












GMP……API PILOT PLANT
PRESENTATION
Pilot plant and scale-up techniques are both integral and critical to drug discovery and development process for new medicinal products. A major decision focuses on that point where the idea or process is advanced from a research oriented program targeted towards commercialization.
The speed of drug discovery has been accelerating at an exponential rate. The past two decades particular have witnessed amazing inventions and innovations in pharmaceutical research, resulting in the ability to produce new drugs faster than even before.
The new drug applications (NDAs) and abbreviated new drug applications (ANDA) are all-time high. The preparation of several clinical batches in the pilot plant provides its personnel with the opportunity to perfect and validate the process. Also different types of laboratories have been motivated to adopt new processes and technologies in an effort to stay at the forefront scientific innovation.
MY PRESENTATION
Pharmaceutical pilot plants that can quickly numerous short-run production lines of multiple batches are essential for ensuring success in the clinical testing and bougainvilleas study phases. Drug formulation research time targets are met by having a well-designed facility with the appropriate equipment mix, to quickly move from the laboratory to the pilot plant scale 1. In pilot plant, a formula is transformed into a viable, robust product by the development of a reliable and practical method of manufacture that effects the orderly transition from laboratory to routine processing in a full scale production facility where as the scale up involves the designing of prototype using the data obtained from the pilot plant model.
Pilot plant studies must includes a close examination of formula to determine its ability to withstand batch-scale and process modifications; it must includes a review of range of relevant processing equipment also availability of raw materials meeting the specification of product and during the scale up efforts in the pilot plant production and process control are evaluated, validated and finalized.
In addition, appropriate records and reports issued to support Good Manufacturing Practices and to provide historical development of the production formulation, process, equipment train, and specifications
A manufacturer’s decision to scale up / scale down a process is ultimately rooted in the economics of the production process, i.e., in the cost of material, personnel, and equipment associated with the process and its control.
When developing technologies, there are a number of steps required between the initial concept and completion of the final production plant. These steps include the development of the commercial process, optimization of the process, scale-up from the bench to a pilot plant, and from the pilot plant to the full scale process. While the ultimate goal is to go directly from process optimization to full scale plant, the pilot plant is generally a necessary step.
Reasons for this critical step include: understanding the potential waste streams, examination of macro-processes, process interactions, process variations, process controls, development of standard operating procedures, etc. The information developed at the pilot plant scale allows for a better understanding of the overall process including side processes. Therefore, this step helps to build the information base so that the technology can be permitted and safely implemented.
Should be versatile pilot plant that is entirely GMP and facilitates the development of API’s in scalable, safe and environmentally friendly ways.
The combination of facilities, experience and flexibility enable an integral Contract Manufacturing service ranging from laboratory to industrial scale; it should manufacture under regulation small amounts of high added value active substances or key intermediate products.
Product quality: Operations that depend on people for executing manual recipes are subject to human variability. How precisely are the operators following the recipe? Processes that are sensitive to variations in processing will result in quality variation. Full recipe automation that controls most of the critical processing operations provides very accurate, repeatable material processing. This leads to very highly consistent product quality.
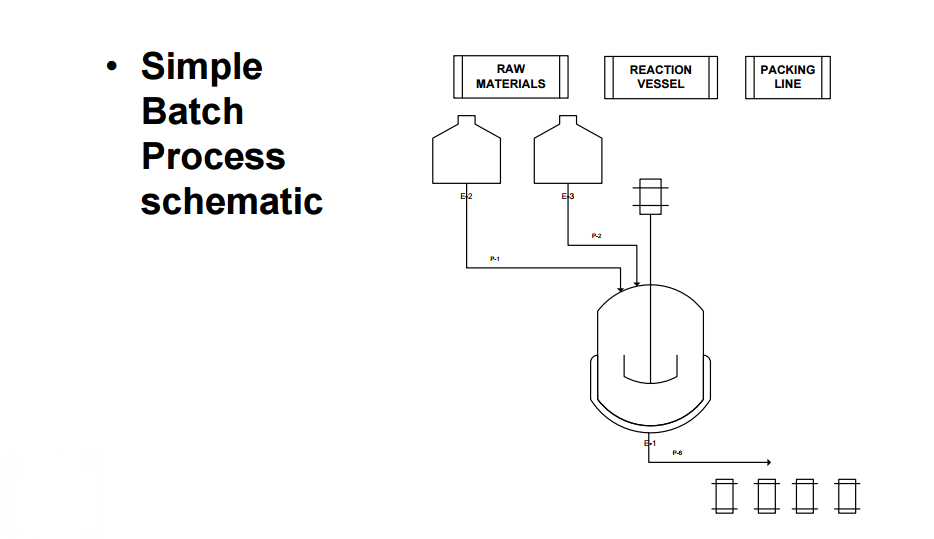
Improved production: Many biotech processes have extremely long cycle times (some up to 6 months), and are very sensitive to processing conditions. It is not uncommon for batches to be lost for unexplained reasons after completing a large portion of the batch cycle time. The longer the batch cycle time and the more sensitive production is to processing conditions, the more batch automation is justified. Imagine losing a batch of very valuable product because the recipe was not precisely followed!
Process optimization: Increasing the product yield can be done by making small changes in processing conditions to improve the chemical conversions or biological growth conditions. Manual control offers a limited ability to finely implement small changes to processing conditions due to the inherent lack of precision in human control. Conversely, computers are very good at controlling conditions precisely. In addition, advanced control capabilities such as model predictive control can greatly improve process optimization. This results in higher product yield and lower production cost. This consideration is highly relevant to pilot plant facilities where part of the goal is to learn how to make the product.
