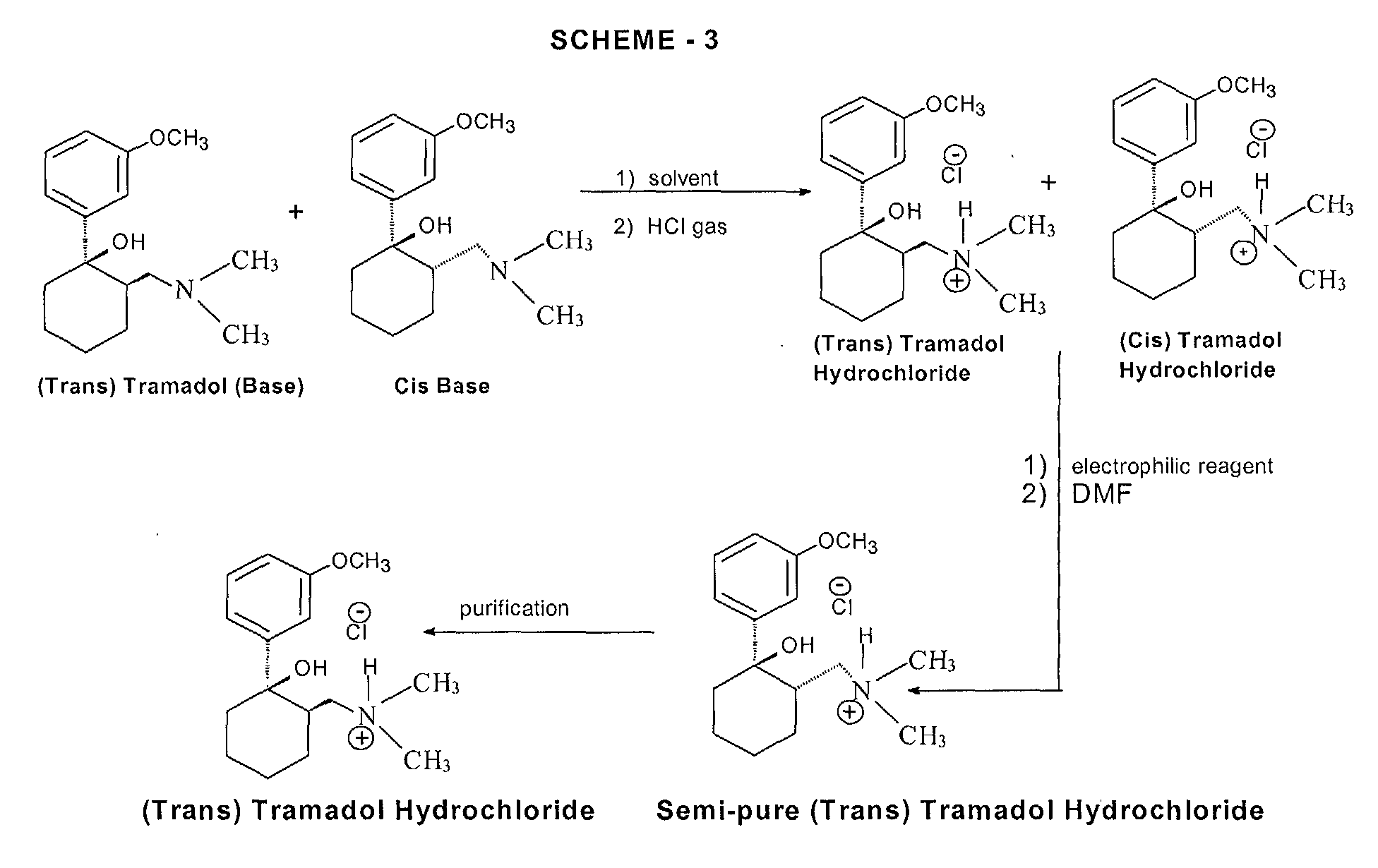
Various processes for the synthesis of tramadol hydrochloride have been described in the prior art. For example, US 3 652 589 and British patent specification no. 992 399 describe the preparation of tramadol hydrochloride. In this method, Grignard reaction of 2-dimethylaminomethyl cyclohexanone (Mannich base) with metabromo-anisole gives an oily mixture of tramadol and the corresponding cis isomer, along with Grignard impurities. This oily reaction mixture is subjected to high vacuum distillation at high temperature to give both the geometric isomers of the product base as an oil. This oil, on acidification with hydrogen chloride gas, furnishes insufficiently pure tramadol hydrochloride as a solid. This must then be purified, by using a halogenated solvent and 1,4-dioxane, to give sufficiently pure tramadol hydrochloride. The main drawback of this process is the use of large quantities of 1,4-dioxane and the need for multiple crystallizations to get sufficiently pure trans isomer hydrochloride (Scheme – 1).
-
The use of dioxane for the separation of tramadol hydrochloride from the corresponding cis isomer has many disadvantages, such as safety hazards by potentially forming explosive peroxides, and it is also a category 1 carcinogen (Kirk and Othmer, 3rd edition, 17, 48). Toxicological studies of dioxane show side effects such as CNS depression, and necrosis of the liver and kidneys. Furthermore, the content of dioxane in the final tramadol hydrochloride has been strictly limited; for example, the German Drug Codex (Deutscher Arzneimittel Codex, DAC (1991)) restricts the level of dioxane in tramadol hydrochloride to 0.5 parts per million (ppm).

