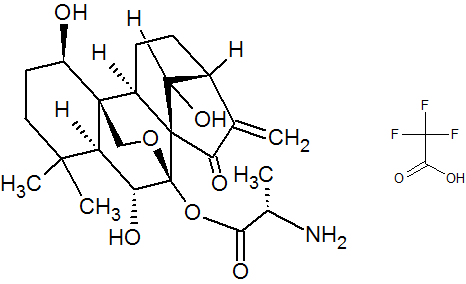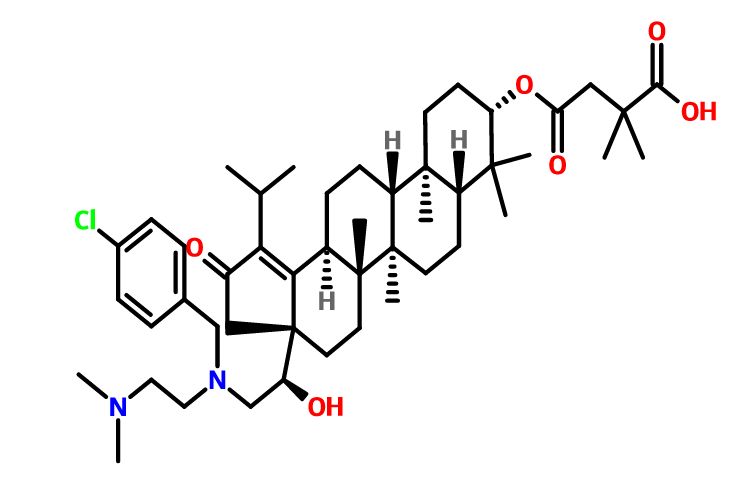


GSK-2838232
4-(((3aR,5aR,5bR,7aR,9S,11aR,11bR,13aS)-3a-((R)-2-((3-chlorobenzyl)(2-(dimethylamino)ethyl)amino)-1-hydroxyethyl)-1-isopropyl-5a,5b,8,8,11a-pentamethyl-2-oxo-3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a-octadecahydro-2H-cyclopenta[a]chrysen-9-yl)oxy)-2,2-dimethyl-4-oxobutanoic acid.
28-Norlup-18-en-21-one, 3-(3-carboxy-3-methyl-1-oxobutoxy)-17-[(1R)-2-[[(4-chlorophenyl)methyl][2-(dimethylamino)ethyl]amino]-1-hydroxyethyl]-, (3β)-
Phase I
Glaxosmithkline Llc INNOVATOR
Mark Andrew HATCHER, Brian Alvin Johns,Michael Tolar Martin, Elie Amine TABET, Jun Tang
A reverse transcriptase inhibitor potentially for the treatment of HIV infection.

GSK-2838232; GSK-8232; 2838232
CAS No. 1443460-91-0
C48H73ClN2O6,809.56
SYNTHESIS
PART 1
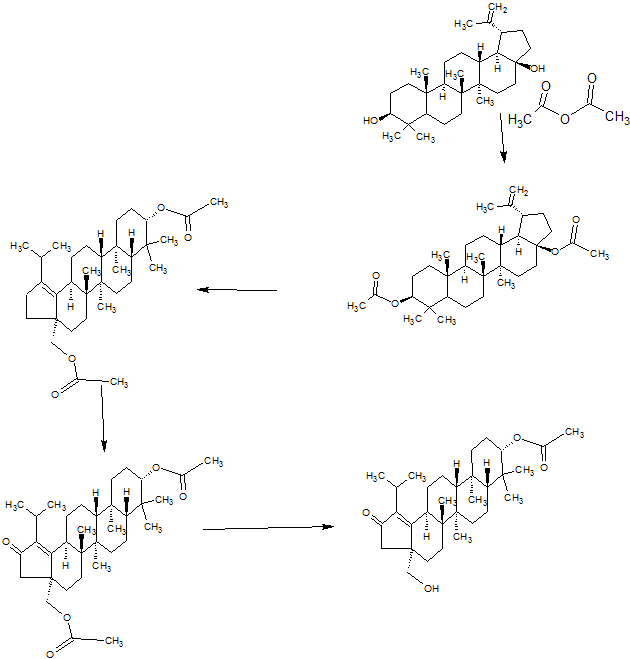
PART2
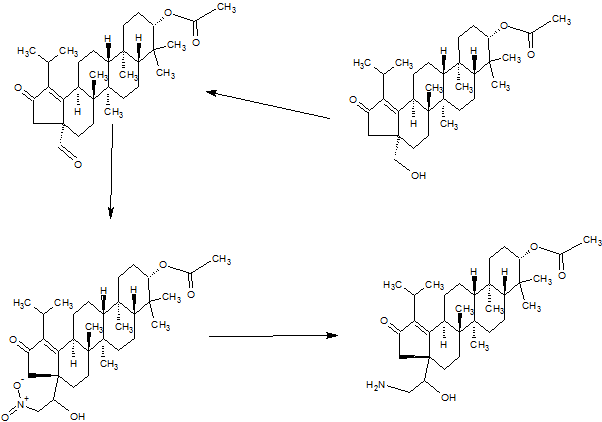