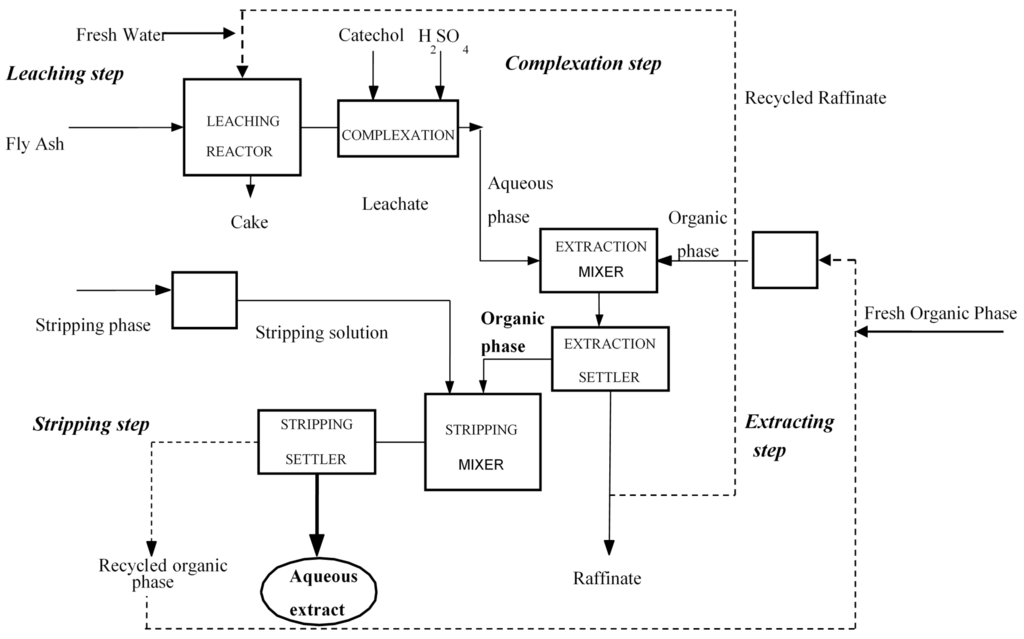
Recordkeeping: A multi-unit recipe control system is capable of collecting detailed records as to how a batch was made and relates all data to a single batch ID. Data of this nature can be very valuable for QA reporting, QA deviation investigations, and process analysis.
Safety: Operators spend less time exposed to chemicals when the process is fully automated as compared to manual control. Less exposure to the process generally results in a safer process.
A good batch historian should be able to collect records for a production run to include the following information: Product and recipe identification
User defined report parameters
Formulation data and relevant changes
Procedural element state changes (Operations, unit procedures, procedures)
Phase state changes
Operator changes
Operator prompts and responses
Operator comments
Equipment acquisitions and releases
Equipment relationships
Campaign creation data (recipe, formula values, equipment, etc.)
Campaign modifications
Campaign execution activity
Controller I/O subsystem events from the Continuous Historian
Process alarms
Process events
Device state changes.
Raw materials
Buildings and facilities. GMPs under the 21 Code of Federal Regulations (CFR) Part 211.42 state that buildings or areas used in the receiving, storage, and handling of raw materials should be of suitable size, construction and location to allow for the proper cleaning, maintenance, and operation (7). The common theme for this section of CFR Parts 210 and 211 is the prevention of errors and contamination. In principle, the requirements for buildings and facilities used in early phase manufacturing are not significantly different than those for later phases or even commercial production. However, there are some areas that are unique to early clinical trial manufacturing.
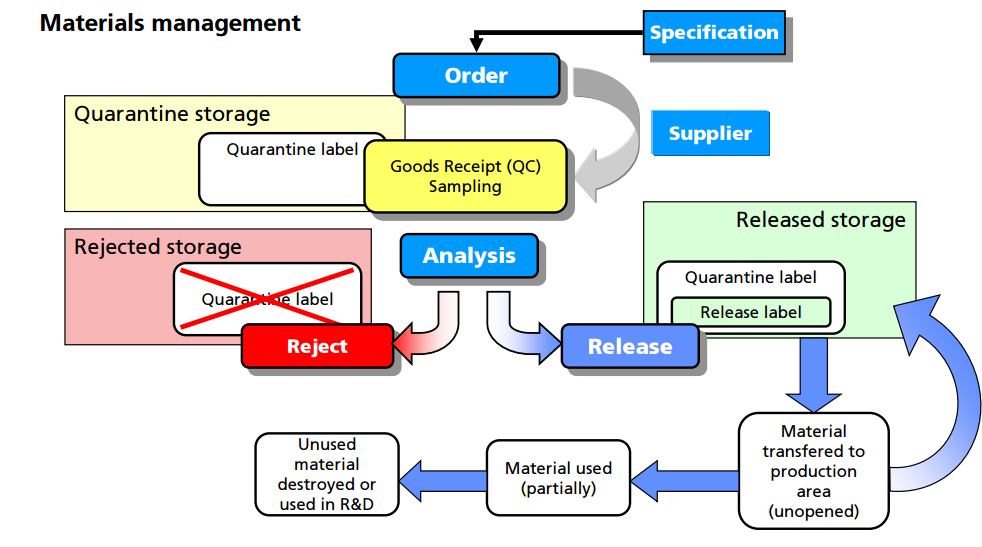
Control of materials. The CFR regulations under Part 211.80 provide good direction with respect to lot identification, inventory, receipt, storage, and destruction of materials (7). The clear intent is to ensure patient safety by establishing controls that prevent errors or cross-contamination and ensure traceability of components from receipt through clinical use. In general, the requirements for the control of materials are identical across all phases of development, so it is important to consider these requirements when designing a GMP facility within a laboratory setting.