-
In another process, disclosed in US patent specification no. 5 414 129, the purification and separation of tramadol hydrochloride is undertaken from a reaction mixture containing the trans and cis isomers, and Grignard reaction side products, in which the reaction mixture is diluted in isopropyl alcohol and acidified with gaseous hydrogen chloride to yield (trans) tramadol hydrochloride (97.8%) and its cis isomer (2.2%), which is itself crystallized twice with isopropyl alcohol to give pure (trans) tramadol hydrochloride (Scheme – 2). This process relies on the use of multiple solvents to separate the isomers (ie butylacetate, 1-butanol, 1-pentanol, primary amyl alcohol mixture, 1-hexanol, cyclohexanol, 1-octanol, 2-ethylhexanol and anisole). The main drawback of this process is therefore in using high boiling solvents; furthermore, the yields of tramadol hydrochloride are still relatively low and the yield of the corresponding cis hydrochloride is relatively high in most cases.

- PCT patent specification no. WO 99/03820 describes a method of preparation of tramadol (base) monohydrate, which involves the reaction of Mannich base with metabromo-anisole (Grignard reaction) to furnish a mixture of tramadol base with its corresponding cis isomer and Grignard impurities. This, on treatment with an equimolar quantity of water and cooling to 0 to -5°C, gives a mixture of tramadol (base) monohydrate with the corresponding cis isomer (crude). It is further purified with ethyl acetate to furnish pure (trans) tramadol (base) monohydrate, which is again treated with hydrochloric acid in the presence of a suitable solvent to give its hydrochloride salt (Scheme – 2). The drawback of this method is that, to get pure (trans) tramadol hydrochloride, first is prepared pure (trans) tramadol (base) monohydrate, involving a two-step process, and this is then converted to its hydrochloride salt. The overall yield is low because of the multiple steps and tedious process involved.
-
More recently, a process for the separation of tramadol hydrochloride from a mixture with its cis isomer, using an electrophilic reagent, has been described in US patent specification no. 5 874 620. The mixture of tramadol hydrochloride with the corresponding cis isomer is reacted with an electrophilic reagent, such as acetic anhydride, thionyl chloride or sodium azide, using an appropriate solvent (dimethylformamide or chlorobenzene) to furnish a mixture of tramadol hydrochloride (93.3 to 98.6%) with the corresponding cis isomer (1.4 to 6.66%), (Scheme – 3). The product thus obtained is further purified in isopropyl alcohol to give pure (trans) tramadol hydrochloride. However, the drawback of this process is that a mixture of tramadol base with its cis isomer is first converted into the hydrochloride salts, and this is further reacted with toxic, hazardous and expensive electrophilic reagents to get semi-pure (trans) tramadol hydrochloride. The content of the cis isomer is sufficiently high to require further purification, and this therefore results in a lower overall yield.


- (1R,2R)-isomer
- (1S,2S)-isomer
- (1R,2S)-isomer
- (1S,2R)-isomer