Zamaporvint
READ ALL AT
https://newdrugapprovals.org/2024/09/15/zamaporvint/
CAS 946414-94-4
Pegylated engineered interleukin-2 (IL-2) with altered receptor binding
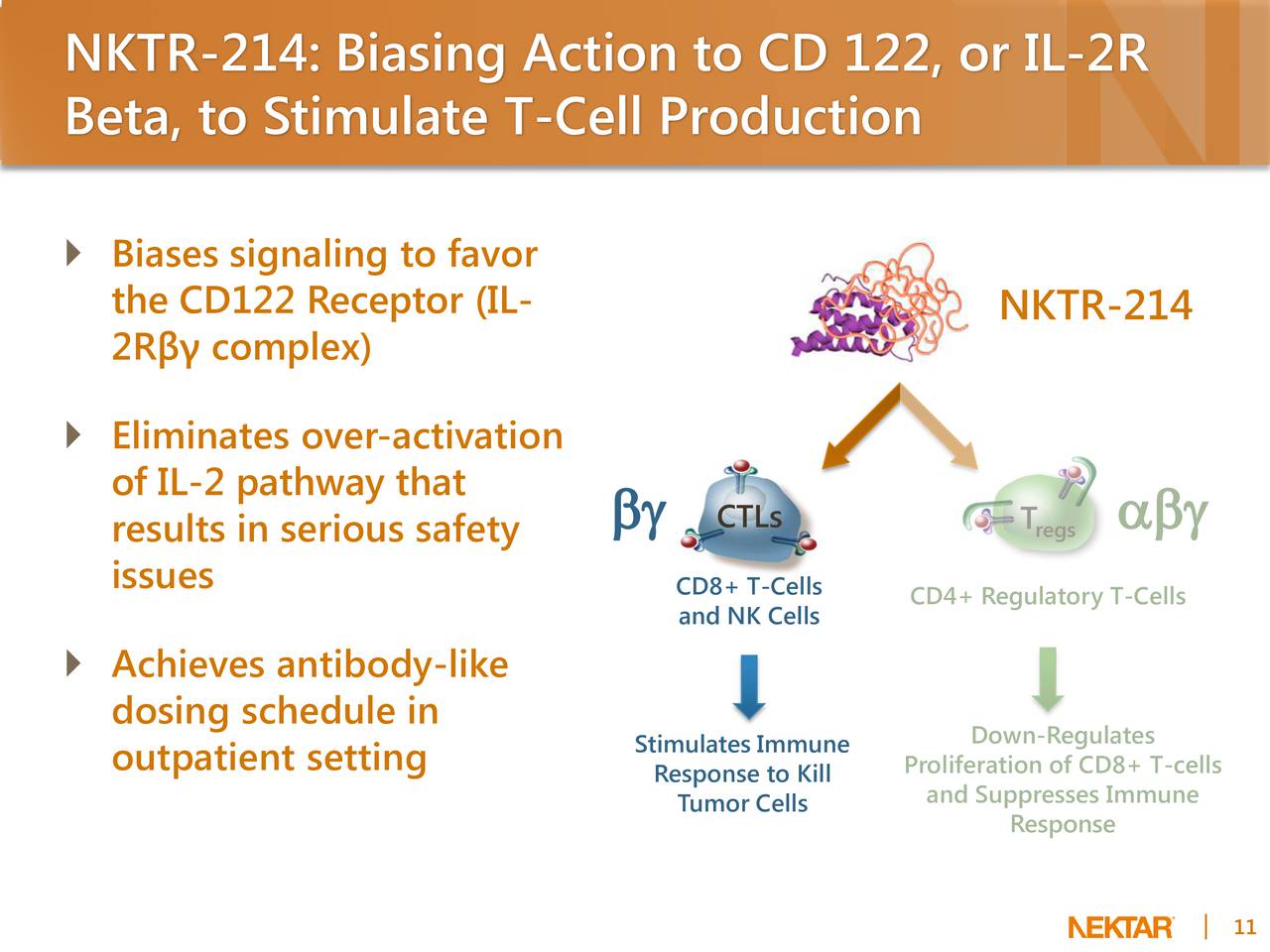
NKTR-214 is a cytokine (investigational agent) that is designed to target CD122, a protein which is found on certain immune cells (known as CD8+ T Cells and Natural Killer Cells) to expand these cells to promote their anti-tumor effects. Nivolumab is a full human monoclonal antibody that binds to a molecule called PD-1 (programmed cell death protein 1) on immune cells and promotes anti-tumor effects.
Sequence Length: 1308, 440, 440, 214, 214multichain; modified (modifications unspecified)
NKTR-214 is a CD122-biased cytokine in phase II clinical trials at the M.D. Anderson Cancer Center for the treatment of advanced sarcoma in combination with nivolumab.
M.D. Anderson Cancer Center, PHASE 2, SARCOMA
RESEARCH FOCUS: Immuno-oncology
DISCOVERED AND WHOLLY OWNED BY NEKTAR
In clinical collaboration with![]()
NKTR-214 is a CD122-biased agonist designed to stimulate the patient’s own immune system to fight cancer. NKTR-214 is designed to grow specific cancer-killing T cells and natural killer (NK) cell populations in the body which fight cancer, which are known as endogenous tumor-infiltrating lymphocytes (TILs). NKTR-214 stimulates these cancer-killing immune cells in the body by targeting CD122 specific receptors found on the surface of these immune cells, known as CD8+ effector T cells and Natural Killer (NK) cells. CD122, which is also known as the Interleukin-2 receptor beta subunit, is a key signaling receptor that is known to increase proliferation of these effector T cells.1 In preclinical studies, treatment with NKTR-214 results in a rapid expansion of these cells and mobilization into the tumor micro-environment. NKTR-214 has an antibody-like dosing regimen similar to the existing checkpoint inhibitor class of approved medicines.
In preclinical studies, NKTR-214 demonstrated a mean ratio of 450:1 within the tumor micro-environment of CD8-positive effector T cells, which promote tumor destruction, compared with CD4-positive regulatory T cells, which are a type of cell that can suppress tumor-killing T cells.2Furthermore, a single dose of NKTR-214 resulted in a 500-fold AUC exposure within the tumor compared with an equivalent dose of the existing IL-2 therapy, enabling, for the first time, an antibody-like dosing regimen for a cytokine.2 In dosing studies in non-human primates, there was no evidence of severe side effects such as low blood pressure or vascular leak syndrome with NKTR-214 at predicted clinical therapeutic doses.2 NKTR-214 has a range of potential uses against multiple tumor types, including melanoma (the most serious type of skin cancer), kidney cancer and non-small cell lung cancer (the most common form of lung cancer).
A Phase 1 study evaluating NKTR-214 as a single agent in patients with locally recurrent or metastatic solid tumors including melanoma, renal cell carcinoma (RCC), bladder, colorectal and other solid tumors is ongoing with patient enrollment complete. Results from this Phase 1 trial were presented at the Society for Immunotherapy of Cancer (SITC) 2016 Annual Meeting and showed encouraging evidence of anti-tumor activity, and a favorable safety and tolerability profile. (Poster #387)
In September 2016, Nektar entered into a clinical collaboration with Bristol-Myers Squibb to evaluate NKTR-214 as a potential combination treatment regimen with Opdivo (nivolumab) in five tumor types and eight potential indications. The Phase 1/2 PIVOT clinical trials, known as PIVOT-02 and PIVOT-04 will enroll up to 260 patients and will evaluate the potential for the combination of Opdivo (nivolumab) and NKTR-214 to show improved and sustained efficacy and tolerability above the current standard of care in melanoma, kidney, triple-negative breast cancer, bladder and non-small cell lung cancer patients.
In May 2017, Nektar entered into a research collaboration with Takeda to explore the combination of NKTR-214 with five oncology compounds from Takeda’s cancer portfolio including a SYK-inhibitor and a proteasome inhibitor. The collaboration will explore the anti-cancer activity of NKTR-214 combined with five different targeted mechanisms in preclinical tumor models of lymphoma, melanoma and colorectal cancer to identify which combination treatment regimens show the most promise for possible advancement into the clinic.
Under the terms of the collaboration, the companies will share costs related to the preclinical studies and each will contribute their respective compounds to the research collaboration. Nektar and Takeda will each maintain global commercial rights to their respective drugs and/or drug candidates.
Additional development plans for NKTR-214 include combination studies with additional checkpoint inhibitors, cell therapies and vaccines.