For example, all materials must be assigned a unique lot number and have proper labeling. An inventory system must provide for tracking each lot of each component with a record for each use. Upon receipt, each lot should be visually examined for appropriate labeling and for evidence of tampering or contamination. Materials should be placed into quarantine or in the approved area or reject area with proper labeling to identify the material and prevent mix-ups with other materials in the storage area. Provision should be made for materials with special storage requirements (e.g., refrigeration, high security). The storage labeling should match the actual conditions that the material is being stored and should include expiry/retest dates for approved materials. Although such labeling is inconvenient for new materials where the expiration or retest date may change as more information is known, this enables personnel to be able to determine quickly whether a particular lot of a material is nearing or exceeding the expiration or retest date. General expiry/retest dates for common materials should be based on manufacturer’s recommendation or the literature.
Finally, there are clear regulatory and environmental requirements for the destruction of expired or rejected materials. It is important to observe regional and international requirements regarding the use of animal sourced materials (12). It is recommended to use materials that are not animal sourced and that there be available certification by the raw material manufacturers that they contain no animal sourced materials. If animal sourced raw materials must be used, then certifications by the raw material manufacturers that they either originate from certified and approved (by regulatory bodies) sources for use in human pharmaceuticals, or that the material has been tested to the level required for acceptance by regulatory agencies (following US, EU, or Japanese guidelines, as applicable) is required.
Direct advantages for customers
- Shorter implementation time for product by determination of the product suitability as well as the necessary process cycle
- Optimized adjustment of the processing times in the production lines (trains) by relatively precise estimation of the drying times
- Definition of effective cleaning processes (CIP/WIP and SIP)
- Definition of the selection criteria based on the weighting of the customer, e.g.: drying time, quality (form of crystal, activity, etc.), cleanout, ability of CIP, price
An overview of further trials and test functions, that can be realized in the new pilot plant facility:
- Product tests for determination of suitability
- Scale-up tests as basis for the extrapolation on production batches regarding drying time, filling degree, crystalline transformation and grain spectrum
- Optimization of the process cycle
- Optimization of the machine
- Data acquisition and analysis
SEE THIS SECTION IN ACTION…………..KEEP WATCHING
Case study 1


Designed and equipped for the manufacturing of solid oral dosage forms, specialized in high-activity substances (cytostatic, cytotoxic, hormonal, hormone inhibitors). It has ancillary areas for the proper management of materials intended for clinical trials of new drugs.
Equipment:
- • Oystar Hüttlin Pilotmix 75T single-pot high shear mixer.
- • Container mixer, 60 and 200 liters containers.
- • Fette 1200iC rotary tablet press, fully equipped for formulation development and manufacturing.
- • Oystar Manesty XL Lab 01 tablet coater, with interchangeable drums.
- • IMA Clinipack blister machine.
- • Quadro Comill sieve
- • Capsunorm 2000 manual capsul filler
- • Sepha press-out deblister
- • Fette Gratex deduster
- • Process control laboratory: Dr. Schleuniger tablet hardness tester, Erweka tablet disintegration tester, Erweka friability tester, Mettler Toledo moisture content determination equipment.
…………………………….
CASE STUDY 2
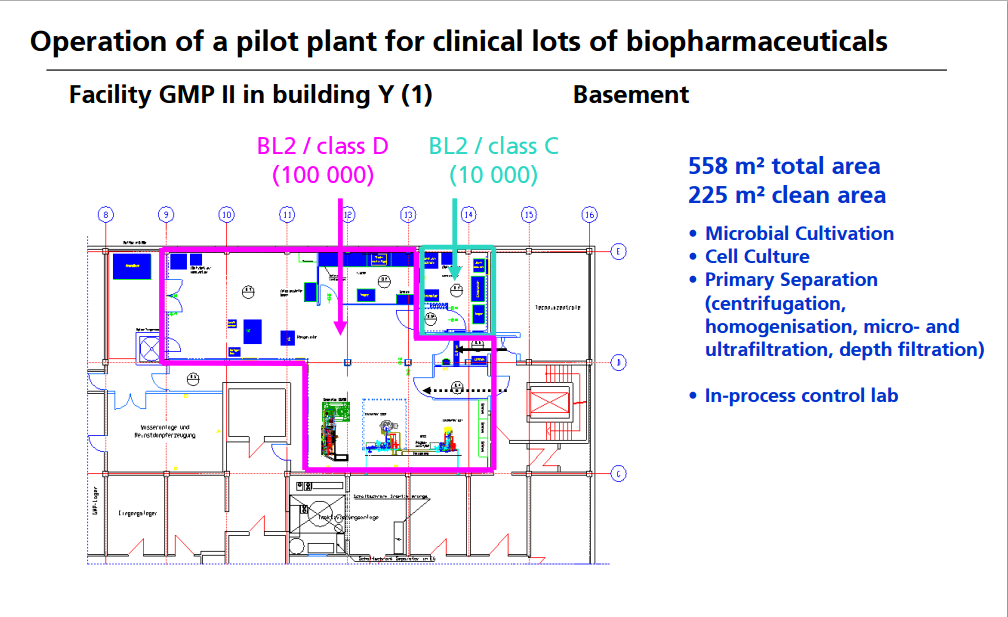
OPERATION OF PILOT PLANT FOR CLINICAL LOTS OF BIOPHARMACEUTICALS
http://www.peq.coppe.ufrj.br/biotec/presentations/Papamichael_RioDeJaneiro2009_secure.pdf



CASE STUDY 3

Good Manufacturing Practices in Active Pharmaceutical Ingredients Development
http://apic.cefic.org/pub/5gmpdev9911.pdf
Example below
3. Introduction Principles basic to the formulation of this guideline are: ·
Development should ensure that all products meet the requirements for quality and purity which they purport or are represented to possess and that the safety of any subject in clinical trials will be guaranteed. ·
During Development all information directly leading to statements on quality of critical intermediates and APIs must be retrievable and/or reconstructable. ·
The system for managing quality should encompass the organisational structure, procedures, processes and resources, as well as activities necessary to ensure confidence that the API will meet its intended specifications for quality and purity. All quality related activities should be defined and documented. Any GMP decision during Development must be based on the principles above.