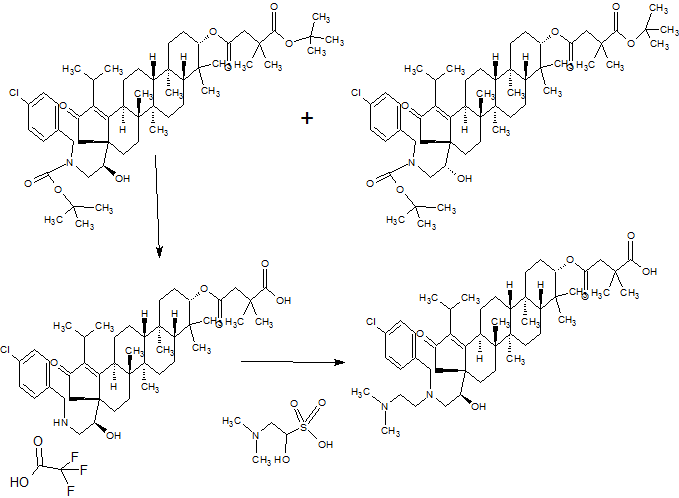
PART3
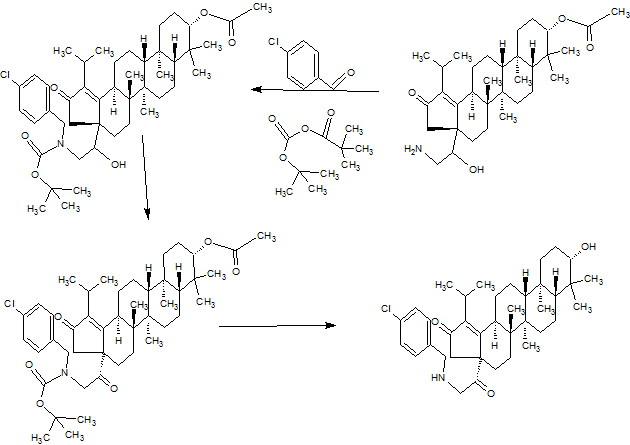
PART 4
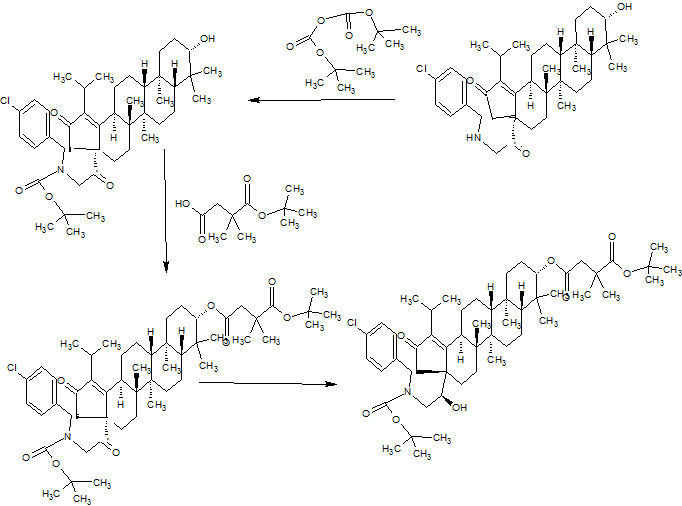
AND UNWANTEDISOMER SHOWN BELOW
PART5

GSK2838232 is a novel human immune virus (HIV) maturation inhibitor being developed for the treatment of chronic HIV infection. GSK2838232 is a betulin derivative
Human immunodeficiency virus type 1 (HIV-1 ) leads to the contraction of acquired immune deficiency disease (AIDS). The number of cases of HIV continues to rise, and currently over twenty-five million individuals worldwide suffer from the virus. Presently, long-term suppression of viral replication with antiretroviral drugs is the only option for treating HIV-1 infection. Indeed, the U.S. Food and Drug Administration has approved twenty-five drugs over six different inhibitor classes, which have been shown to greatly increase patient survival and quality of life.