The dose-escalation stage of the Excel Phase 1/2 study is designed to evaluate safety, efficacy, and define the recommended Phase 2 dose of NKTR-214 in approximately 20 patients with solid tumors. In addition to a determination of the recommended Phase 2 dose, the study will assess preliminary anti-tumor activity, including objective response rate (ORR). The immunologic effect of NKTR-214 on tumor-infiltrating lymphocytes (TILs) and other immune infiltrating cells in both blood and tumor tissue will also be assessed. Enrollment in the dose escalation study is completed. More information on the Excel Phase 1/2 study can be found on clinicaltrials.gov.
![]()
The dose escalation stage of the PIVOT program (PIVOT-02 Phase 1/2 study) is underway and will determine the recommended Phase 2 dose of NKTR-214 administered in combination with nivolumab. The study is first evaluating the clinical benefit, safety, and tolerability of combining NKTR-214 with nivolumab in approximately 30 patients with melanoma, renal cell carcinoma or non-small cell lung cancer. Once the recommended Phase 2 dose is achieved, the study will expand into additional patients for each tumor type. The second phase of the expansion cohorts in the PIVOT program (PIVOT-04 Phase 2 study) will evaluate safety and efficacy of the combination in up to 260 patients, in five tumor types and eight indications, including first and second-line melanoma, second-line renal cell carcinoma in immune-oncology therapy (IO) naïve and IO-relapsed patients, second-line non-small cell lung cancer in IO-naïve and IO-relapsed patients, first-line urothelial carcinoma, and second-line triple negative breast cancer. This study is expected to initiate in the second quarter of 2017.
Information on the PIVOT-02 study can be found on clinicaltrials.gov.