During the development of an API the required level of GMP control increases. Using these guidelines, the appropriate standard may be implemented according to the intended use of the API. Firms should apply proper judgement, to discern which aspects need to be addressed during different development stages (non-clinical, clinical, scale-up from laboratory to pilot plant to manufacturing site).
Suppliers of APIs and/or critical intermediates to pharmaceutical firms should be notified on the intended use of the materials, in order to apply appropriate GMPs. The matrix (section 8) should be used in conjunction with text in section 7, as is only intended as an initial guide. READ MORE AT…. http://apic.cefic.org/pub/5gmpdev9911.pdf
CASE STUDY 4
http://www.steroglass.it/doc_area_download/ita/process/20LT_PILOT_PLANT.pdf
![]()

CASE STUDY 5

Health Canada
http://www.hc-sc.gc.ca/dhp-mps/compli-conform/gmp-bpf/question/gmp-bpf-eng.php
The Good Manufacturing Practices questions and answers (GMP Q&A) presented below have been updated following the issuance of the “Good Manufacturing Practices Guidelines, 2009 Edition Version 2 (GUI-0001)“.
This Q&A list will be updated on a regular basis.
Sanitation – C.02.007 & C.02.008
Raw Material Testing – C.02.009 & C.02.010
Manufacturing Control – C.02.011 & C.02.012
Quality Control Department – C.02.013, C.02.014 & C.02.015
Packaging Material Testing – C.02.016 & C.02.017
Finished Product Testing – C.02.018 & C.02.019
Records – C.02.020, C.02.021, C.02.022, C.02.023 & C.02.024
Stability – C.02.027 & C.02.028
CASE STUDY 6
![]()
http://www.pharmtech.com/early-development-gmps-drug-product-manufacturing-small-molecules-industry-perspective-part-iii?rel=canonical
Also
- Part VII GMPs for Small Molecule Drugs in Early Development Workshop Summary
- Early Development GMPs for Small-Molecule Specifications: An Industry Perspective (Part V)
- Early Development GMPs for Stability (Part IV)
- GMPs for Method Validation in Early Development: An Industry Perspective (Part II)
- GMPs for Small-Molecule Drugs in Early Development (Part I)
- GMPs for Small-Molecule Drugs in Early Development: Workshop Summary-Part VI
CASE STUDY 7

http://www.niper.gov.in/tdc_2013.pdf
CASE STUDY 8
Multi-kilo scale-up under GMP conditions
Examples of flow processes being used to produce exceptionally large amounts of material are becoming increasingly common as industrial researchers become more knowledgeable about the benefits of continuous reactions. The above examples from academic groups serve to illustrate that reactions optimized in small reactors processing tens to hundreds of mg hour−1 of material can be scaled up to several grams per hour. Projects in process chemistry are often time-sensitive, however, and production of multiple kg of material may be needed in a short amount of time. An example of how the efficient scaling of a flow reaction can save time and reduce waste is provided by a group of researchers at Eli Lilly in their kg synthesis of a key drug intermediate under GMP conditions . In batch, ketoamide 13 was condensed with NH4OAc and cyclized to form imidazole 14 at 100 °C in butanol on a 1 gram scale. However, side product formation became a significant problem on multiple runs at a 250 g scale. It was proposed that this was due to slow heat up times of the reactor with increasing scale, as lower temperatures seemed to favour increased degradation over productivecyclization. Upon switching to a 4.51 mL flow reactor, another optimization was carried out which identified methanol as a superior solvent that had been neglected in batch screening due to its low boiling point at atmospheric pressure. Scale-up to a 7.14 L reactor proceeded smoothly without the need for reoptimization, and running on this scale with a residence time of 90 minutes for a six-day continuous run provided 29.2 kg of product after recrystallization (approximately 207 g hour−1). The adoption of a flow protocol by a group of industrial researchers in a scale-up with time constraints demonstrates both the effectiveness and maturity of flow chemistry. While the given reaction was used to produce kilograms of material for a deadline, continuous operation without further optimization could produce over 1 metric tonne of product per year in a reactor that fits into a GC oven.
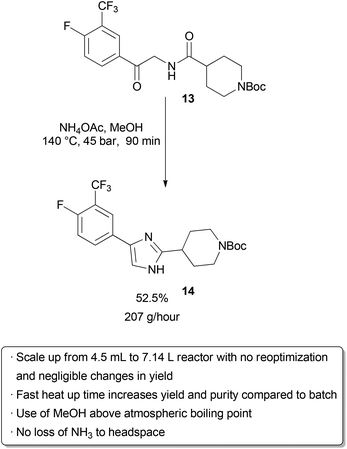 |
||
| Scheme 20 Kilogram-scale synthesis of an imidazole API precursor. | ||
…………………………….
Definitions
Plant: A plant is a place where an industrial or manufacturing process takes place. It may also be expressed as a place where the 5 M’s that are; man, materials, money, method and materials are brought together for the manufacture of products.
Pilot Plant: A part of a manufacturing industry where a laboratory scale formula is transformed into a viable product by development of reliable practical procedures of manufacturing.
Scale-Up: This is the art of designing a prototype based on the information or data obtained from a pilot plant model.
cGMP: current Good Manufacturing Processes refer to an established system of ensuring that products are consistently produced and controlled according to quality standards. It is designed to minimize risk involved in any industrial design. GMP covers all aspects of production from the starting materials, premises and equipment to the training and personal hygiene of staff within industries. Detailed, written procedures are essential for each process that could affect the quality of the finished product. There must be a system to provide documented proof that correct procedures are consistently followed at each step in the manufacturing process every time a product is made.
