The ICH Q11 Guideline describing approaches to developing and understanding the manufacturing process of drug substances was finalised in May 2012. Since then the pharmaceutical industry and the drug substance manufacturers had time to get familiar with the principles outlined in this guideline. However, experience has shown that there is some need for clarification. Thus the Q11 Implementation Working Group recently issued a Questions and Answers Document.

The ICH Q11 Guideline describes approaches to developing and understanding the manufacturing process of drug substances. It was finalised in May 2012 and since then the pharmaceutical industry and the drug substance manufacturers had time to get familiar with the principles outlined in this guideline. However, experiences during implementation of these principles within this 4 years period have shown that there is need for clarification in particular with regard to the selection and justification of starting materials.
On 30 November 2016 the ICH published a Questions and Answers document “Development and Manufacture of Drug Substances (Chemical Entities and Biotechnological/Biological Entities)” which was developed by the Q11 Implementation Working Group. This document aims at addressing the most important ambiguities with respect to starting materials and at promoting a harmonised approach for their selection and justification as well as the information that should be provided in marketing authorisation applications and/or Drug Master Files.
In the following some examples of questions and answers from this document:
Question:
ICH Q11 states that “A starting material is incorporated as a significant structural fragment into the structure of the drug substance.” Why then are intermediates used late in the synthesis, which clearly contain significant structural fragments, often not acceptable as starting materials?
Answer:
The selection principle about “significant structural fragment” has frequently been misinterpreted as meaning that the proposed starting material should be structurally similar to the drug substance. However, as stated in ICH Q11, the principle is intended to help distinguish between reagents, catalysts, solvents, or other raw materials (which do not contribute a “significant structural fragment” to the molecular structure of the drug substance) from materials that do. … The presence of a “significant structural fragment” should not be the sole basis for of starting material selection. Starting materials justified solely on the basis that they are a “significant structural fragment” probably will not be accepted as starting materials by regulatory authorities, as the other principles for the appropriate selection of a proposed starting material also require consideration.
Question:
Do the ICH Q11 general principles for selection of starting materials apply to processes where multiple chemical transformations are run without isolation of intermediates?
Answer:
Yes. The ICH Q11 general principles apply to processes where multiple chemical transformations are run without isolation of intermediates. In the absence of such isolations (e.g., crystallization, precipitations), other unit operations (e.g., extraction, distillation, the use of scavenging agents) should be in place to adequately control impurities and be described in the application. The drug substance synthetic process should include appropriate unit operations that purge impurities.
The ICH Q11 general principles also apply for sequential chemical transformations run continuously. Non isolated intermediates are generally not considered appropriate starting materials.
Question:
Is a “starting material” as described in ICH Q11 the same as an “API starting material” as described in ICH Q7?
Answer:
Yes. ICH Q11 states that the Good Manufacturing Practice (GMP) provisions described in ICH Q7 apply to each branch of the drug substance manufacturing process beginning with the first use of a “starting material”. ICH Q7 states that appropriate GMP (as defined in that guidance) should be applied to the manufacturing steps immediately after “API starting materials” are entered into the process … . Because ICH Q11 sets the applicability of ICH Q7 as beginning with the “starting material”, and ICH Q7 sets the applicability of ICH Q7 as beginning with the “API starting material”, these two terms are intended to refer to the same material.
ICH Q7 states that an “API Starting Material” is a raw material, intermediate, or an API that is used in the production of an API. ICH Q7 provides guidance regarding good manufacturing practices for the drug substance; however, it does not provide specific guidance on the selection and justification of starting materials. When a chemical, including one that is also a drug substance, is proposed to be a starting material, all ICH Q11 general principles still need to be considered.
With the recent publication of this draft Q&A Document with the complete title “Development and Manufacture of Drug Substances (Chemical Entities and Biotechnological/Biological Entities) Questions and Answers (regarding the selection and justification of starting materials)” on the ICH website it reached Step 2b of the ICH Process and now enters the consultation period. Comments may be provided by e-mailing to the ICH Secretariat at admin@ich.org.


