
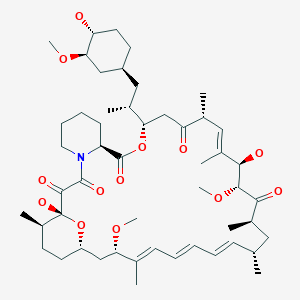
Rapamycin (Sirolimus)
(3S,6R,7E,9R,10R,12R,14S,15E,17E,19E,21S,23S,26R,27R,34aS)-9,10,12,13,14,21,22,23,24,25, 26,27,32,33,34,34a-Hexadecahydro-9,27-dihydroxy-3-[(1R)-2-[(1S,3R,4R)-4-hydroxy-3-methoxycyclohexyl]-1-methylethyl]-10,21-dimethoxy-6,8,12,14,20,26-hexamethyl-23,27-epoxy-3H-pyrido[2,1-c][1,4]oxaazacyclohentriacontine-1,5,11,28,29(4H,6H,31H)-pentone
Wyeth Pharmaceuticals (Originator)
M.Wt:914.18
Formula:C51H79NO13
53123-88-9 cas no
Antifungal and immunosuppressant. Specific inhibitor of mTOR (mammalian target of Rapamycin). Complexes with FKBP-12 and binds mTOR inhibiting its activity. Inhibits interleukin-2-induced phosphorylation and activation of p70 S6 kinase. Induces autophagy in yeast and mammalian cell lines.
Rapamycin is a triene macrolide antibiotic, which demonstrates anti-fungal, anti-inflammatory, anti-tumor and immunosuppressive properties. Rapamycin has been shown to block T-cell activation and proliferation, as well as, the activation of p70 S6 kinase and exhibits strong binding to FK-506 binding proteins. Rapamycin also inhibits the activity of the protein, mTOR, (mammalian target of rapamycin) which functions in a signaling pathway to promote tumor growth. Rapamycin binds to a receptor protein (FKBP12) and the rapamycin/FKB12 complex then binds to mTOR and prevents interaction of mTOR with target proteins in this signaling pathway. Rapamycin name is derived from the native word for Easter Island, Rapi Nui.
- (-)-Rapamycin
- Antibiotic AY 22989
- AY 22989
- AY-22989
- CCRIS 9024
- HSDB 7284
- NSC 226080
- Rapammune
- Rapamune
- Rapamycin
- SILA 9268A
- Sirolimus
- UNII-W36ZG6FT64
- WY-090217
- A 8167
A macrolide compound obtained from Streptomyces hygroscopicus that acts by selectively blocking the transcriptional activation of cytokines thereby inhibiting cytokine production. It is bioactive only when bound to IMMUNOPHILINS. Sirolimus is a potent immunosuppressant and possesses both antifungal and antineoplastic properties.
Sirolimus (INN/USAN), also known as rapamycin, is an immunosuppressant drug used to prevent rejection in organ transplantation; it is especially useful in kidney transplants. It prevents activation of T cells and B cells by inhibiting their response to interleukin-2 (IL-2). Sirolimus is also used as a coronary stent coating. Sirolimus works, in part, by eliminating old and abnormal white blood cells.[citation needed] Sirolimus is effective in mice with autoimmunity and in children with a rare condition called autoimmune lymphoproliferative syndrome (ALPS).
 sirolimus
sirolimus
A macrolide, sirolimus was discovered by Brazilian researchers as a product of the bacterium Streptomyces hygroscopicus in a soil sample fromEaster Island[1] — an island also known as Rapa Nui.[2] It was approved by the FDA in September 1999 and is marketed under the trade nameRapamune by Pfizer (formerly by Wyeth).
Sirolimus was originally developed as an antifungal agent. However, this use was abandoned when it was discovered to have potent immunosuppressive and antiproliferative properties. It has since been shown to prolong the life of mice and might also be useful in the treatment of certain cancers.
Unlike the similarly named tacrolimus, sirolimus is not a calcineurin inhibitor, but it has a similar suppressive effect on the immune system. Sirolimus inhibits the response tointerleukin-2 (IL-2), and thereby blocks activation of T and B cells. In contrast, tacrolimus inhibits the secretion of IL-2.
The mode of action of sirolimus is to bind the cytosolic protein FK-binding protein 12(FKBP12) in a manner similar to tacrolimus. Unlike the tacrolimus-FKBP12 complex which inhibits calcineurin (PP2B), the sirolimus-FKBP12 complex inhibits themammalian target of rapamycin (mTOR, rapamycin being an older name for sirolimus) pathway by directly binding the mTOR Complex1 (mTORC1).
mTOR has also been called FRAP (FKBP-rapamycin associated protein), RAFT (rapamycin and FKBP target), RAPT1, or SEP. The earlier names FRAP and RAFT were coined to reflect the fact that sirolimus must bind FKBP12 first, and only the FKBP12-sirolimus complex can bind mTOR. However, mTOR is now the widely accepted name, since Tor was first discovered via genetic and molecular studies of sirolimus-resistant mutants of Saccharomyces cerevisiae that identified FKBP12, Tor1, and Tor2 as the targets of sirolimus and provided robust support that the FKBP12-sirolimus complex binds to and inhibits Tor1 and Tor2.
 rapamycin
rapamycin
Unlike the similarly named tacrolimus, sirolimus is not a calcineurin inhibitor, but it has a similar suppressive effect on the immune system. Sirolimus inhibits the response to interleukin-2 (IL-2), and thereby blocks activation of T and B cells. In contrast, tacrolimus inhibits the secretion of IL-2.
The mode of action of sirolimus is to bind the cytosolic protein FK-binding protein 12 (FKBP12) in a manner similar to tacrolimus. Unlike the tacrolimus-FKBP12 complex which inhibits calcineurin (PP2B), the sirolimus-FKBP12 complex inhibits the mammalian target of rapamycin(mTOR, rapamycin being an older name for sirolimus) pathway by directly binding the mTOR Complex1 (mTORC1).