SCALING UP FROM PILOT PLANTS
When scaling up, it is of utmost importance to consider all aspects of risk and futuristic expansion. The pilot plant is usually a costly apparatus and therefore the decision of building it is always a hard one. The function of a pilot plant is not just to prove that the laboratory experiments work, but;
- To test technologies that are about to be implemented on industrial plants before establishment
- To evaluate performance specifications before the actual installation of industrial plant.
- Evaluation of reliability of mathematical models within real environment.
- Economic considerations for production involving process optimization and automated control systems.
GMP GENERAL PRACTISES
Facilities and Equipment Systems
- Ø Cleaning and maintenance
- Ø Facility layout and air handling systems for prevention of cross-contamination (e.g. Penicillin, beta-lactams, steroids, hormones, cytotoxic, etc.)
- Ø Specifically designed areas for the manufacturing operations performed by the firm to prevent contamination or mix-ups.
Facilities
- Ø General air handling systems
- Ø Control system for implementing changes in the building
- Ø Lighting, potable water, washing and toilet facilities, sewage and refuse disposal
- Ø Sanitation of the building, use of rodenticides, fungicides, insecticides, cleaning and sanitizing agents.
GMP FOR PLANT DESIGN
The application of GMP to plant design is primary to the establishment of such plants. Regulatory boards have precedence over these operations helping to establish a proper and functional system in plant design.
Design Review
l Conceptual drawings;
From plant design drawings which are inspected and approved by cGMP regulatory bodies (such as Department of Petroleum Resources in Nigeria), approvals are issued depending on adherence to specifications such as muster points, proper spacing of fuel sources from combustion units and other more elaborate considerations.
l Proposed plant layouts;
A choice of location for plant and layout play an important role on environmental impact. Hence, environmental impact assessment is a major part of GMP. Industries must be located at least 100M from closest residential quarter (depending of materials processed in plant).
l Flow diagrams for facility
For optimization and efficiency purposes, flow diagrams for complete refinery process are important for review with intent to ensure they conform to GMP
l Critical systems and areas
Some areas in a plant may require extra safety precautions in operations. The cGMP makes provision for such special considerations with the creation of customized set of operational guidelines that ensure safety and wellness of staff and environment alike.
cGMP EXAMPLE: FOOD PROCESSING PLANT
Outlined below are the cGMP considerations in the establishment and handling of a food processing plant.
Safety of Water
1. Process water is safe, if private supply should be tested at least annually.
2. Backflow prevention by an air gap or back flow prevention device. Sinks that are used to prepare food must have an air-gap.
Food Contact Surface
1. Designed, maintained, and installed so that it is easy to clean and to withstand the use, environment, and cleaning compounds.
2. If cleaning is necessary to protect against microorganisms, food-contact surfaces shall be cleaned in this sequence: wash with detergent, rinse with clear water, and then use an approved sanitizer. The sanitizer used shall be approved for use on food-contact surfaces. UA three-compartment ware washing sink or other equivalent methods shall be used for this purpose.
3. Gloves shall be clean/sanitary. Outer garments suitable.
Prevention of Cross-Contamination
1. Food handlers use good hygienic practices; hands shall be washed before starting work, after absence from work station, or when they become contamination (such as with eating or smoking).
2. Signs shall be posted in processing rooms and other appropriate areas directing employees that handle unprotected food, food-contact surfaces, food packaging materials to wash their hands prior to starting to work, after each absence from the work station, and whenever hands may become contaminated.
3. Plant design so that the potential for contamination of food, food-contact surfaces, or packaging materials is reduced to the extent possible.
4. Physical separation of raw and finished products.
Hand Washing Sinks and Toilet Facilities
1. Hand washing sinks, properly equipped, shall be conveniently located to exposed food processing areas. Ware washing sinks shall not be used for this purpose.
2. Adequate supply of hot and cold water under pressure.
3. Toilet facilities; adequate and accessible, self-closing doors.
4. Sewage disposal system shall be installed and maintained according to State law.
Protection from Adulteration (Food, Food Contact Surfaces, and Packaging Materials)
1. Food processing equipment designed to preclude contamination with lubricants, fuel, metal fragments, contaminated water, or other sources of contamination.
2. Food processed so that production methods to not contaminate the product.
3. Raw materials, works-in-process, filling, assembly, packaging, and storage and transportation conducted so that food is not contaminated.
4. Protection from drip and condensate overhead.
5. Ventilation adequate and air not blown on food or food-contact surfaces.
6. Lights adequately shielded.
7. Compressed air or gas mechanically introduced adequately filtered.
Scope of services
- Engineering support
- Representation of the construction owner (equipment, construction: supervision of general contractors, GMP concept draft)
- Basic and detailed design
- Support during the implementation phase
- Clean room planning (incl. lab areas)
- Construction management
- Qualification
- Validation support
Toxic Items: Labelling, Use, and Storage
1. Products used approved and used according to product’s label.
2. Sanitizer used on food-contact surfaces must be approved for that use.
3. Shall be securely stored, so unauthorized use is prevented.
Personnel Disease Control
1. Food handler, who has illness or open lesion, or other source of microbiological contamination that presents a reasonable possibility of contamination of food, food-contact surfaces, or packaging materials shall be excluded from such operations.
2. Adequate training in food protection, dangers of poor personal hygiene, and unsanitary practices shall be provided.
3. Management shall provide adequate supervision and competent training to ensure compliance with these provisions.
Pest Control
1. Management shall provide an adequate pest control program so that pests are excluded from the plant.
2. Program shall ensure that only approved pesticides are used and applied per the product’s label.
Plant Construction and Design
1. Walls, floors, and ceilings constructed so that they can be adequately cleaned and kept in good repair.
2. Adequate lighting provided.
3. Adequate ventilation or controls to minimize odours and vapours.
4. Adequate screening or protection of outer openings.
5. Grounds maintained free of litre, weeds, and pooling water.
6. Roads, yards, and parking lots maintained so that food is not contaminated.
Equipment
1. Equipment, utensils, and seams on equipment – adequately cleanable, properly maintained, designed, and made of safe materials.