The dose escalation stage of the PROPEL program will determine the recommended Phase 2 dose of NKTR-214 administered in combination with anti-PD-L1 agent, atezolizumab or anti-PD-1 agent, pembrolizumab. The study will evaluate the clinical benefit, safety and tolerability of combining NKTR-214 with atezolizumab or pembrolizumab and will enroll patients into two separate arms concurrently. The first arm will evaluate an every three-week dose regimen of NKTR-214 in combination with atezolizumab in up to 30 patients in approved treatment settings of atezolizumab, including patients with non-small cell lung cancer or bladder cancer. The second arm will evaluate an every three-week dose regimen of NKTR-214 in combination with pembrolizumab in up to 30 patients in approved treatment settings of pembrolizumab, including patients with melanoma, non-small cell lung cancer or bladder cancer.
Information on the PROPEL study can be found on clinicaltrials.gov.
References
1Boyman, J., et al., Nature Reviews Immunology, 2012, 12, 180-190.
2Charych, D., et al., Clin Can Res; 22(3) February 1, 2016
http://www.nektar.com/application/files/7714/7887/7212/2016_SITC_NKTR-214-clinical_poster.pdf
https://www.google.co.in/patents/WO2015125159A1?cl=en
| Inventors | Murali Krishna Addepalli, Deborah H. Charych, Seema Kantak, Steven Robert Lee |
| Applicant | Nektar Therapeutics (India) Pvt. Ltd., Nektar Therapeutics |
////////////946414-94-4, BMS 936558, MDX 1106, NKTR 214, ONO 4538, Opdivio, NIVOLUMAB, PHASE 2
AMISELIMOD
UNII-358M5150LY; CAS 942399-20-4; 358M5150LY; MT-1303; Amiselimod, MT-1303
| Molecular Formula: | C19H30F3NO3 |
|---|---|
| Molecular Weight: | 377.448 g/mol |
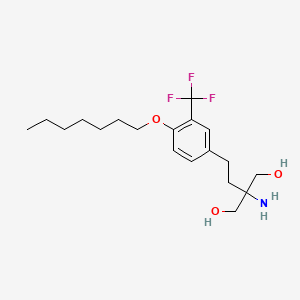
2-amino-2-[2-[4-heptoxy-3-(trifluoromethyl)phenyl]ethyl]propane-1,3-diol
Phase II Crohn’s disease; Multiple sclerosis; Plaque psoriasis
Amiselimod, also known as MT1303, is a potent and selective immunosuppressant and sphingosine 1 phosphate receptor modulator. Amiselimod may be potentially useful for treatment of multiple sclerosis; inflammatory diseases; autoimmune diseases; psoriasis and inflammatory bowel diseases. Amiselimod is currently being developed by Mitsubishi Tanabe Pharma Corporation
Mitsubishi Tanabe is developing amiselimod, an oral sphingosine-1-phosphate (S1P) receptor antagonist, for treating autoimmune diseases, primarily multiple sclerosis, psoriasis and inflammatory bowel diseases, including Crohn’s disease.
EU states expire 2026, and
Expire in the US in June 2030 with US154 extension.
| Inventors | Masatoshi Kiuchi, Kaoru Marukawa, Nobutaka Kobayashi, Kunio Sugahara |
| Applicant | Mitsubishi Tanabe Pharma Corporation |
In recent years, calcineurin inhibitors such as cyclosporine FK 506 have been used to suppress rejection of patients receiving organ transplantation. While doing it, certain calcineurin inhibitors like cyclosporin can cause harmful side effects such as nephrotoxicity, hepatotoxicity, neurotoxicity, etc. For this reason, in order to suppress rejection reaction in transplant patients, development of drugs with higher safety and higher effectiveness is advanced.
[0003] Patent Documents 1 to 3 are useful as inhibitors of (acute or chronic) rejection in organ or bone marrow transplantation and also useful as therapeutic agents for various autoimmune diseases such as psoriasis and Behcet’s disease and rheumatic diseases 2 aminopropane 1, 3 dioly intermediates are disclosed.
[0004] One of these compounds, 2-amino-2- [2- (4-octylphenel) propane] 1, 3 diol hydrochloride (hereinafter sometimes referred to as FTY 720) is useful for renal transplantation It is currently under clinical development as an inhibitor of rejection reaction. FTY 720 is phosphorylated by sphingosine kinase in vivo in the form of phosphorylated FTY 720 [hereinafter sometimes referred to as FTY 720-P]. For example, 2 amino-2-phosphoryloxymethyl 4- (4-octafil-el) butanol. FTY720 – P has four types of S1 P receptors (hereinafter referred to as S1 P receptors) among five kinds of sphingosine – 1 – phosphate (hereinafter sometimes referred to as S1P) receptors It acts as an aggroove on the body (other than S1P2) (Non-Patent Document 1).
[0005] It has recently been reported that S1P1 among the S1P receptors is essential for the export of mature lymphocytes with thymus and secondary lymphoid tissue forces. FTY720 – P downregulates S1P1 on lymphocytes by acting as S1P1 ghost. As a result, the transfer of mature lymphocytes from the thymus and secondary lymphatic tissues is inhibited, and the circulating adult lymphocytes in the blood are isolated in the secondary lymphatic tissue to exert an immunosuppressive effect Has been suggested (
Non-Patent Document 2).
[0006] On the other hand, conventional 2-aminopropane 1, 3 dioly compounds are concerned as transient bradycardia expression as a side effect, and in order to solve this problem, 2-aminopropane 1, 3 diiori Many new compounds have been reported by geometrically modifying compounds. Among them, as a compound having a substituent on the benzene ring possessed by FTY 720, Patent Document 4 discloses an aminopropenol derivative as a S1P receptor modulator with a phosphate group, Patent Documents 5 and 6 are both S1P Discloses an amino-propanol derivative as a receptor modulator. However, trihaloalkyl groups such as trifluoromethyl groups are not disclosed as substituents on the benzene ring among them. In any case, it is currently the case that it has not yet reached a satisfactory level of safety as a pharmaceutical.