mTOR has also been called FRAP (FKBP-rapamycin associated protein), RAFT (rapamycin and FKBP target), RAPT1, or SEP. The earlier names FRAP and RAFT were coined to reflect the fact that sirolimus must bind FKBP12 first, and only the FKBP12-sirolimus complex can bind mTOR. However, mTOR is now the widely accepted name, since Tor was first discovered via genetic and molecular studies of sirolimus-resistant mutants of Saccharomyces cerevisiae that identified FKBP12, Tor1, and Tor2 as the targets of sirolimus and provided robust support that the FKBP12-sirolimus complex binds to and inhibits Tor1 and Tor2.
 SIROLIMUS
SIROLIMUS
Rapamycin and its preparation are described in US Patent No. 3,929,992, issued December 30, 1975. Alternatively, rapamycin may be purchased commercially [Rapamune®, Wyeth].
Rapamycin (Sirolimus) is a 31-member natural macrocyclic lactone [C51H79N1O13; MWt=914.2] produced by Streptomyces hygroscopicus and found in the 1970s (U.S. Pat. No. 3,929,992; 3,993,749). Rapamycin (structure shown below) was approved by the Food and Drug Administration (FDA) for the prophylaxis of renal transplant rejection in 1999.
Rapamycin resembles tacrolimus (binds to the same intracellular binding protein or immunophilin known as FKBP-12) but differs in its mechanism of action. Whereas tacrolimus and cyclosporine inhibit T-cell activation by blocking lymphokine (e.g., IL2) gene transcription, sirolimus inhibits T-cell activation and T lymphocyte proliferation by binding to mammalian target of rapamycin (mTOR). Rapamycin can act in synergy with cyclosporine or tacrolimus in suppressing the immune system.
Rapamycin is also useful in preventing or treating systemic lupus erythematosus [U.S. Pat. No. 5,078,999], pulmonary inflammation [U.S. Pat. No. 5,080,899], insulin dependent diabetes mellitus [U.S. Pat. No. 5,321,009], skin disorders, such as psoriasis [U.S. Pat. No. 5,286,730], bowel disorders [U.S. Pat. No. 5,286,731], smooth muscle cell proliferation and intimal thickening following vascular injury [U.S. Pat. Nos. 5,288,711 and 5,516,781], adult T-cell leukemia/lymphoma [European Patent Application 525,960 A1], ocular inflammation [U.S. Pat. No. 5,387,589], malignant carcinomas [U.S. Pat. No. 5,206,018], cardiac inflammatory disease [U.S. Pat. No. 5,496,832], anemia [U.S. Pat. No. 5,561,138] and increase neurite outgrowth [Parker, E. M. et al, Neuropharmacology 39, 1913-1919, 2000].
Although rapamycin can be used to treat various disease conditions, the utility of the compound as a pharmaceutical drug has been limited by its very low and variable bioavailability and its high immunosuppressive potency and potential high toxicity. Also, rapamycin is only very slightly soluble in water. To overcome these problems, prodrugs and analogues of the compound have been synthesized. Water soluble prodrugs prepared by derivatizing rapamycin positions 31 and 42 (formerly positions 28 and 40) of the rapamycin structure to form glycinate, propionate, and pyrrolidino butyrate prodrugs have been described (U.S. Pat. No. 4,650,803). Some of the analogues of rapamycin described in the art include monoacyl and diacyl analogues (U.S. Pat. No. 4,316,885), acetal analogues (U.S. Pat. No. 5,151,413), silyl ethers (U.S. Pat. No. 5,120,842), hydroxyesters (U.S. Pat. No. 5,362,718), as well as alkyl, aryl, alkenyl, and alkynyl analogues (U.S. Pat. Nos. 5,665,772; 5,258,389; 6,384,046; WO 97/35575).
………………………………………….
Synthesis
http://www.google.co.in/patents/US3929992
PREPARATION
CUT PASTE FROM TEXT
In one embodiment of this invention rapamycin is prepared in the followingmanner: 4
A suitable fermenter is charged with production meis reached in the fermentation mixture after 2-8 days,
usually after about 5 days, as determined by the cup plate method and Candida albicans as the test organism. The mycelium is harvested by filtration with diatomaceous earth. Rapamycin is then extracted from the mycelium with a water-miscible solvent, for example a lower alkanol, preferably methanol or ethanol. The latter extract is then concentrated, preferably under reduced pressure, and the resulting aqueous phase is extracted with a water-immiscible solvent. A preferred water-immiscible solvent for this purpose is methylene dichloride although chloroform, carbon tetrachloride, benzene, n-butanol and the like may also be used. The latter extract is concentrated, preferably under reduced pressure, to afford the crude product as an oil.
The product may be purified further by a variety of methods. Among the preferred methods of purification is to dissolve the crude product in a substantially nonpolar, first solvent, for example petroleum ether or hexane, and to treat the resulting solution with a suit able absorbent, for example charcoal or silica gel, so that the antibiotic becomes absorbed on the absorbant. The absorbant is then separated and washed or eluted with a second solvent more polar than the first solvent, for example ethyl acetate, methylene dichloride, or a mixture of methylene dichloride and ether (preferred). Thereafter, concentration of the wash solution or eluate affords substantially pure rapamycin. Further purification is obtained by partial precipitation with a nonpolar solvent, for example, petroleum ether, hexane, pentane and the like, from a solution of the rapamycin in a more polar solvent, for example, ether, ethyl acetate, benzene and the like. Still-further purification is obtained by column chromatography, preferably employing silica gel, and by crystallization of the rapamycin from ether.