extra info…………
A PRESENTATION
Ever since the FDA issued its landmark guidance Pharmaceutical GMPs-A Risk Based Approach in 2004, the industry has been struggling with how to demonstrate process understanding as a basis for quality. Bolstered by guidance from ICH, specifically Q6-Q10, the pieces have long been in place to build a solution that is philosophically consistent with these best practice principles. Even so, the evolution to process understanding as a basis for quality has been slow. Pressure to accelerate this transformation spiked in 2011 when the FDA issued its new guidance on process validation that basically mandated the core components of ICH Q6-10 as part of Stages 1 and 2. To be fair, enforcement has been uneven and that fact has further impeded adoption, with the compliance inspectors themselves struggling to acquire the necessary skills to fully evaluate statistical arguments of process control and predictability.
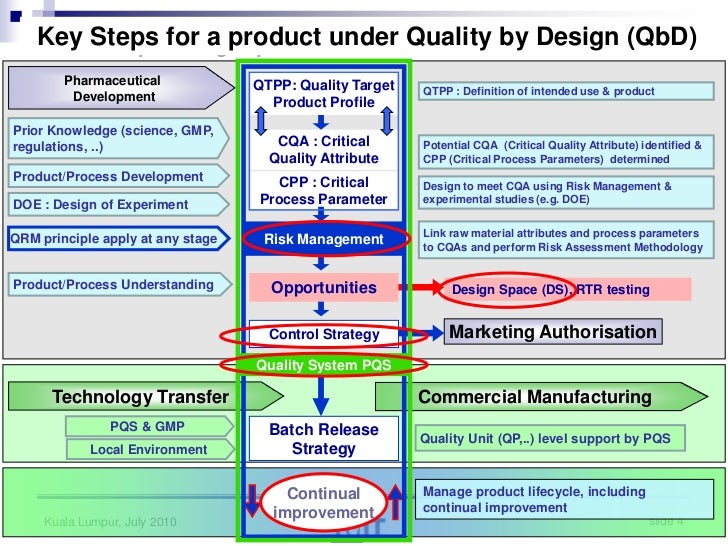
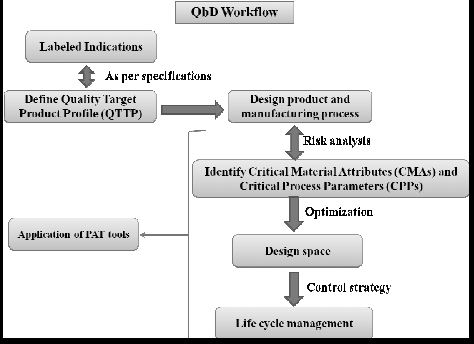
One area debated since 2008 is the application of GMPs and demonstration of control for drug substances. Drug substance suppliers and drug product manufacturers have used the tenets of ICH Q7A as the foundation for deciding where GMPs can be reasonably implemented, to establish the final intermediate (FI) and the regulatory starting material (RSM). However, the ability to support the quality of the drug substance has a profound impact on the ability to defend the drug product quality. In the last few years it has become apparent that it was not reasonable to apply the same requirements for drug products to drug substances because the processes can be markedly different. In response to this need, the ICH issued a new guidance; Q11: Development and Manufacture of Drug Substances (Chemical Entities and Biotechnological/Biological Entities). The key ICH documents that impact Q11 are shown in Figure 1.
Figure 1. Guidances Impacting ICH Q11.
The FDA formally adopted ICHQ11 in November 2012 and its purpose is two-fold. First, it offers guidance on the information to provide in Module 3 of the Common Technical Document (CTD) Sections 3.2.S.2.2 – 3.2.S.2.6 (ICH M4Q). Second, and perhaps most importantly, it attempts to clarify the concepts defined in the ICH guidelines on Pharmaceutical Development (Q8), Quality Risk Management (Q9), and Pharmaceutical Quality System (Q10) as they pertain to the development and manufacture of drug substances.