Patent Document 1: International Publication Pamphlet WO 94 Z 08943
Patent Document 2: International Publication Pamphlet WO 96 Z 06068
Patent Document 3: International Publication Pamphlet W 0 98 z 45 429
Patent Document 4: International Publication Pamphlet WO 02 Z 076995
Patent document 5: International public non-fret WO 2004 Z 096752
Patent Document 6: International Publication Pamphlet WO 2004 Z 110979
Non-patent document 1: Science, 2002, 296, 346-349
Non-patent document 2: Nature, 2004, 427, 355-360
Reference Example 3
5 bromo 2 heptyloxybenzonitrile
(3- 1) 5 Synthesis of bromo-2 heptyloxybenzonitrile (Reference Example Compound 3- 1)
1-Heptanol (1.55 g) was dissolved in N, N dimethylformamide (24 ml) and sodium hydride (0.321 g) was added at room temperature. After stirring for 1 hour, 5 bromo-2 fluoborosyl-tolyl (2.43 g) was added and the mixture was further stirred for 50 minutes. The reaction solution was poured into water, extracted with ethyl acetate, washed with water, saturated brine, dried over anhydrous sodium sulfate, and the solvent was distilled off under reduced pressure. After eliminating the 5 bromo 2 fluconate benzonitrile as a raw material, the reaction was carried out again under the same conditions and purification was carried out by silica gel column chromatography (hexane: ethyl acetate = 50: 1 to 5: 1) to obtain the desired product (3.10 g ) As a colorless oil.
– NMR (CDCl 3) δ (ppm): 0.89 (3H, t, J = 6.4 Hz), 1.24-1.35 (6H, m
J = 8.8 Hz), 1.48 (2H, quint, J = 7.2 Hz), 1.84 7.59 (1 H, dd, J = 8.8, 2.4 Hz), 7.65 (1 H, d, J = 2.4 Hz).
Example 1
2 Amino 2- [2- (4-heptyloxy-3 trifluoromethylph enyl) propane-1, 3-diol hydrochloride
(1 – 1) {2, 2 Dimethyl 5- [2- (4 hydroxy 3 trifluoromethylfuethyl) ethyl] 1,3 dioxane 5 mercaptothenylboronic acid t butyl ester (synthesis compound 1 1)
Reference Example Compound 2-5 (70.3 g) was dissolved in tetrahydrofuran (500 ml), t-butoxycallium (13.Og) was added, and the mixture was stirred for 1 hour. To the mixed solution was dropwise added a solution of the compound of Reference Example 1 (15.Og) in tetrahydrofuran (100 ml) under ice cooling, followed by stirring for 2 hours under ice cooling. Water was added to the reaction solution, the mixture was extracted with ethyl acetate, washed with water, saturated brine, dried with anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure. The residue was purified by silica gel column chromatography (hexane: ethyl acetate = 3: D to obtain 31. Og of a pale yellow oily matter.) The geometric isomer ratio of the obtained product was (E : Z = 1: 6).
This pale yellow oil was dissolved in ethyl acetate (200 ml), 10% palladium carbon (3.00 g) was added, and the mixture was stirred under a hydrogen atmosphere at room temperature for 7 hours. After purging the inside of the reaction vessel with nitrogen, the solution was filtered and the filtrate was concentrated. The residue was washed with diisopropyl ether to obtain the desired product (2.2 g) as a colorless powder.
1 H-NMR (CDCl 3) δ (ppm): 1. 43 (3H, s), 1.44 (3H, s), 1. 47 (9H, s), 1
(2H, m), 91- 1. 98 (2H, m), 2. 50-2.66 (2H, m), 3. 69 (2H, d, J = Il. 6 Hz), 3. 89 J = 8.2 Hz), 7. 22 (1 H, dd J = 8 Hz), 5. 02 (1 H, brs), 5. 52 . 2, 1. 7 Hz), 7. 29 (1 H, d, J = l. 7 Hz).
(1-2) {2,2 Dimethyl-5- [2- (4heptyloxy-3 trifluoromethyl) ethyl] 1,3 dioxane 5-mercaptobutyric acid t-butyl ester Synthesis (compound 1 2)
Compound 1-1 (510 mg) was dissolved in N, N dimethylformamide (10 ml), potassium carbonate (506 mg) and n-heptyl bromide (0.235 ml) were added and stirred at 80 ° C. for 2 hours. Water was added to the reaction solution, the mixture was extracted with ethyl acetate, washed with water and saturated brine, dried with anhydrous sulfuric acid
The resultant was dried with GENSCHUM and the solvent was distilled off under reduced pressure to obtain the desired product (640 mg) as a colorless oil.
– NMR (CDCl 3) δ (ppm): 0.89 (3H, t, J = 6.8 Hz), l.30-1.37 (6H, m
(2H, m), 1.91-1.98 (2H, m), 1.42-1.50 (2H, m), 1.42 (3H, s), 1.44 (3H, s), 1.47 J = 16.6 Hz), 4.00 (2H, t, J = 6.4 Hz), 4.9 8 (2H, d, J = 11.6 Hz), 3.69 1 H, brs), 6.88 (1 H, d, J = 8.5 Hz), 7.26 – 7.29 (1 H, m), 7.35 (1 H, d, J = 1.5 Hz).
(1-3) Synthesis of 2-amino-2- [2- (4heptyloxy 3 trifluoromethyl) ethyl] propane 1, 3 diol hydrochloride (Compound 1- 3)
Compound 12 (640 mg) was dissolved in ethanol (15 ml), concentrated hydrochloric acid (3 ml) was caught and stirred at 80 ° C. for 2 hours. The reaction solution was concentrated, and the residue was washed with ethyl ether to give the desired product (492 mg) as a white powder.
MS (ESI) m / z: 378 [M + H]
– NMR (DMSO-d) δ (ppm): 0.86 (3H,
6 t, J = 6.8 Hz), 1.24 – 1.39 (6
(4H, m), 3.51 (4H, d, J = 5. lHz), 4.06 (2H, m), 1.39-1.46 (2H, m), 1.68-1.78 (4H, m), 2.55-2.22 , 7.32 (2H, t, J = 5.1 Hz), 7.18 (1 H, d, J = 8.4 Hz), 7.42 – 7.45 (2 H, m), 7.76 (3 H, brs;).
PATENT
WO 2009119858
JP 2011136905
WO 2017188357
PATENT
WO-2018021517