In another preferred embodiment of this invention a first stage inoculum of S treptomyces hygroscopicus NRRL 5491 is prepared in small batches in a medium containing soybean flour, glucose, ammonium sulfate, and calcium carbonate incubated at about 25C at pH 7.l-7.3 for 24 hrs. with agitation, preferably on a gyrotary shaker. The growth thus obtained is used to inoculate a number of somewhat larger batches of the same medium as described above which are incubated at about 25C and pH 7.1-7.3 for 18 hrs. with agitation, preferably on a reciprocating’shaker, to obtain a sec- “ond stagc inoculum which is used to inoculate the production stage fermenters.
6 5.86′.2.-The fermenters are inoculated with the second stage inoculum described above and incubated at about 25C with’ agitationand aeration while controlling and ‘mai’ntaining the mixture at approximately pH 6.0 by
addition offa base, for example, sodium hydroxide, potassium hydroxide or preferably ammonium hydroxide, as required from time to time. Addition of a source -of assimilable carbon, preferably glucose, is started when theconcentrationof the latter in the broth has dropped to about 0.5% wt/vol, normally about 48 hrs after. the start of fermentation, and is maintained until the end ofthe particular run. In this manner a fermentation broth containing about 60 ug/ml of rapamycin as determined by the assay method described above is obtained in 45 days, when fermentation is stopped.
‘ Filtration of the’mycelium, mixing the latter with a watef-miscible ‘lower’ alkanol, preferably methanol, followed by extraction with a halogenated aliphatic hydrocarbon, preferably trichloroethane, and evaporation of the solvents yields a first oily residue. This first oily residue is dissolved in a lower aliphatic ketone, preferably acetone, filtered from insoluble impurities, the filtrate evaporated to yield a second oily residue which is extractedjwith a water-miscible lower alkanol,
preferably methanol, and the latter extract is evaporated to yield crude rapamycin as a third oily residue. This third oily residue is dissolved in a mixture of a lower aliphatic ketone and a lower aliphatic hydrocarbon, preferably acetone-hexane, an absorbent such as charcoal or preferably silica gel is added to adsorb the rapamycin, the latter is eluted from the adsorbate with a similar but more polar solvent mixture, for example a mixture as above but containing a higher proportion of the aliphatic ketone, the eluates are evaporated and the residue is crystallized from diethyl ether, to yield pure crystalline rapamycin. In this manner a total of 45-5 8% of the rapamycin initially present in the fermentation mixture is recovered as pure crystalline rapamycin.
CHARACTERIZATION solvent systems; for example, ether-hexane 40:60 (Rf 0.42), ‘isopropyl alcoholvbenzene 15:85 (Rf= 0.5) and ethanol-benzene 20:80 (Rf f 0.43);
d. rapamycin obtained from four successive fermentation batchesgave the following values on repeated The production stage fermenters are equipped with 7 devices for controlling and maintaining pH at a predetermined level and for continuous metered addition of elemental analyses:
AVER- e. rapamycin exhibits the following characteristic absorption maxima in its ultraviolet absorption spectrum ethanol):
f. the infrared absorption spectrum of rapamycin in chloroform is reproduced in FIG. 1 and shows characteristic absorption bands at 3560, 3430, 1730, 1705 and 1630-1610 cm;
Further infrared absorption bands are characterized by the following data given in reciprocal centimeters with (s) denoting a strong, (m) denoting a medium, and (w) denoting a weak intensity band. This classification is arbitrarily selected in such a manner that a band is denoted as strong (s) if its peak absorption is more than two-thirds of the background in the same region; medium (m) if its peak is between one-third and twothirds of the background in the same region; and weak (w) if its peak is less than one-third of the background in the same region.
2990 cm (m) 1158 cm” (m) 2955 cm (s) 1129 cm (s) 2919 cm (s) 1080 cm (s) 2858 cm (s) 1060 cm (s) 2815 cm (m) 1040 cm (m) 1440 cm (s) 1020 crn’ (m) 1365 cm (m) 978 cm” (s) 1316 cm (in) 905 cm (m) 1272 cm (m) 888 cm” (w) 1178 cm (s) 866 cm- (w) g. the nuclear magnetic resonance spectrum of rapamycinin deuterochloroform is reproduced in FIG. 2; SEE PATENT
CLAIMS
l. Rapamycin, an antibiotic which a. is a colourless, crystalline compound with a melting point of 183 to l8SC, after recrystallization from ether;
b. is soluble in ether, chloroform, acetone, methanol and dimethylformamide, very sparingly soluble in hexane and petroleum ether and substantially insoluble in water;
c. shows a uniform spot on thin layer plates of silica gel”,
d. has a characteristic elemental analysis of about C,
e. exhibits the following characteristic absorption maxima in its ultraviolet absorption spectrum (95% ff has ‘a characteristic infrared absorption spectrum shown in accompanying FIG. 1; SEE PATENT
……………………………………………..
Rapamycin synthetic studies. 1. Construction of the C(27)-C(42) subunit. Tetrahedron Lett 1994, 35, 28, 4907