However, additional therapies are still required because of undesirable drug-drug interactions; drug-food interactions; non-adherence to therapy; and drug resistance due to mutation of the enzyme target.
Currently, almost all HIV positive patients are treated with therapeutic regimens of antiretroviral drug combinations termed, highly active antiretroviral therapy (“HAART”). However, HAART therapies are often complex because a combination of different drugs must be administered often daily to the patient to avoid the rapid emergence of drug-resistant HIV-1 variants. Despite the positive impact of HAART on patient survival, drug resistance can still occur. The emergence of multidrug-resistant HIV-1 isolates has serious clinical consequences and must be suppressed with a new drug regimen, known as salvage therapy.
Current guidelines recommend that salvage therapy includes at least two, and preferably three, fully active drugs. Typically, first-line therapies combine three to four drugs targeting the viral enzymes reverse transcriptase and protease. One option for salvage therapy is to administer different combinations of drugs from the same mechanistic class that remain active against the resistant isolates.
However, the options for this approach are often limited, as resistant mutations frequently confer broad cross-resistance to different drugs in the same class.
Alternative therapeutic strategies have recently become available with the development of fusion, entry, and integrase inhibitors. However, resistance to all three new drug classes has already been reported both in the lab and in patients. Sustained successful treatment of HIV-1 -infected patients with antiretroviral drugs will therefore require the continued development of new and improved drugs with new targets and mechanisms of action.
Presently, long-term suppression of viral replication with antiretroviral drugs is the only option for treating HIV-1 infection. To date, a number of approved drugs have been shown to greatly increase patient survival. However, therapeutic regimens known as highly active antiretroviral therapy (HAART) are often complex because a combination of different drugs must be administered to the patient to avoid the rapid emergence of drug-resistant HIV-1 variants. Despite the positive impact of HAART on patient survival, drug resistance can still occur.
The HIV Gag polyprotein precursor (Pr55Gag), which is composed of four protein domains – matrix (MA), capsid (CA), nucleocapsid (NC) and p6 – and two spacer peptides, SP1 and SP2, represents a new therapeutic target. Although the cleavage of the Gag polyprotein plays a central role in the progression of infectious virus particle production, to date, no antiretroviral drug has been approved for this mechanism.
In most cell types, assembly occurs at the plasma membrane, and the
MA domain of Gag mediates membrane binding. Assembly is completed by budding of the immature particle from the cell. Concomitant with particle release, the virally encoded PR cleaves Gag into the four mature protein domains, MA, CA, NC and p6, and the two spacer peptides, SP1 and SP2. Gag-Pol is also cleaved by PR, liberating the viral enzymes PR, RT and IN. Gag proteolytic processing induces a
morphological rearrangement within the particle, known as maturation. Maturation converts the immature, donut-shaped particle to the mature virion, which contains a condensed conical core composed of a CA shell surrounding the viral RNA genome in a complex with NC and the viral enzymes RT and IN. Maturation prepares the virus for infection of a new cell and is absolutely essential for particle infectivity.