Patent Document 1: WO2007 / 069712
[Chemical formula 3]


PATENTS
////////////AMISELIMOD, Phase II, Crohn’s disease, Multiple sclerosis, Plaque psoriasis, MT-1303, MT1303, MT 1303, Mitsubishi Tanabe Pharma Corporation, Mitsubishi , JAPAN, PHASE 2
CCCCCCCOC1=C(C=C(C=C1)CCC(CO)(CO)N)C(F)(F)F
ACT-334441
Cenerimod
UNII-Y333RS1786; Y333RS1786
S1P receptor 1 agonist
CAS 1262414-04-9
Chemical Formula: C25H31N3O5
Exact Mass: 453.22637
Martin Bolli, Cyrille Lescop, Boris Mathys,Keith Morrison, Claus Mueller, Oliver Nayler,Beat Steiner,
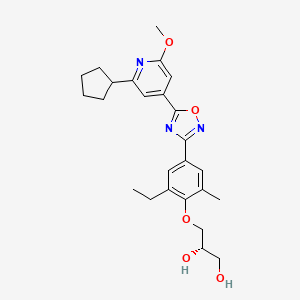
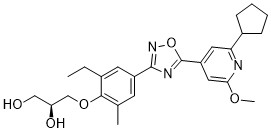
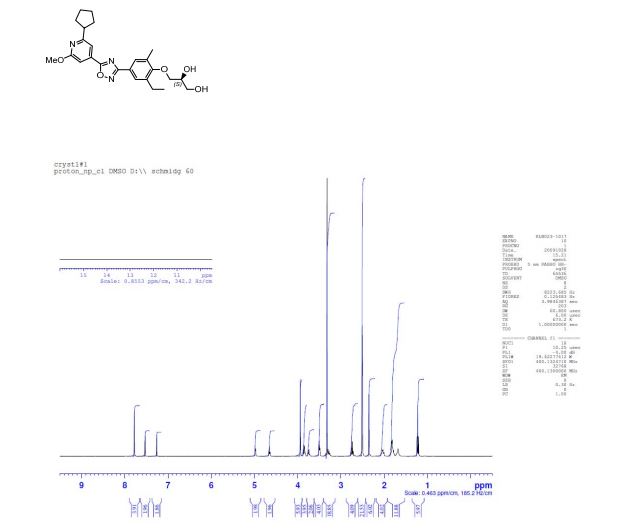
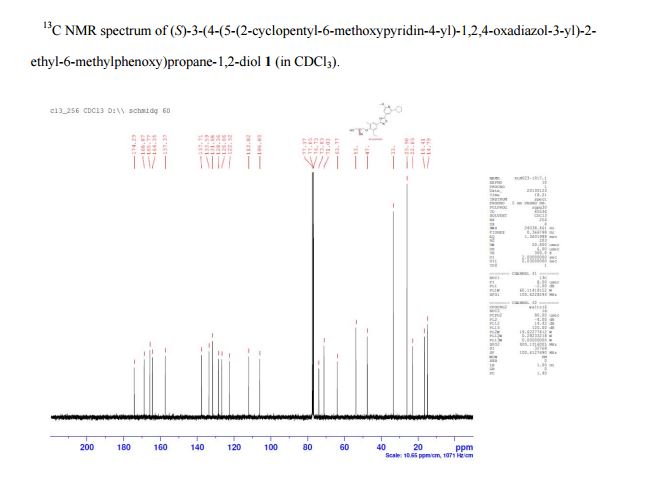
(S)-3-(4-(5-(2-cyclopentyl-6-methoxypyridin-4-yl)-1,2,4-oxadiazol-3-yl)-2-ethyl-6-methylphenoxy)propane-1,2-diol
(2S)-3-[4-[5-(2-cyclopentyl-6-methoxypyridin-4-yl)-1,2,4-oxadiazol-3-yl]-2-ethyl-6-methylphenoxy]propane-1,2-diol
(S)-3-(4-(5-(2-Cyclopentyl-6-methoxypyridin-4-yl)-1,2,4-oxadiazol-3-yl)-2-ethyl-6-methylphenoxy)propane-1,2-diol
(S)-3-{4-[5-(2-Cyclopentyl-6-methoxy-pyridin-4-yl)-[1,2,4]oxadiazol-3-yl]-2-ethyl-6-methyl-phenoxy}-propane-1,2-diol
| Mechanism Of Action | Sphingosine 1 phosphate receptor modulator |
|---|---|
| Who Atc Codes | L03A-X (Other immunostimulants) |
| Ephmra Codes | L3A (Immunostimulating Agents Excluding Interferons) |
| Indication | Systemic Lupus Erythematosus |
Cenerimod is a potent and orally active immunomodulator, exhibited EC50 value of 2.7 nM. Cenerimod is an agonist for the G protein-coupled receptor S1 P1/EDG1 and has a powerful and long-lasting immunomodulating effect which is achieved by reducing the number of circulating and infiltrating T- and B-lymphocytes, without affecting their maturation, memory, or expansion. Cenerimod may be useful for prevention or treatment of diseases associated with an activated immune system

CENERIMOD
ACT-334441; lysosphingolipid receptor agonist – Actelion; S1P1 receptor modulator – Actelion; Second selective S1P1 receptor agonist – Actelion; Sphingosine 1 phosphate receptor modulators – Actelion; Sphingosine 1-phosphate receptor 1 agonists – Actelion
| # | Nct Number | Title | Recruitment | Conditions | Interventions | Phase | |
|---|---|---|---|---|---|---|---|
| 1 | NCT02472795 | Clinical Study to Investigate the Biological Activity, Safety, Tolerability, and Pharmacokinetics of ACT-334441 in Subjects With Systemic Lupus Erythematosus | Recruiting | Systemic Lupus Erythematosus | Drug: ACT-334441|Drug: Placebo | Phase 2 Actelion | |
| 2 | NCT02479204 | Drug Interaction Study of ACT-334441 With Cardiovascular Medications in Healthy Subjects | Suspended | Healthy Subjects | Drug: ACT-334441 2 mg|Drug: ACT-334441 4 mg|Drug: placebo|Drug: atenolol|Drug: diltiazem ER | Phase 1 Actelion |