A partial synthesis of rapamycin has been reported: The condensation of sulfone (I) with epoxide (II) by means of butyllithium followed by desulfonation with Na/Hg gives the partially protected diol (III), which is treated with methanesulfonyl chloride and NaH to afford the epoxide (IV). Ring opening of epoxide (IV) with LiI and BF3.Et2O followed by protection of the resulting alcohol with PMBOC(NH)CCl3 yields the primary iodo compound (V). The condensation of (V) with the fully protected dihydroxyaldehyde (VI) (see later) by means of butyllithium in THF/HMPT gives the fully protected trihydroxyketone (VII), which is hydrolyzed with camphorsulfonic acid (CSA) to the corresponding gemdiol and reprotected with pivaloyl chloride (the primary alcohol) and tert-butyldimethylsilyl trifluoromethanesulfonate (the secondary alcohol), yielding a new fully protected trihydroxyketone (VIII). Elimination of the pivaloyl group with DIBAL and the dithiane group with MeI/CaCO3 affords the hydroxyketone (IX), which is finally oxidized with oxalyl chloride to the ketoaldehyde (X), the C(27)-C(42) fragment [the C(12)-C(15) fragment with the C(12)-substituent based on the IUPAC nomenclature recommendations]. The fully protected dihydroxyaldehyde (VI) is obtained as follows: The reaction of methyl 3-hydroxy-2(R)-methylpropionate (XI) with BPSCl followed by reduction with LiBH4 to the corresponding alcohol and oxidation with oxalyl chloride gives the aldehyde (XII), which is protected with propane-1,3-dithiol and BF3.Et2O to afford the dithiane compound (XIII). Elimination of the silyl group with TBAF followed by esterification with tosyl chloride, reaction with NaI and, finally, with sodium phenylsulfinate gives the sulfone (XIV), which is condensed with the partially protected dihydroxyaldehyde (XV), oxidized with oxalyl chloride and desulfonated with Al/Hg to afford the dithianyl ketone (XVI). The reaction of (XVI) with lithium hexamethyldisilylazane gives the corresponding enolate, which is treated with dimethyllithium cuprate to yield the fully protected unsaturated dihydroxyaldehyde (VI).
……………………………………………
http://www.google.com/patents/US8088789
JUT HAVE A LOOK
……………………………
The Ley Synthesis of Rapamycin
Rapamycin (3) is used clinically as an immunosuppressive agent. The synthesis of 3 (Angew. Chem. Int. Ed. 2007, 46, 591. DOI: 10.1002/anie.200604053) by Steven V. Ley of the University of Cambridge was based on the assembly and subsequent coupling of the iododiene 1 and the stannyl alkene 2.