Bevirimat (PA-457) is a maturation inhibitor that inhibits the final step in the processing of Gag, the conversion of capsid-SP1 (p25) to capsid, which is required for the formation of infectious viral particles. Bevirimat has activity against ART-resistant and wild-type HIV, and has shown synergy with antiretrovirals from all classes. Bevirimat reduced HIV viral load by a mean of 1.3 logi0/mL in patients who achieved trough levels of >= 20 μg/mL and who did not have any of the key baseline Gag polymorphisms at Q369, V370 or T371. However, Bevirimat users with Gag polymorphisms at Q369, V370 or T371 demonstrated significantly lower load reductions than patients without Gag polymorphisms at these sites.
Other examples of maturation inhibitors can be found in PCT Patent
Application No. WO201 1/100308, “Derivatives of Betulin”; PCT Patent Application No. PCT/US2012/024288, “Novel Anti-HIV Compounds and Methods of Use Thereof ; Chinese PCT Application No. PCT/CN201 1/001302, “Carbonyl Derivatives of Betulin”; Chinese PCT Application No. PCT/CN201 1/001303, “Methylene Derivatives of Betulin”; Chinese PCT Application Nos. PCT/CN201 1/002105 and PCT/CN201 1/002159, “Propenoate Derivatives of Betulin”. Maturation inhibitors in the prior art leave open gaps in the areas of polymorphism coverage whereby potency against a broad range of clinically relevant gag sequences is extremely important, along with overall potency including the clinically relevant protein adjusted antiviral activity that will be required for robust efficacy in long term durability trials. To date, no maturation inhibitor has achieved an optimal balance of these properties.


PATENT
WO 2013090664
https://www.google.com/patents/WO2013090664A1?cl=en
Example 17: Compound 50
4-(((3aR, 5aR, 5bR, 7aR, 9S, 11aR, 11bR, 13aS)-3a-((S)-1-Acetoxy-2-((4- chlorobenzyl)amino)ethyl)-1-isopropyl-5a, 5b, 8, 8, 11 a-pentamethyl-2-oxo- 3, 3a, 4, 5, 5a, 5b, 6, 7, 7a, 8,9, 10, 11, 11a, 11b, 12, 13, 13a-octadecahydro-2H- cyclopenta[a]chrysen-9-yl)oxy)-2,2-dimethyl-4-oxobutanoic acid
[00241] The title compound was made in a similar manner to Example 16 and isolated as a TFA salt. 1H NMR (400MHz ,CHLOROFORM-d) δ = 7.49 – 7.30 (m, 4 H), 5.85 – 5.71 (m, 1 H), 4.59 – 4.40 (m, 1 H), 4.31 – 4.03 (m, 2 H), 3.41 – 2.79 (m, 4 H), 2.79 – 2.50 (m, 2 H), 2.37 (d, J = 18.1 Hz, 2 H), 2.02 – 0.63 (m, 49 H); LC/MS: m/z calculated 779.5, found 780.3 (M+1 )+.