The human immune system is designed to defend the body against foreign micro-organisms and substances that cause infection or disease. Complex regulatory mechanisms ensure that the immune response is targeted against the intruding substance or organism and not against the host. In some cases, these control mechanisms are unregulated and autoimmune responses can develop. A consequence of the uncontrolled inflammatory response is severe organ, cell, tissue or joint damage. With current treatment, the whole immune system is usually suppressed and the body’s ability to react to infections is also severely compromised. Typical drugs in this class include azathioprine, chlorambucil, cyclophosphamide, cyclosporin, or methotrexate. Corticosteroids which reduce inflammation and suppress the immune response, may cause side effects when used in long term treatment. Nonsteroidal anti-inflammatory drugs (NSAIDs) can reduce pain and inflammation, however, they exhibit considerable side effects. Alternative treatments include agents that activate or block cytokine signaling.
Orally active compounds with immunomodulating properties, without compromising immune responses and with reduced side effects would significantly improve current treatments of uncontrolled inflammatory diseases.
In the field of organ transplantation the host immune response must be suppressed to prevent organ rejection. Organ transplant recipients can experience some rejection even when they are taking immunosuppressive drugs. Rejection occurs most frequently in the first few weeks after transplantation, but rejection episodes can also happen months or even years after transplantation. Combinations of up to three or four medications are commonly used to give maximum protection against rejection while minimizing side effects. Current standard drugs used to treat the rejection of transplanted organs interfere with discrete intracellular pathways in the activation of T-type or B-type white blood cells. Examples of such drugs are cyclosporin, daclizumab, basiliximab, everolimus, or FK506, which interfere with cytokine release or signaling; azathioprine or leflunomide, which inhibit nucleotide synthesis; or 15-deoxyspergualin, an inhibitor of leukocyte differentiation.
The beneficial effects of broad immunosuppressive therapies relate to their effects; however, the generalized immunosuppression which these drugs produce diminishes the immune system’s defense against infection and malignancies. Furthermore, standard immunosuppressive drugs are often used at high dosages and can cause or accelerate organ damage.
SYNTHESIS
PATENT
https://www.google.com/patents/WO2011007324A1?cl=zh
The human immune system is designed to defend the body against foreign microorganisms and substances that cause infection or disease. Complex regulatory mechanisms ensure that the immune response is targeted against the intruding substance or organism and not against the host. In some cases, these control mechanisms are unregulated and autoimmune responses can develop. A consequence of the uncontrolled inflammatory response is severe organ, cell, tissue or joint damage. With current treatment, the whole immune system is usually suppressed and the body’s ability to react to infections is also severely compromised. Typical drugs in this class include azathioprine, chlorambucil, cyclophosphamide, cyclosporin, or methotrexate. Corticosteroids which reduce inflammation and suppress the immune response, may cause side effects when used in long term treatment. Nonsteroidal anti-inflammatory drugs (NSAIDs) can reduce pain and inflammation, however, they exhibit considerable side effects. Alternative treatments include agents that activate or block cytokine signaling.
Orally active compounds with immunomodulating properties, without compromising immune responses and with reduced side effects would significantly improve current treatments of uncontrolled inflammatory diseases.
In the field of organ transplantation the host immune response must be suppressed to prevent organ rejection. Organ transplant recipients can experience some rejection even when they are taking immunosuppressive drugs. Rejection occurs most frequently in the first few weeks after transplantation, but rejection episodes can also happen months or even years after transplantation. Combinations of up to three or four medications are commonly used to give maximum protection against rejection while minimizing side effects. Current standard drugs used to treat the rejection of transplanted organs interfere with discrete intracellular pathways in the activation of T-type or B-type white blood cells. Examples of such drugs are cyclosporin, daclizumab, basiliximab, everolimus, or FK506, which interfere with cytokine release or signaling; azathioprine or leflunomide, which inhibit nucleotide synthesis; or 15-deoxyspergualin, an inhibitor of leukocyte differentiation.
The beneficial effects of broad immunosuppressive therapies relate to their effects; however, the generalized immunosuppression which these drugs produce diminishes the immune system’s defense against infection and malignancies. Furthermore, standard immunosuppressive drugs are often used at high dosages and can cause or accelerate organ damage.
Description of the invention
The present invention provides novel compounds of Formula (I) that are agonists for the G protein-coupled receptor S1 P1/EDG1 and have a powerful and long-lasting immunomodulating effect which is achieved by reducing the number of circulating and infiltrating T- and B-lymphocytes, without affecting their maturation, memory, or expansion. The reduction of circulating T- / B-lymphocytes as a result of S1 P1/EDG1 agonism, possibly in combination with the observed improvement of endothelial cell layer function associated with S1 P1/EDG1 activation, makes such compounds useful to treat uncontrolled inflammatory diseases and to improve vascular functionality. Prior art document WO 2008/029371 discloses compounds that act as S1 P1/EDG1 receptor agonists and show an immunomodulating effect as described above. Unexpectedly, it has been found that the compounds of the present invention have a reduced potential to constrict airway tissue/vessels when compared to compounds of the prior art document WO 2008/029371. The compounds of the present invention therefore demonstrate superiority with respect to their safety profile, e.g. a lower risk of bronchoconstriction.
Examples of WO 2008/029371 , which are considered closest prior art analogues are shown in Figure 1.

Figure 1 : Structure of Examples of prior art document WO 2008/029371 , which are considered closest analogues to the compounds of the present invention.
The data on the constriction of rat trachea rings compiled in Table 1 illustrate the superiority of the compounds of the present invention as compared to compounds of prior art document WO 2008/029371.
For instance, the compounds of Example 1 and 6 of the present invention show a significantly reduced potential to constrict rat trachea rings when compared to the compounds of prior art Examples 222 and 226 of WO 2008/029371 , respectively. Furthermore, the compounds of Example 1 and 6 of the present invention also show a reduced potential to constrict rat trachea rings when compared to the compounds of prior art Examples 196 and 204 of WO 2008/029371 , respectively. These data demonstrate that compounds wherein R1 represents 3-pentyl and R2 represents methoxy are superior compared to the closest prior art compounds of WO 2008/029371 , i.e. the compounds wherein R1 represents an isobutyl and R2 represents methoxy or wherein R1represents methyl and R2 represents 3-pentyl. Moreover, also the compound of Example 16 of the present invention, wherein R1 is 3-methyl-but-1-yl and R2 is methoxy, exhibits a markedly reduced potential to constrict rat trachea rings when compared to its closest analogue prior art Example 226 of WO 2008/029371 wherein R1 is isobutyl and R2 is methoxy.
The unexpected superiority of the compounds of the present invention is also evident from the observation that the compounds of Example 2 and 7 of the present invention show a markedly reduced potential to constrict rat trachea rings when compared to the compounds of prior art Examples 229 and 233 of WO 2008/029371 , respectively. This proves that compounds wherein R1represents cyclopentyl and R2 represents methoxy are superior compared to the closest prior art compounds of WO 2008/029371 , i.e. the compounds wherein R1 represents methyl and R2 represents cyclopentyl.
Also, the compound of Example 3 of the present invention exhibits the same low potential to constrict rat trachea rings as its S-enantiomer, i.e. the compound of Example 2 of the present invention, indicating that the configuration at this position has no significant effect on trachea constriction. Furthermore, also Example 21 of the present invention exhibits the same low potential to constrict rat trachea rings as present Example 2, which differs from Example 21 only by the linker A (forming a 5-pyridin-4-yl-[1 ,2,4]oxadiazole instead of a 3- pyridin-4-yl-[1 ,2,4]oxadiazole). This indicates that also the nature of the oxadiazole is not critical regarding trachea constriction.
Table 1 : Rat trachea constriction in % of the constriction induced by 50 mM KCI. n.d. = not determined. For experimental details and further data see Example 33.