The lactone of 1 was prepared by Fe-mediated cyclocarbonylation of the alkenyl epoxide 5, following the protocol developed in the Ley group.

The cyclohexane of 2 was constructed by SnCl4-mediated cyclization of the allyl stannane 9, again employing a procedure developed in the Ley group. Hydroboration delivered the aldehyde 11, which was crotylated with 12, following the H. C. Brown method. The alcohol so produced (not illustrated) was used to direct the diastereoselectivity of epoxidation, then removed, to give 13. Coupling with 14 then led to 2.

Combination of 1 with 2 led to 15, which was condensed with catechol to give the macrocycle 16. Exposure of 16 to base effected Dieckmann cyclization, to deliver the ring-contracted macrolactone 17, which was carried on to (-)-rapamycin (3).

|
|
……………………………….
Total Synthesis of Rapamycin
Angewandte Chemie International Edition

PREVIEW THIS ARTICLE WITH READCUBE

http://www.readcube.com/articles/10.1002%2Fanie.200604053?r3_referer=wol
……………………..

Ley, Maddess, Tackett, Watanabe, Brennan, Spilling, Scott and Osborn. ACIEE, 2006, EarlyView. DOI:10.1002/anie.200604053.
It’s been in the works for quite a while, but Steve Ley’s synthesis of Rapamycin has just been published. This complex beast has a multitude of biological activities, including an interesting immunosuppressive profile, resulting in clinical usage following organ transplantation. So, unsurprisingly, it’s been the target of many projects, with complete total syntheses published by Smith, Danishefsky, Schreiber and KCN.
So what makes this one different? Well, it does have one of the most interesting macrocyclisations I’ve seen since Jamison’s paper, and a very nice demonstration of the BDA-aldol methodology. The overall strategy is also impressive, so on with the retro:

First stop is the BDA-aldol; this type of chemistry is interesting, because the protecting group for the diol is also the stereo-directing group. The stereochemistry for this comes from a glycolic acid, and has been usedin this manner by the group before. The result is as impressive as ever, with a high yield, and presumably a very high d.r. (no mention of actual numbers).

The rest of the fragment synthesis was completed in a succinct and competent manner, but using relatively well known chemistry. However, I was especially impressed with the macrocyclisation I mentioned:

Tethering the free ends of the linear precursor with a simple etherification/esterification onto catechol gave then a macrocycle holding the desired reaction centres together. Treatment of this with base then induces a Dieckmann-condensation type cyclisation to deliver the desired macrocycle. Of course, at this stage, only a few more steps were required to complete the molecule, and end an era of the Wiffen Lab.
………………………………
Drugs Fut 1999, 24(1): 22
DOI: 10.1358/dof.1999.024.01.474036