2. Refrigerators and freezers equipped with adequate thermometer.
3. Instruments and control devices – accurate and maintained.
4. Compressed air or gas designed/treated so that food is not contaminated.
Equipment. Most equipment used to manufacture early GMP drug product is be managed under a qualification, preventive maintenance, and calibration program for the GMP facility. However, in early development, there may occasionally be a need to use equipment that is not part of such a program. Rather than performing a comprehensive qualification for a piece of equipment not expected to be frequently used, an organization may choose to qualify it for a single step or campaign. Documentation from an installation qualification/operational qualification (IQ/OQ) and or performance verification at the proposed operating condition is sufficient. For example, if solution preparation needs a mixer with a rotation speed of 75 rpm, then documentation in the batch record using a calibrated tachometer to verify that the mixer was operating at 75 rpm will suffice.
The use of dedicated or disposable equipment or product contact parts may be preferable to following standard cleaning procedures to ensure equipment is clean and acceptable for use. However, not all equipment or equipment parts are disposable or may have a substantial cost that makes disposal prohibitive. In that case, the product contact parts could be dedicated to a specific drug substance for use in drug product manufacture. Dedicating product contact parts to a compound may be costly and may be avoided in some cases by carefully considering product changeover and effective cleaning methods when purchasing equipment.
Another item to consider with respect to equipment, is that the more complicated the equipment is to run or maintain, the less desirable it might be for early GMP batches. In most cases, simple equipment is adequate and will uses less material and consume less total time for preparation, operation, and cleaning activities.
Weights and Measures
1. Scales used to measure net weight of contents shall be designed so they can be calibrated.
2. Products in interstate commerce – net weights/measurements also in metric.
CONCLUSION
Plant establishment is an activity that has kept rising from the inception of the industrial revolution until date. Giving rise to increase in raw material demand, increased pollution levels, higher energy demand, and overall greater economic output. As history and record keeping has served for an even longer period, it becomes necessary for adaptation to be made to avoid incidents and accidents that have occurred previously and also those that can be anticipated without actual devastating effect.
The development of the GMP is as a result of observed challenges in industry and environment over years of industrialization. It becomes necessary to upset these poor trends that have developed as a result of industrialization by so doing increasing the pros and reducing the cons.
GMP protects consumer, produce, equipment, and conserves the processes as a whole, leading to a more efficient sustainable process defining a new standard for yields and profit and eliminating the tendency of compromise made by industrialists to increase overall profits at the risk of staff and environment.

Batch documentation and execution
Batch record documentation preparation. Manufacturing documentation is a basic requirement for all phases of clinical development. 21 CFR Parts 211.186 and 211.188 describe master production and batch production records, respectively (7). The stated purpose of the master production record is to “assure uniformity from batch to batch.” Although the record assurance is important for a commercial validated manufacturing process, it does not necessarily apply to clinical-development batches. Material properties, manufacturing scale, and quality target product profile frequently change from batch to batch. Therefore, batch production records are the appropriate documentation for clinical trial supplies. Batch production records for Phase 1 materials should minimally include:
- Name, strength, and description of the dosage form
- A complete list of active and inactive ingredients, including weight or measure per dosage unit and total weight or measure per unit
- Theoretical batch size (number of units)
- Manufacturing and control instructions.
These minimum requirements are consistent with the FDA Guidance for Industry: cGMP for Early Phase Investigational Drugs, which requires a record of manufacturing that details the materials, equipment, procedures used and any problems encountered during manufacturing (2). The records should allow for the replication of the process. On this basis, there is flexibility in the manner for which documentation of batch activities can occur, provided that the documentation allows for the post execution review by the quality unit and for the retention of these records.
Batch documentation approvals. Review and approval of executed batch records by the Quality unit is required per 21 CFR Part 211.192 (7). This review and approval is required for all stages of clinical manufacturing. Pre-approvals of batch records should be governed by internal procedures as there is no requirement in CFR 21 that the Quality unit pre-approves the batch record (though this is highly recommended in order to minimize the chance of errors). Indeed, Table I shows that pre-approval of batch records by the Quality Unit is practiced by all 10 companies that participated in the IQ Consortium’s drug-product manufacturing survey related to early development. Batch records must be retained for at least 1 year after the expiration of the batch according to CFR Part 211.180, but many companies keep their GMP records archived for longer terms.
Room clearance. 21 CFR Part 211.130 requires inspection of packaging and labeling facilities immediately before use to ensure that all drug products from previous operations have been removed. This inspection should be documented and can be performed by any qualified individual.
Although line clearance for bulk manufacture is not specifically mentioned in the CFR, it is expected that a room clearance be performed. At a minimum, this clearance should be performed prior to the initiation of a new batch (i.e., prior to batch materials entering a processing room).
Hold time. During the early stages of development, final dosage form release testing confirms product quality and support establishment of hold times later in the clinical development. There is no requirement to establish hold times for work in process in early development. Specific formulation and stability experience, which is usually limited at this stage of development, should be leveraged to assess any substantial variations from expected batch processing times. The data gathered from these batches and subsequent development can be used to help establish hold times for future batches. (Exceptions to this approach may include solution or suspension preparations used in solid dosage form manufacturing, where procedures typically govern allowable hold times to ensure the absence of microbial contamination in the final product.)