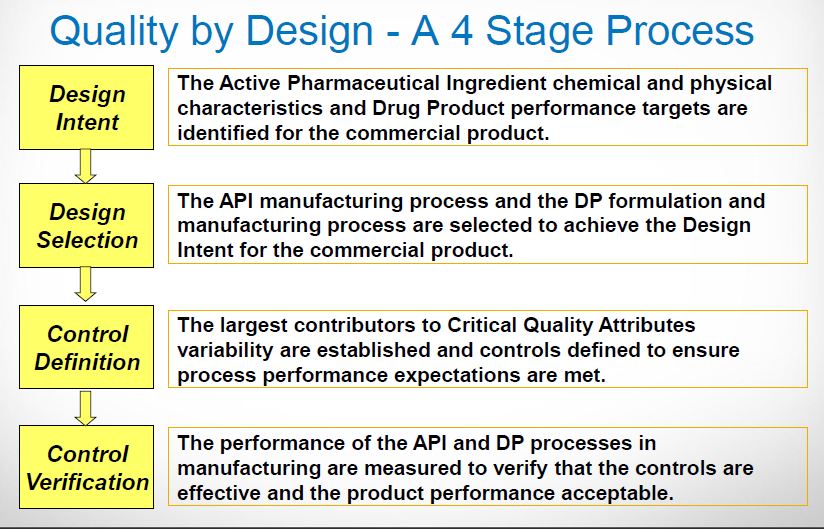
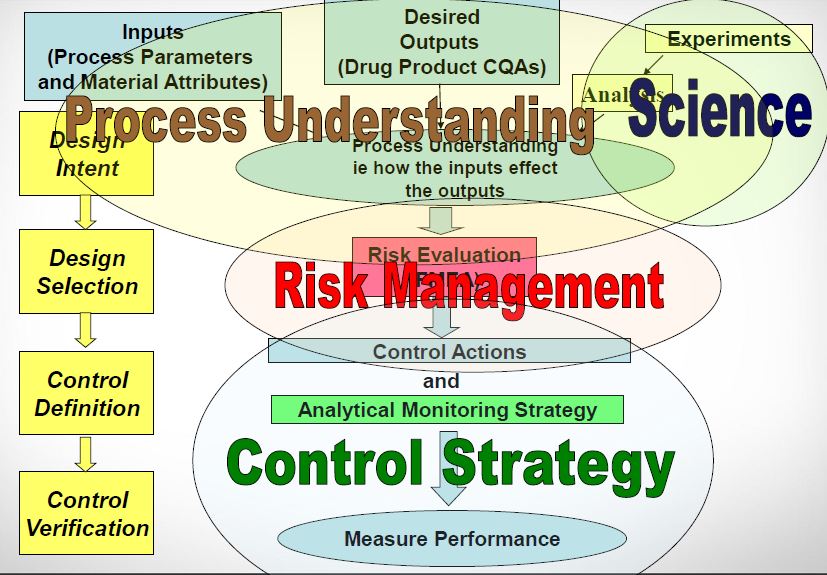
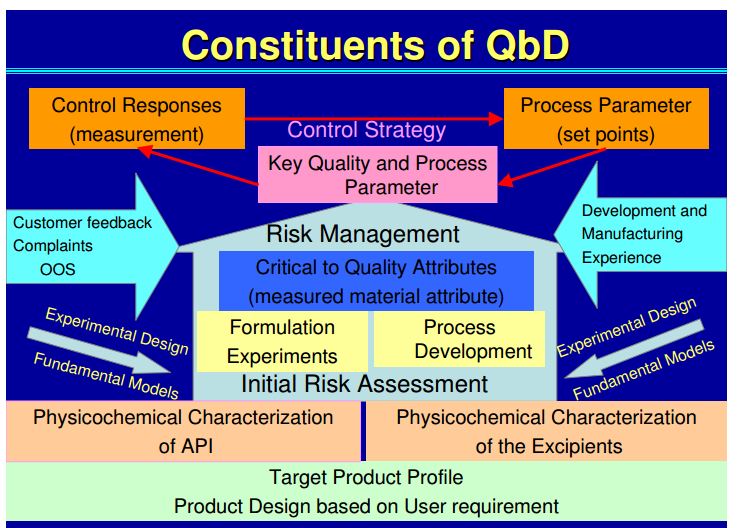
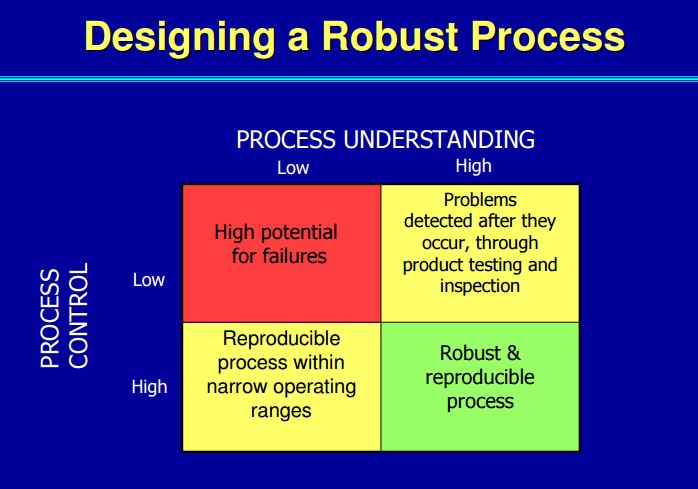
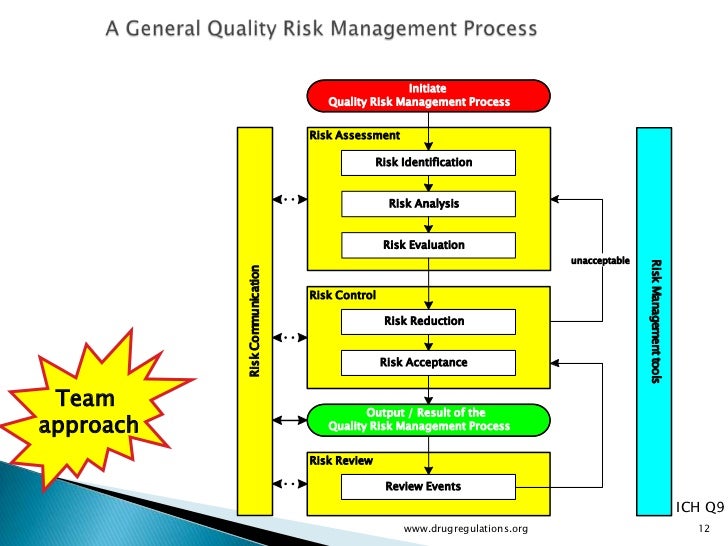
What makes ICH Q11 so important is its emphasis on control strategy. This concept was introduced in ICH Q10 as “a planned set of controls, derived from current product and process understanding that assures process performance and product quality.”