REFERENCES
- Vézina C, Kudelski A, Sehgal SN (October 1975). “Rapamycin (AY-22,989), a new antifungal antibiotic”. J. Antibiot. 28 (10): 721–6. doi:10.7164/antibiotics.28.721. PMID 1102508.
- Pritchard DI (2005). “Sourcing a chemical succession for cyclosporin from parasites and human pathogens”. Drug Discovery Today 10 (10): 688–691. doi:10.1016/S1359-6446(05)03395-7. PMID 15896681.
5. Hybrid inhibitors of phosphatidylinositol 3-kinase (PI3K) and the mammalian target of rapamycin (mTOR): design, synthesis, and superior antitumor activity of novel wortmannin-rapamycin conjugates.
Ayral-Kaloustian S, Gu J, Lucas J, Cinque M, Gaydos C, Zask A, Chaudhary I, Wang J, Di L, Young M, Ruppen M, Mansour TS, Gibbons JJ, Yu K.
J Med Chem. 2010 Jan 14;53(1):452-9. doi: 10.1021/jm901427g.
8 Total synthesis of rapamycin.
Ley SV, Tackett MN, Maddess ML, Anderson JC, Brennan PE, Cappi MW, Heer JP, Helgen C, Kori M, Kouklovsky C, Marsden SP, Norman J, Osborn DP, Palomero MA, Pavey JB, Pinel C, Robinson LA, Schnaubelt J, Scott JS, Spilling CD, Watanabe H, Wesson KE, Willis MC.
Chemistry. 2009;15(12):2874-914. doi: 10.1002/chem.200801656.
13 Total synthesis of rapamycin.
Maddess ML, Tackett MN, Watanabe H, Brennan PE, Spilling CD, Scott JS, Osborn DP, Ley SV.
Angew Chem Int Ed Engl. 2007;46(4):591-7. No abstract available.
16 CCI-779 Wyeth.
Elit L.
Curr Opin Investig Drugs. 2002 Aug;3(8):1249-53. Review.
17 Everolimus. Novartis.
Dumont FJ.
Curr Opin Investig Drugs. 2001 Sep;2(9):1220-34. Review.
18 Kuo et al (1992) Rapamycin selectively inhibits interleukin-2 activation of p70 S6 kinase. Nature 358 70. PMID:1614535.
19 Huang et al (2003) Rapamycins: mechanism of action and cellular resistance. Cancer Biol.Ther. 2 221. PMID:12878853.
20 Kobayashi et al (2007) Rapamycin, a specific inhibitor of the mammalian target of rapamycin, suppresses lymphangiogenesis and lymphatic metastasis. Cancer Sci. 98 726. PMID: 17425689.
21 Fleming et al (2011) Chemical modulators of autophagy as biological probes and potential therapeutics. 7 9. PMID:21164513.
22 J Am Chem Soc1993,115,(10):4419
23 Tetrahedron Lett1994,35,(28):4911
24 Chemistry (Weinheim)1995,1,(5):318
24
 SIROLIMUS
SIROLIMUS
FEMALE FERTILITY
http://amcrasto.theeurekamoments.com/2013/02/11/immunosuppressant-drug-rapamycin-helps-preserving-female-fertility/
PATENTS
| Canada |
2293793 |
APPROVED2006-07-11 |
EXP 2018-06-11 |
| Canada |
2103571 |
2003-04-29 |
2012-02-21 |
| United States |
5989591 |
1998-09-11 |
2018-09-11 |
| United States |
5212155 |
1993-05-18 |
2010-05-18 |
| WO1998054308A2 * |
May 28, 1998 |
Dec 3, 1998 |
Biotica Tech Ltd |
Polyketides and their synthesis and use |
| EP0589703A1 * |
Sep 23, 1993 |
Mar 30, 1994 |
American Home Products Corporation |
Proline derivative of rapamycin, production and application thereof |
| US20010039338 * |
Jun 7, 2001 |
Nov 8, 2001 |
American Home Products Corporation |
Regioselective synthesis of rapamycin derivatives |
| WO2007067560A2 * |
Dec 6, 2006 |
Jun 14, 2007 |
Clifford William Coughlin |
Scalable process for the preparation of a rapamycin 42-ester from a rapamycin 42-ester boronate |
| WO2012131019A1 |
Mar 30, 2012 |
Oct 4, 2012 |
Sandoz Ag |
Regioselective acylation of rapamycin at the c-42 position |
| US7622578 |
Dec 6, 2006 |
Nov 24, 2009 |
Wyeth |
Scalable process for the preparation of a rapamycin 42-ester from a rapamycin 42-ester boronate |
| US3929992 |
Apr 12, 1974 |
Dec 30, 1975 |
Ayerst Mckenna & Harrison |
Rapamycin and process of preparation |
| US5646160 |
May 26, 1995 |
Jul 8, 1997 |
American Home Products Corporation |
Method of treating hyperproliferative vascular disease with rapamycin and mycophenolic acid |
| US5665772 |
Sep 24, 1993 |
Sep 9, 1997 |
Sandoz Ltd. |
O-alkylated rapamycin derivatives and their use, particularly as immunosuppressants |
| US5728710 |
Jul 16, 1993 |
Mar 17, 1998 |
Smithkline Beecham Corporation |
Rapamycin derivatives |
| US5957975 |
Dec 15, 1997 |
Sep 28, 1999 |
The Centre National De La Recherche Scientifique |
Stent having a programmed pattern of in vivo degradation |
| US5985890 |
Jun 5, 1996 |
Nov 16, 1999 |
Novartis Ag |
Rapamycin derivatives |
| US6001998 |
Oct 13, 1995 |
Dec 14, 1999 |
Pfizer Inc |
Macrocyclic lactone compounds and their production process |
| US6015815 |
Sep 24, 1998 |
Jan 18, 2000 |
Abbott Laboratories |
Tetrazole-containing rapamycin analogs with shortened half-lives |
| US6187568 |
Aug 20, 1999 |
Feb 13, 2001 |
Pfizer Inc |
Macrocyclic lactone compounds and their production process |
| US6273913 |
Apr 16, 1998 |
Aug 14, 2001 |
Cordis Corporation |
Modified stent useful for delivery of drugs along stent strut |
| US6585764 |
Jun 4, 2001 |
Jul 1, 2003 |
Cordis Corporation |
Stent with therapeutically active dosage of rapamycin coated thereon |
| US6641611 |
Nov 26, 2001 |
Nov 4, 2003 |
Swaminathan Jayaraman |
Therapeutic coating for an intravascular implant |
| US6805703 |
Sep 18, 2001 |
Oct 19, 2004 |
Scimed Life Systems, Inc. |
Protective membrane for reconfiguring a workpiece |
| US7025734 |
Sep 28, 2001 |
Apr 11, 2006 |
Advanced Cardiovascular Systmes, Inc. |
Guidewire with chemical sensing capabilities |
| US7056942 |