Example 18: Compound 51
4-(((3aR, 5aR, 5bR, 7aR, 9S, 11aR, 11bR, 13aS)-3a-((R)-2-((4-Chlorobenzyl)(2- (dimethylamino)ethyl)amino)-1-hydroxyethyl)-1-isopropyl-5a,5b,8,8, 11a-pe
2-0X0-3, 3a, 4, 5, 5a, 5b, 6, 7, 7a, 8,9, 10, 11, 11a, 11b, 12, 13, 13a-octadecahydro-2H- cyclopenta[a]chrysen-9-yl)oxy)-2,2-dimethyl-4-oxobutanoic acid
[00242] To a solution of 2-(dimethylamino)acetaldehyde, hydrochloride (6.75 g, 54.6 mmol) in methanol (20 ml_) was added 4-
(((3aR,5aR,5bR,7aR,9S, 1 1 aR, 1 1 bR, 13aS)-3a-((R)-2-((4-chlorobenzyl)amino)-1 – hydroxyethyl)-1 -isopropyl-5a,5b,8,8, 1 1 a-pentamethyl-2-oxo- 3,3a,4,5,5a,5b,6,7,7a,8,9,10,1 1 ,1 1 a,1 1 b,12,13,13a-octadecahydro-2H- cyclopenta[a]chrysen-9-yl)oxy)-2,2-dimethyl-4-oxobutanoic acid , Trifluoroacetic acid salt (46) (9.5 g, 10.92 mmol). The pH was adjusted to 7-8 with Et3N. The reaction mixture was stirred at rt for 2 h. Sodium cyanoborohydride (0.686 g, 10.92 mmol) was then added and the mixture was stirred at rt overnight. After the reaction was complete, water (15 ml_) and EtOAc (15 ml_) were added, and then the organic phase was removed and concentrated under reduced presure. The product was extracted with EtOAc (80 ml_x3), the combined organic phase was washed with brine, dried, and concentrated. The product was purified by flash chromatography (DCM:EtOAc=2: 1 to 1 : 1 , then DCM:MeOH=100: 1 to 20: 1 ) to give 4- (((3aR,5aR,5bR,7aR,9S, 1 1 aR, 1 1 bR, 13aS)-3a-((R)-2-((4-chlorobenzyl)(2- (dimethylamino)ethyl)amino)-1 -hydroxyethyl)-1 -isopropyl-5a,5b,8,8, 1 1 a-pentamethyl- 2-0X0-3, 3a,4, 5, 5a, 5b, 6, 7, 7a, 8, 9, 10, 1 1 , 1 1 a, 1 1 b, 12, 13, 13a-octadecahydro-2H- cyclopenta[a]chrysen-9-yl)oxy)-2,2-dimethyl-4-oxobutanoic acid (51 ) (6 g, 7.41 mmol, 67.9 % yield) as white solid. Multiple batches of this material (were combined 95 g), dissolved in 600 mL of dichloromethane and washed with NaHC03 (400 ml_*3) and the organic phase was dried over Na2S04, filtered and concentrated. The solids were washed with a mixture of EtOAc: petroleum ether (600 mL), and filtered followed by lyophilization to provide the final title compound 62 g as a white solid. 1H NMR (400MHz ,METHANOL-d4) δ = 7.47 – 7.29 (m, 4 H), 4.48 (dd, J = 5.8, 10.3 Hz, 1 H), 4.15 – 4.04 (m, 1 H), 3.80 (d, J = 13.8 Hz, 1 H), 3.57 (d, J = 14.1 Hz, 1 H), 3.21 – 2.82 (m, 5 H), 2.72 – 2.41 (m, 9 H), 2.37 – 2.05 (m, 4 H), 2.05 – 0.74 (m, 45 H);
LC/MS: m/z calculated 808.5, found 809.5 (M+1 )+.
REFERENCES
Hatcher, Mark Andrew; Johns, Brian Alvin; Martin, Michael Tolar; Tabet, Elie Amine; Tang, Jun. Preparation of betulin derivatives for the treatment of HIV, PCT Int. Appl. (2013), WO 2013090664 A1 20130620.

////////GSK-2838232, 1443460-91-0, GSK 2838232, GSK-8232, 2838232, treatment of HIV, phase1
O=C(C1)C(C(C)C)=C2[C@@]1([C@@H](O)CN(CCN(C)C)CC3=CC=CC(Cl)=C3)CC[C@]4(C)[C@]2([H])CC[C@@]5([H])[C@@]4(C)CC[C@]6([H])[C@]5(C)CC[C@H](OC(CC(C)(C)C(O)=O)=O)C6(C)C













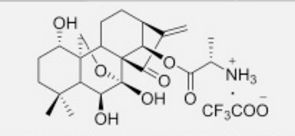
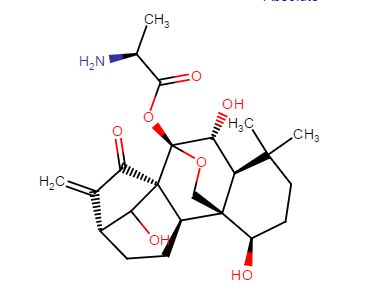 .CF3COOH
.CF3COOH