Change control. Changes to raw materials, processes, and products during early development are inevitable. It is not required that these changes be controlled by a central system but rather may be appropriately documented in technical reports and manufacturing batch records. Any changes in manufacturing process from a previous batch should be captured as part of the batch record documentation and communicated to affected areas. The rationale for these changes should also be documented as this serves as a source for development history reports and for updating regulatory filings. The authors recommend that those changes that could affect a regulatory filing be captured in a formal system.
Process changes. Process parameters should be recorded but do not need to be predetermined because processes may not be fixed or established in early development. Given the limited API availability in early development, a clinical batch is often the first time a product is manufactured at a particular scale or using a particular process train. Therefore, process changes should be expected. Process trains and operating parameters must be documented in the batch record but changes should not trigger an exception report or CAPA. Changes should be documented as an operational note or modification to the batch record in real time. Such changes driven by technical observations should not require prior approval by the Quality unit, but should have the appropriate scientific justification (via formulator/scientist) or the appropriate flexibility built into the batch record to allow for the changes. This documentation should be available for Quality review prior to product disposition.
Calculation of yield. Actual yields should be calculated for major processing steps to further process understanding and enable optimization of processes. Expected yield tolerances are not always applicable to early development manufacture. At this stage of early development, when formulation and process knowledge is extremely limited, there may be no technical basis for setting yield tolerances and, therefore, this yield may not be an indicator of the quality of the final product.
In-process controls and R&D sampling. In-process tests and controls should follow basic requirements of GMPS to document consistency of the batch. For capsule products, these requirements may include capsule weights and physical inspection. For tablet products, compression force or tablet hardness and weights should be monitored together with appearance. R&D sampling, defined as samples taken for purposes of furthering process understanding but not utilized for batch disposition decisions, is a normal part of all phases of clinical manufacturing. In early development manufacturing, a sampling plan is required for in-process control tests, but not for R&D samples. However, for the purpose of material accountability, R&D sampling should be documented as part of batch execution. For these samples, testing results may be managed separately, and are not required to be included in regulatory documentation.
Facilities and equipment
Regardless of the scale of manufacturing, the facility used for manufacturing clinical trial supplies must meet the basic GMP requirements as described in the regulations and guidance documents. Below are three scenarios for early development and the advantages of each as pertaining to early development. The first involves a pilot plant facility designed and equipped for routine GMP operations. The second scenario aims to establish a GMP area within a laboratory environment. The third example focuses on conducting GMP manufacturing or leveraging the practice of pharmacy in close proximity to the clinical site.
GMP facility for drug-product manufacture. The traditional approach in GMP drug-product manufacture is to use a dedicated facility (often called a pilot plant) for early phase clinical trials. Advantages of this approach include that the quality systems for the facility (i.e., maintenance, calibration, cleaning, change management, CAPA, and documentation) are well defined, and that training and other activities required for maintaining GMP compliance are centralized. Other drivers to use a pilot plant in early development may be the need for specialized equipment, or larger batch sizes in special situations.
GMP area within a laboratory setting. In some cases, it may be advantageous to establish a GMP area within a “laboratory setting” (i.e., a drug-development facility not dedicated to the production of clinical supplies) for the manufacture of drug product in early development. The rationale for this approach might be to avoid the significant investment in setting up a dedicated facility and to create simpler, more flexible systems that meet GMP requirements but are tailored for the specific activity envisioned. Examples where this approach might be considered include the need for special containment not available in the pilot-plant; the need to work with radioactive or hazardous materials, use of controlled substances and the production of “one-off manufactured” product used for proof of concept. The business rationale should be documented and approved by the manufacturing and Quality groups. As long as the appropriate GMP controls are maintained, especially as related to operator safety, cleaning, and prevention of cross-contamination, there is no compliance barrier to using “lab-type” facilities for the manufacturing of early phase clinical batches. Before GMP manufacturing is initiated, however, a risk assessment should be conducted and documented. Inclusion of representatives from Quality, analytical, clinical manufacturing, product development, and environmental health and safety would be prudent. When selecting/designing an early development clinical manufacturing facility, consideration should be made for the receipt, storage, dispensing, and movement of materials. The manufacturing processes in the nondedicated area must protect the product, patient, and the manufacturing operators.
Additionally, companies should consider what items are appropriate for the manufacture. For example, the use of a certified laminar flow hood may be a better choice for manufacturing than a fume hood, because the former is designed to prevent contamination of the product, protect the operator, and the laboratory environment. In addition, with the appropriate cleaning, a laminar flow hood can more easily be used for multiple products. Small scale/manual equipment or procedures may be the best approach because the space is likely to be limited. With a small batch size, the use of small scale or manual equipment/procedures will minimize yield loss. Additional measures to be assessed include appropriate gowning and operator personal protection devices, area and operator monitoring for potent or radiolabeled drug exposure, and so forth.
Documentation of the facility preparation, product manufacture, and the return of the facility to the previous state, if needed, is recommended. This documentation should describe the rationale for the manufacture in the nondedicated area, risk assessment, preparation of the area, cleaning procedures, and list of responsible persons. This documentation can reference existing procedures or standard operating procedures (SOPs) along with documents associated with the meetings and preparation for the manufacture of the batch. Batch records and cleaning records should be part of the documentation and should follow the company’s data-retention policy.
Receipt and approval
Specifications. It is a GMP requirement that all raw materials for the manufacture of drug product have appropriate specifications to ensure quality. The compendial requirements should be used for setting specifications provided the material is listed in at least one pharmaceutical compendium (e.g., US, European, and Japanese Pharmacopeias). It is important that the use of materials meeting the requirements of a single compendium is acceptable for use in early phase clinical studies conducted in the US, Europe, and Japan. For example, a material that meets USP criteria and is used in the manufacture of a drug product should be acceptable for use in early clinical studies in the European Union. In the absence of a pharmaceutical compendium monograph, the vendor specification and/or alternative compendial specifications such as USP’s Food Chemical Codex should guide specification setting. In any case, the sponsor is responsible for the establishment of appropriate specifications. Therefore, it is the authors’ position that good practice is to have at least a basic understanding of the manufacture, chemistry, and toxicology of the materials to guide appropriate specification setting.