result obtained at a compound concentration of 300 nM.
The compounds of the present invention can be utilized alone or in combination with standard drugs inhibiting T-cell activation, to provide a new immunomodulating therapy with a reduced propensity for infections when compared to standard immunosuppressive therapy. Furthermore, the compounds of the present invention can be used in combination with reduced dosages of traditional immunosuppressant therapies, to provide on the one hand effective immunomodulating activity, while on the other hand reducing end organ damage associated with higher doses of standard immunosuppressive drugs. The observation of improved endothelial cell layer function associated with S1 P1/EDG1 activation provides additional benefits of compounds to improve vascular function.
The nucleotide sequence and the amino acid sequence for the human S1 P1/EDG1 receptor are known in the art and are published in e.g.: HIa, T., and Maciag, T., J. Biol
Chem. 265 (1990), 9308-9313; WO 91/15583 published 17 October 1991 ; WO 99/46277 published 16 September 1999. The potency and efficacy of the compounds of Formula (I) are assessed using a GTPγS assay to determine EC5O values and by measuring the circulating lymphocytes in the rat after oral administration, respectively (see in experimental part). i) In a first embodiment, the invention relates to pyridine compounds of the Formula (I),

Formula (I)
WO 2013175397
https://www.google.com/patents/WO2013175397A1?cl=en
Pyridine-4-yl derivatives of formula (PD),

Formula (PD) A represents

(the asterisks indicate the bond that is linked to the pyridine group of Formula (PD));
Ra represents 3-pentyl, 3-methyl-but-1-yl, cyclopentyl, or cyclohexyl;
Rb represents methoxy;
Rc represents 2,3-dihydroxypropoxy, -OCH2-CH(OH)-CH2-NHCO-CH2OH,
-OCH2-CH(OH)-CH2N(CH3)-CO-CH2OH, -NHS02CH3, or -NHS02CH2CH3; and
Rd represents ethyl or chloro.)
disclosed in WO201 1007324, have immunomodulating activity through their S1 P1/EDG1 receptor agonistic activity. Therefore, those pyridine-4-yl derivatives are useful for prevention and / or treatment of diseases or disorders associated with an activated immune system, including rejection of transplanted organs such as kidney, liver, heart, lung, pancreas, cornea, and skin; graft-versus-host diseases brought about by stem cell transplantation; autoimmune syndromes including rheumatoid arthritis, multiple sclerosis, inflammatory bowel diseases such as Crohn’s disease and ulcerative colitis, psoriasis, psoriatic arthritis, thyroiditis such as Hashimoto’s thyroiditis, uveo-retinitis; atopic diseases such as rhinitis, conjunctivitis, dermatitis; asthma; type I diabetes; post-infectious autoimmune diseases including rheumatic fever and post-infectious glomerulonephritis; solid cancers and tumor metastasis. 2-Cyclopentyl-6-methoxy-isonicotinic acid, which is also disclosed in WO201 1007324, is a useful intermediate for the synthesis of the pyridine-4-yl derivatives of formula (PD), wherein Ra is a cyclopentyl group.
In the process described in WO201 1007324, 2-cyclopentyl-6-methoxy-isonicotinic acid was prepared according to the following reaction scheme 1 :

Compound D Compound E
Rieke Zinc: cyclopentylzinc bromide;
PdCI2(dppf)dcm: 1 ,1 ‘-Bis(diphenylphosphino)ferrocene-palladium(ll)dichloride
dichloromethane complex
However, the abovementioned process has drawbacks for larger scale, i.e. industrial scale synthesis of 2-cyclopentyl-6-methoxy-isonicotinic acid, for the following reasons:
a) The commercially available starting material, 2,6-dichloro-isonicotinic acid (Compound A) is expensive.
b) The conversion of Compound C to Compound D is cost-intensive. The reaction has to be performed under protective atmosphere with expensive palladium catalysts and highly reactive and expensive Rieke zinc complex. Such synthesis steps are expensive to scale up and it was therefore highly desired to find alternative synthesis methods.
Even though Goldsworthy, J. Chem. Soc. 1934, 377-378 discloses the preparation of 1 -cyclopentylethanone, which is a key building block in the new process of the present invention, by using ethyl 1 -acetoacetate as a starting material, this synthesis was far from being suitable in an industrial process. The reported yield was low (see also under “Referential Examples” below). Scheme 2