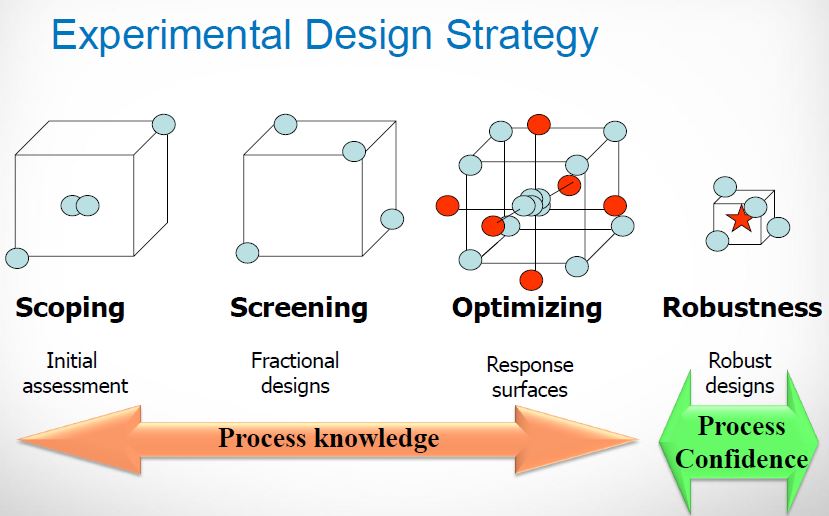
Within the drug product world, the control strategy concept has been elusive as industry grapples with moving from a sample-and-test concept of quality to one of process understanding and behavior. This concept is even more removed for drug substance manufacturers and, in some cases, is more difficult to implement. But Q11 is much more than a mere framework for control strategy. The guidance is structured very similarly to the concepts discussed in the new 2011 Process Validation guidance. Looking closely, Q11 addresses:
• Product Design/Risk Assessment/CQA Determination
• Defining the Design Space and establishing a control strategy
• Process validation and analysis
• Information required for Sections 3.2.S.2.2 – 3.2.S.2.6 of the eCTD
• Lifecycle management
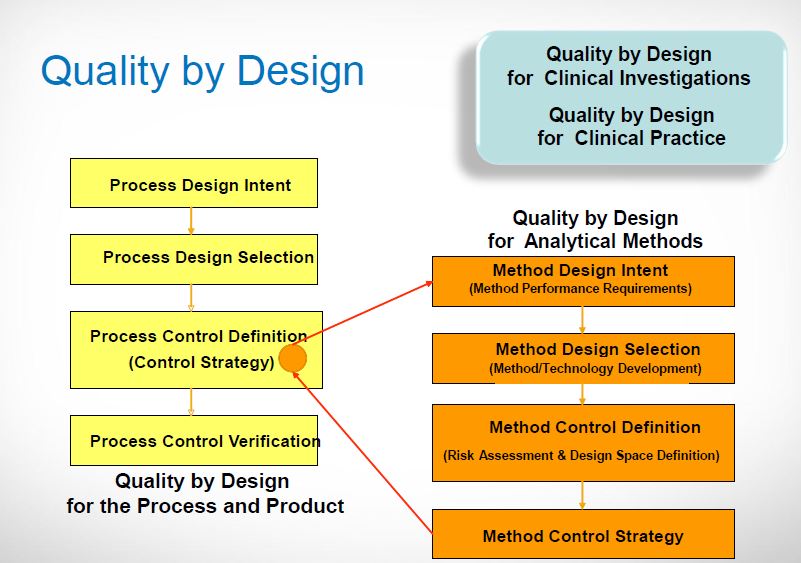
Product design/Risk assessment/CQA determination
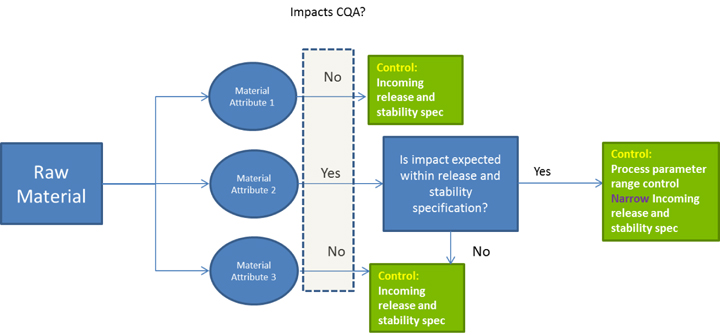
Within the context of process development, the guidance defines similar considerations to those defined in the Stage 1 activity of Process Validation. Understanding the quality linkage between the drug substance’s physical, chemical, and microbiological characteristics, and the final drug products’ Quality Target Product Profile (QTPP), is the primary objective of the product and process design phase. The product’s QTPP is comprised of the final product Critical to Quality Attributes (CQAs). Identifying the raw material characteristics of the drug substance that can impact the drug product is a critical first step in developing a defensible control strategy. Employing risk analysis tools at the outset can help focus the process development activities upon the unit operations that have the potential to impact the final product’s CQAs. In the case of biological drug substances, any knowledge regarding mechanism of action and biological characterization, such as studies that evaluate structure-function relationships, can contribute to the assessment of risk for some product attributes.