Jan 16, 2004 |
Jun 6, 2006 |
Teva Pharmaceutical Industries Ltd. |
Carvedilol |
| US7820812 * |
Jul 23, 2007 |
Oct 26, 2010 |
Abbott Laboratories |
Methods of manufacturing crystalline forms of rapamycin analogs |
| US20010027340 |
Jun 4, 2001 |
Oct 4, 2001 |
Carol Wright |
Stent with therapeutically active dosage of rapamycin coated thereon |
| US20010029351 |
May 7, 2001 |
Oct 11, 2001 |
Robert Falotico |
Drug combinations and delivery devices for the prevention and treatment of vascular disease |
| US20020005206 |
May 7, 2001 |
Jan 17, 2002 |
Robert Falotico |
Antiproliferative drug and delivery device |
| US20020007213 |
May 7, 2001 |
Jan 17, 2002 |
Robert Falotico |
Drug/drug delivery systems for the prevention and treatment of vascular disease |
| US20020082680 |
Sep 7, 2001 |
Jun 27, 2002 |
Shanley John F. |
Expandable medical device for delivery of beneficial agent |
| US20020123505 |
Sep 10, 2001 |
Sep 5, 2002 |
Mollison Karl W. |
Medical devices containing rapamycin analogs |
| US20030129215 |
Sep 6, 2002 |
Jul 10, 2003 |
T-Ram, Inc. |
Medical devices containing rapamycin analogs |
| US20040072857 |
Jul 2, 2003 |
Apr 15, 2004 |
Jacob Waugh |
Polymerized and modified rapamycins and their use in coating medical prostheses |
| US20050033417 |
Jul 1, 2004 |
Feb 10, 2005 |
John Borges |
Coating for controlled release of a therapeutic agent |
| US20050101624 |
Nov 12, 2003 |
May 12, 2005 |
Betts Ronald E. |
42-O-alkoxyalkyl rapamycin derivatives and compositions comprising same |
| US20050152842 |
Dec 22, 2004 |
Jul 14, 2005 |
Chun Li |
Poly (L-glutamic acid) paramagnetic material complex and use as a biodegradable MRI contrast agent |
| US20050175660 |
Oct 29, 2004 |
Aug 11, 2005 |
Mollison Karl W. |
Medical devices containing rapamycin analogs |
| US20050208095 |
Nov 22, 2004 |
Sep 22, 2005 |
Angiotech International Ag |
Polymer compositions and methods for their use |
| US20050209244 |
Feb 27, 2003 |
Sep 22, 2005 |
Prescott Margaret F |
N{5-[4-(4-methyl-piperazino-methyl)-benzoylamido]-2-methylphenyl}-4-(3-pyridyl)-2-pyrimidine-amine coated stents |
| US20050239178 |
Apr 25, 2005 |
Oct 27, 2005 |
Wyeth |
Labeling of rapamycin using rapamycin-specific methylases |
| US20060094744 |
Sep 28, 2005 |
May 4, 2006 |
Maryanoff Cynthia A |
Pharmaceutical dosage forms of stable amorphous rapamycin like compounds |
| US20060229711 |
Apr 4, 2006 |
Oct 12, 2006 |
Elixir Medical Corporation |
Degradable implantable medical devices |
| US20070015697 |
Nov 1, 2005 |
Jan 18, 2007 |
Peyman Gholam A |
Enhanced ocular neuroprotection and neurostimulation |
| US20070059336 |
Feb 27, 2006 |
Mar 15, 2007 |
Allergan, Inc. |
Anti-angiogenic sustained release intraocular implants and related methods |
| US20070207186 |
Mar 3, 2007 |
Sep 6, 2007 |
Scanlon John J |
Tear and abrasion resistant expanded material and reinforcement |
| US20080086198 |
May 24, 2007 |
Apr 10, 2008 |
Gary Owens |
Nanoporous stents with enhanced cellular adhesion and reduced neointimal formation |
| EP1236478A1 |
Feb 27, 2002 |
Sep 4, 2002 |
Medtronic Ave, Inc. |
Peroxisome proliferator-activated receptor gamma ligand eluting medical device |
| EP1588727A1 |
Apr 20, 2005 |
Oct 26, 2005 |
Cordis Corporation |
Drug/drug delivery systems for the prevention and treatment of vascular disease |
| WO1993016189A1 |
Feb 11, 1993 |
Aug 19, 1993 |
Pfizer |
Novel macrocyclic lactones and a productive strain thereof |
| WO1994009010A1 |
Sep 24, 1993 |
Apr 28, 1994 |
Sandoz Ag |
O-alkylated rapamycin derivatives and their use, particularly as immunosuppressants |
| WO1996041807A1 |
Jun 5, 1996 |
Dec 27, 1996 |
Sylvain Cottens |
Rapamycin derivatives |
| WO1998007415A2 |
Aug 18, 1997 |
Feb 26, 1998 |
Ciba Geigy Ag |
Methods for prevention of cellular proliferation and restenosis |
| WO2001087263A2 |
May 14, 2001 |
Nov 22, 2001 |
Cordis Corp |
Delivery systems for treatment of vascular disease |
| WO2001087342A2 |
May 14, 2001 |
Nov 22, 2001 |
Cordis Corp |
Delivery devices for treatment of vascular disease |
| WO2001087372A1 |
Apr 25, 2001 |
Nov 22, 2001 |
Cordis Corp |
Drug combinations useful for prevention of restenosis |
| WO2001087373A1 |
May 14, 2001 |
Nov 22, 2001 |
Cordis Corp |
Delivery devices for treatment of vascular disease |
| WO2001087374A1 |
May 14, 2001 |
Nov 22, 2001 |
Cordis Corp |
Delivery systems for treatment of vascular disease |
| WO2001087375A1 |
May 14, 2001 |
Nov 22, 2001 |
Cordis Corp |
Delivery devices for treatment of vascular disease |
| WO2001087376A1 |
May 14, 2001 |
Nov 22, 2001 |
Cordis Corp |
Drug/drug delivery systems for the prevention and treatment of vascular disease |
| WO2002056790A2 |
Dec 18, 2001 |
Jul 25, 2002 |
Avantec Vascular Corp |
Delivery of therapeutic capable agents |
| WO2002065947A2 |
Feb 18, 2002 |
Aug 29, 2002 |
Jomed Gmbh |
Implants with fk506 for prophylaxis and treatment of restonoses |
| WO2003064383A2 |
Feb 3, 2003 |
Aug 7, 2003 |
Ariad Gene Therapeutics Inc |
Phosphorus-containing compounds & uses thereof |
| WO2006116716A2 |
Apr 27, 2006 |
Nov 2, 2006 |
William A Dunn |
Materials and methods for enhanced degradation of mutant proteins associated with human disease |