Material testing and evaluation. The minimum testing required for incoming materials is visual inspection and identification. However, as mentioned above, the appropriate tests should be determined for the material based on the knowledge of the manufacture, chemistry, and toxicology. If the vendor is qualified, then the certificate of analysis may be acceptable in conjunction with the visual inspection and identification testing (see “Vendor Qualification” section below).
Approval for use. Ideally, manufacture of a bulk drug product should begin with approved material specifications and with materials that are fully tested and released. However, there are circumstances where it may not be feasible to start manufacture with approved specifications and fully tested and released materials, including API. Manufacturing prior to final release (sometimes called manufacturing “at risk”) may be acceptable, however, because the quality system ensures that all specifications are approved, test results are within specifications, and all relevant documents are in place before the product is released for administration to humans. The “risk” must lie fully with the manufacturer and not with the patient.
Vendor qualification. Vendors supplying excipients, raw materials, or API must be qualified by the sponsor. Appropriate qualification should depend on the stage of development and an internal risk assessment. For, example if a vendor has a history of supplying the pharmaceutical industry and the material is to be used in early development, a paper assessment (e.g., a questionnaire) should be sufficient. If a supplier does not have a history of supplying the pharmaceutical industry, a risk assessment should be performed and depending on the outcome a site audit may be required prior to accepting material for use.
Ideally, vendors should be qualified prior to using raw materials for manufacture. However, it is acceptable for qualification to proceed in parallel as long as documentation/risk assessments are available prior to product release and as in the previous section all risk lies with the manufacturer and not the patient.
A production mixing unit is usually not geometrically similar to the mixer used for process development. Such differences can make scale-up from the laboratory or pilot plant challenging. A solution to these problems is to systematically calculate and evaluate mixing characteristics for each geometry change.
Geometric similarity is often used in mixing scale-up because it greatly simplifies design calculations. Geometric similarity means that a single ratio between small scale and large scale applies to every length dimension (see figure). With geometric similarity, all of the length dimensions in the large-scale equipment are set by the corresponding dimensions in the small-scale equipment. The only remaining variable for scale-up to large-scale mixing is the rotational speed — one or more mixing characteristics, such as tip speed, can be duplicated by the appropriate selection of a large-scale mixer speed.

The two most popular and effective geometric scale-up methods are equal tip speed and equal power per volume. Equal tip speed results when the small-scale mixer speed is multiplied by the inverse geometric ratio of the impeller diameters to get the large-scale mixer speed:
N2 = N1(D1/D2)
Equal power per volume involves a similar calculation, except the geometry ratio is raised to the two-thirds power:
N2 = N1(D1/D2)(2/3)
This expression for power per volume only applies strictly for turbulent conditions, where the power number is constant, but is approximately correct for transition-flow mixing.
Avoid mix-ups
As we have seen, taking successive steps allows the development of alternative solutions to scale-up. Similar methods can be used to scale-down process problems for investigation in a pilot-plant or laboratory simulation. Here, too, non-geometric similarity often is a problem. Such scale-down calculations should help pinpoint appropriate operating speeds to test in the small-scale mixer.
In any scale-up or scale-down evaluation, some variables can be held constant while others must change. For example, even with geometric similarity, scale-up will result in less surface per volume because surface area increases as the length squared and volume increases as length cubed. Similarly, keeping blend time constant rarely is practical with any significant scale change. Larger tanks take longer to blend than smaller ones. Also, Reynolds number is expected to increase as size increases. In addition, standard operating speeds or available impeller sizes may necessitate a final adjustment to the scale-up calculations.
Rules for scale-up always have exceptions but understanding the effects of scale-up, especially non-geometric scale-up, can provide valuable guidance. Indeed, appreciation of the tradeoffs involved in non-geometric scale-up may be crucial for success with large-scale mixing processes.
REFERENCES
2 http://apic.cefic.org/pub/5gmpdev9911.pdf
“ICH Q7a. Good Manufacturing Practice for Active Pharmaceutical Ingredients” (Draft 6, October 19th, 1999, section 19).
“ICH Q6a. Specifications: test procedures and acceptance criteria for new drug substances and new drug products: chemical substances”.
“Good Manufacturing Practices for Active Pharmaceutical Ingredients” (EFPIA / CEFIC Guideline, August, 1996).
“Quality Management System for Active Pharmaceutical Ingredients Manufacturers” (APIC/CEFIC May 1998).
“Good Manufacturing Practices Guide for Bulk Pharmaceutical Excipients”, The International Pharmaceutical Excipients Council (October 1995).
“21 Code of Federal Regulations, parts 210 to 211”, U.S. Food & Drug Administration. “Guide to inspection of Bulk Pharmaceutical Chemicals”, U.S. Food & Drug Administration, (Revised Edition: May 1994).
“Guidance for Industry. ANDAs: Impurities in Drug Substances”, U.S. Food and Drug Administration, CDER (June 1998).
“Guideline on the Preparation of Investigational New Drug Products”, U.S. Food & Drug Administration, CDER (March 1991).
“EC Guides to GMP, Annex 13: Manufacture of Investigational Medicinal Products” (Revised Dec. 1996).
“GMP Compliance during Development”, David J. DeTora. Drug Information Journal, 33, 769-776, 1999.
FDA Guidance documents on internet address: http://www.fda.gov/cder/guidance /index.htm
EMEA Guidance documents on internet address: http://www.eudra.org.
………………..
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE



 Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com


जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
////////