Drug substance CQAs typically include those properties or characteristics that affect identity, purity, biological activity, and stability of the final drug product. In the case of biotechnological/biological products, most of the CQAs of the drug product are associated with the drug substance and thus are a direct result of the design of the drug substance or its manufacturing process. When considering CQAs for the drug substance, it is important to not overlook the impact of impurities because of their potential impact on drug product safety. For chemical entities, these include organic impurities (including potentially mutagenic impurities), inorganic impurities such as metal residues, and residual solvents.
For biotechnological/biological products, impurities may be process-related or product-related (see ICH Q6B). Process-related impurities include: cell substrate-derived impurities (e.g., Host Cell Proteins [HCP] and DNA); cell culture-derived impurities (e.g., media components); and downstream-derived impurities (e.g., column leachable). Determining CQAs for biotechnology/biological products should also include consideration of contaminants, as defined in Q6B, including all adventitiously introduced materials not intended to be part of the manufacturing process (e.g., viral, bacterial, or mycoplasma contamination).
Defining the design space and establishing a control strategy
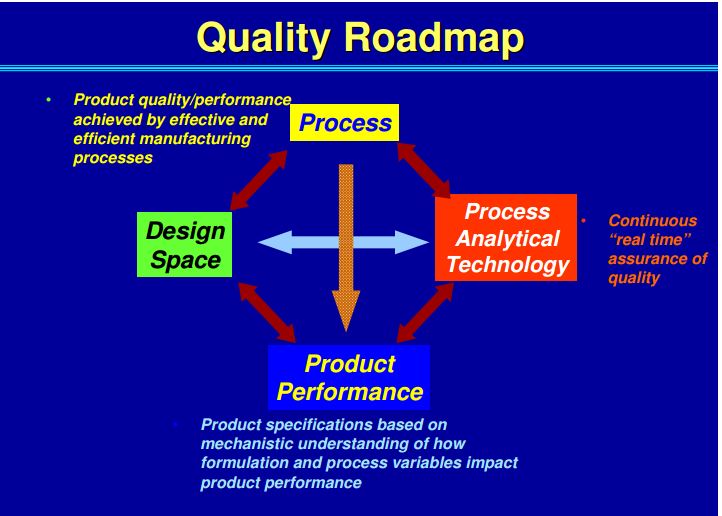
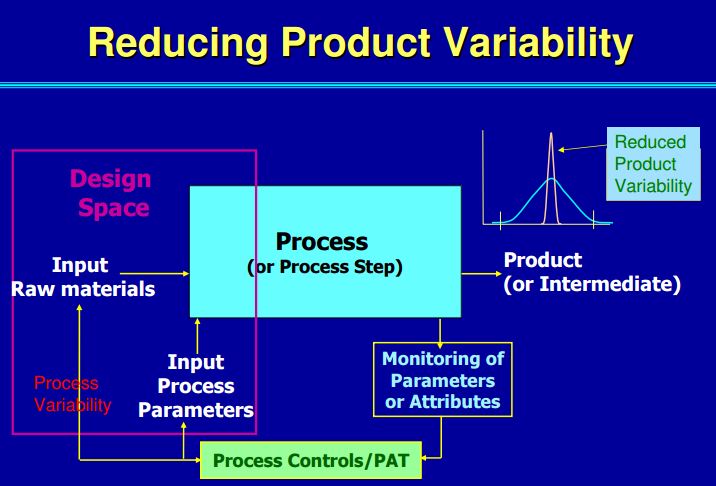
ICH Q8 describes a tiered approach to establishing final processing conditions that consists of moving from the knowledge space to the process design space and finally the control space. ICH Q8 and Q11 define the Design Space as “the multidimensional combination and interaction of input variables (e.g., material attributes) and process parameters that have been demonstrated to provide assurance of quality.” In the drug product world the terminology typically applied to the design space is the Proven Acceptable Range (PAR) that used to equate to the validated range.