A plaque, written in Brazilian Portuguese, commemorating the discovery of sirolimus on Easter Island, near Rano Kau
mTOR inhibitor
temsirolimus (CCI-779), everolimus (RAD001), deforolimus (AP23573), AP21967, biolimus, AP23102, zotarolimus (ABT 578), sirolimus (Rapamune), and tacrolimus (Prograf).













 The presentation will load below
The presentation will load below




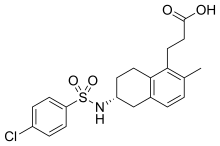
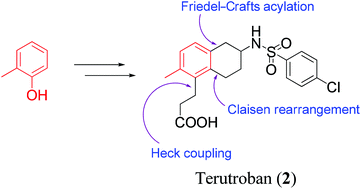
![3-[(6R)-6-[(4-chlorophenyl)sulfonylamino]-2-methyl-5,6,7,8-tetrahydronaphthalen-1-yl]propanoic acid NMR spectra analysis, Chemical CAS NO. 165538-40-9 NMR spectral analysis, 3-[(6R)-6-[(4-chlorophenyl)sulfonylamino]-2-methyl-5,6,7,8-tetrahydronaphthalen-1-yl]propanoic acid H-NMR spectrum](http://pic11.molbase.net/nmr/nmr_image/2014-11-09/001/775/1775661_1h.png)
![3-[(6R)-6-[(4-chlorophenyl)sulfonylamino]-2-methyl-5,6,7,8-tetrahydronaphthalen-1-yl]propanoic acid NMR spectra analysis, Chemical CAS NO. 165538-40-9 NMR spectral analysis, 3-[(6R)-6-[(4-chlorophenyl)sulfonylamino]-2-methyl-5,6,7,8-tetrahydronaphthalen-1-yl]propanoic acid C-NMR spectrum](http://pic11.molbase.net/nmr/nmr_image/2014-11-09/001/775/1775661_13c.png)





)/Images/CSIRlogo.gif)
)/Images/iictlogo.gif)
)/Images/iict70logo.gif)
 DR AV RAMA RAO
DR AV RAMA RAO Professor J. R. Falck
Professor J. R. Falck Professor L. F. Tietze
Professor L. F. Tietze