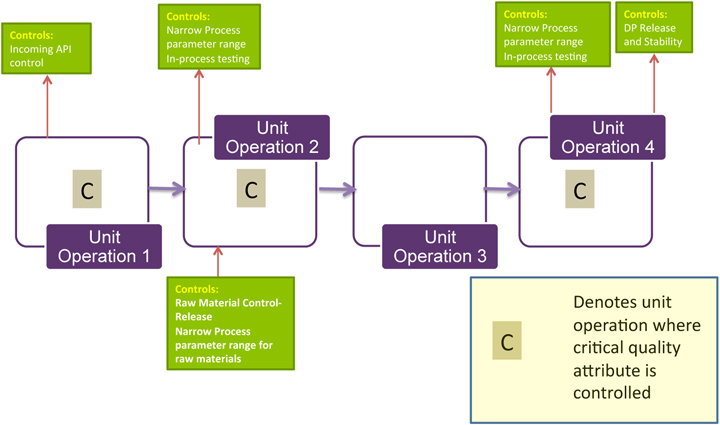
Here is why this is important: the ability to accurately assess the significance and effect of the variability of material attributes and process parameters on drug substance CQAs, and hence the limits of a design space, depends on the extent of process and product understanding. The challenge with drug substance processes is where to apply the characterization. ICH Q7A recognizes that upstream of the RSM does not require GMP control. The design space can be developed based on a combination of prior knowledge, first principles, and/or empirical understanding of the process. A design space might be determined per unit operation (e.g., reaction, crystallization, distillation, purification), or a combination of selected unit operations should generally be selected based on their impact on CQAs.
In developing a control strategy, both upstream and downstream factors should be considered. Starting material characteristics, in-process testing, and critical process parameters variation control are the key elements in a defensible control strategy. For in-process and release testing criteria the resolution of the measurement tool should be considered before making any conclusions.
Process validation
ICH Q11’s description of process validation mimics the same description in ICH Q7A but offers up an alternative for continuous verification that mirrors the concepts in ICH Q8 and the new process validation guidance. As mentioned, the enforcement of the new guidance by the FDA has been uneven, but positioning the process validation to satisfy the new guidance requires the drug substance manufacturer to formally implement characterization and validation standards, just as a drug product manufacturer would be required to do.
Life-cycle management
The quality system elements and management responsibilities described in ICH Q10 are intended to encourage the use of science-based and risk-based approaches at each lifecycle stage, thereby promoting continual improvement across the entire product lifecycle. There should be a systematic approach to managing knowledge related to both drug substance and its manufacturing process throughout the lifecycle. This knowledge management should include but not be limited to process development activities, technology transfer activities to internal sites and contract manufacturers, process validation studies over the lifecycle of the drug substance, and change management activities.
Conclusion
The new ICH Q11 guidance represents the most recent example of the FDA’s commitment to the principles of QbD to define an integrated framework for implementing the principles of ICH Q6-Q10. Although the guidance does not mandate adopting ICH Q8, the considerations required to create a defensible control strategy require a much higher level of process understanding than the conventional approach of sample and test, once the foundation of product development. Defining the requirements is another example of where the FDA is going in terms of expectations for drug substance and drug product understanding. If effectively enforced, this can be a significant step forward, pushing the industry toward a QbD philosophy for process and product development.
/////////Selection, justification, starting materials, ICH Q11 , ich, qbd








 SAKSHI MODGIL
SAKSHI MODGIL