ethyl 1 -acetylcyclo- 1-cyclopentyl- pentanecarboxylate ethanone
Besides the early work by Goldsworthy there are several recent examples for the preparation of 1 -cyclopentylethanone described in the literature. Such examples include:
1 ) Addition of methyl lithium to a N-cyclopentanecarbonyl-N,0-dimethylhydroxylamine at -78°C in a yield of 77%. US2006/199853 A1 , 2006 and US2006/223884 A1 , 2006.
2) Addition of methyl lithium to a cyclopentyl carboxylic acid in diethylether at -78°C in a yield of 81 %. J. Am. Chem. Soc, 1983, 105, 4008-4017.
3) Addition of methylmagnesiumbromide to cyclopentanecarbonitrile.
Bull. Soc. Chim. Fr., 1967, 3722-3729.
4) Oxidation of 1 -cyclopentylethanol with chromtrioxide. US5001 140 A1 , 1991.
WO2009/71707 A1 , 2009.
5) Addition of cyclopentylmagnesium bromide to acetic anhydride at -78 °C with a yield of 54%. WO2004/74270 A2, 2004.
6) Synthesis of 1-cyclopentylethanone in 5 steps from cyclopentanone. Zhang, Pang; Li, Lian-chu, Synth. Commun., 1986, 16, 957-966.
However, the processes described in the above-listed publications are not efficient for scale-up since they require cryogenic temperatures, expensive starting materials, toxic reagents or many steps. The lack of an efficient process to manufacture 1 -cyclopentylethanone is further also mirrored by the difficulty in sourcing this compound on kilogram scale for a reasonable price and delivery time. Therefore, the purpose of the present invention is to provide a new, efficient and cost effective process for the preparation of 2-cyclopentyl-6-methoxy-isonicotinic acid, which is suitable for industrial scale synthesis.
Patent
Disclosed in WO2011007324, have immunomodulating activity through their S1P1/EDG1 receptor agonistic activity. Therefore, those pyridine-4-yl derivatives are useful for prevention and/or treatment of diseases or disorders associated with an activated immune system, including rejection of transplanted organs such as kidney, liver, heart, lung, pancreas, cornea, and skin; graft-versus-host diseases brought about by stem cell transplantation; autoimmune syndromes including rheumatoid arthritis, multiple sclerosis, inflammatory bowel diseases such as Crohn’s disease and ulcerative colitis, psoriasis, psoriatic arthritis, thyroiditis such as Hashimoto’s thyroiditis, uveo-retinitis; atopic diseases such as rhinitis, conjunctivitis, dermatitis; asthma; type I diabetes; post-infectious autoimmune diseases including rheumatic fever and post-infectious glomerulonephritis; solid cancers and tumor metastasis. 2-Cyclopentyl-6-methoxy-isonicotinic acid, which is also disclosed in WO2011007324, is a useful intermediate for the synthesis of the pyridine-4-yl derivatives of formula (PD), wherein Ra is a cyclopentyl group.
 Rieke Zinc: cyclopentylzinc bromide;
Rieke Zinc: cyclopentylzinc bromide;
PdCl2(dppf)dcm: 1,1′-Bis(diphenylphosphino)ferrocene-palladium(II)dichloride dichloromethane complex


PATENT
https://www.google.com/patents/US8658675
Martin Bolli, Cyrille Lescop, Boris Mathys,Keith Morrison, Claus Mueller, Oliver Nayler,Beat Steiner,
novel compounds of Formula (I) that are agonists for the G protein-coupled receptor S1P1/EDG1 and have a powerful and long-lasting immunomodulating effect which is achieved by reducing the number of circulating and infiltrating T- and B-lymphocytes, without affecting their maturation, memory, or expansion. The reduction of circulating T-/B-lymphocytes as a result of S1P1/EDG1 agonism, possibly in combination with the observed improvement of endothelial cell layer function associated with S1P1/EDG1 activation, makes such compounds useful to treat uncontrolled inflammatory diseases and to improve vascular functionality. Prior art document WO 2008/029371 discloses compounds that act as S1P1/EDG1 receptor agonists and show an immunomodulating effect as described above. Unexpectedly, it has been found that the compounds of the present invention have a reduced potential to constrict airway tissue/vessels when compared to compounds of the prior art document WO 2008/029371. The compounds of the present invention therefore demonstrate superiority with respect to their safety profile, e.g. a lower risk of bronchoconstriction.
Examples of WO 2008/029371, which are considered closest prior art analogues are shown in FIG. 1.


The data on the constriction of rat trachea rings compiled in Table 1 illustrate the superiority of the compounds of the present invention as compared to compounds of prior art document WO 2008/029371.
For instance, the compounds of Example 1 and 6 of the present invention show a significantly reduced potential to constrict rat trachea rings when compared to the compounds of prior art Examples 222 and 226 of WO 2008/029371, respectively. Furthermore, the compounds of Example 1 and 6 of the present invention also show a reduced potential to constrict rat trachea rings when compared to the compounds of prior art Examples 196 and 204 of WO 2008/029371, respectively. These data demonstrate that compounds wherein R1 represents 3-pentyl and R2represents methoxy are superior compared to the closest prior art compounds of WO 2008/029371, i.e. the compounds wherein R1 represents an isobutyl and R2represents methoxy or wherein R1 represents methyl and R2 represents 3-pentyl. Moreover, also the compound of Example 16 of the present invention, wherein R1is 3-methyl-but-1-yl and R2 is methoxy, exhibits a markedly reduced potential to constrict rat trachea rings when compared to its closest analogue prior art Example 226 of WO 2008/029371 wherein R1 is isobutyl and R2 is methoxy.
The unexpected superiority of the compounds of the present invention is also evident from the observation that the compounds of Example 2 and 7 of the present invention show a markedly reduced potential to constrict rat trachea rings when compared to the compounds of prior art Examples 229 and 233 of WO 2008/029371, respectively. This proves that compounds wherein R1 represents cyclopentyl and R2 represents methoxy are superior compared to the closest prior art compounds of WO 2008/029371, i.e. the compounds wherein R1represents methyl and R2 represents cyclopentyl.
Preparation of Intermediates2-Chloro-6-methyl-isonicotinic acid
The title compound and its ethyl ester are commercially available.
2-(1-Ethyl-propyl)-6-methoxy-isonicotinic acid
a) To a solution of 2,6-dichloroisonicotinic acid (200 g, 1.04 mol) in methanol (3 L), 32% aq. NaOH (770 mL) is added. The stirred mixture becomes warm (34° C.) and is then heated to 70° C. for 4 h before it is cooled to rt. The mixture is neutralised by adding 32% aq. HCl (100 mL) and 25% aq. HCl (700 mL). The mixture is stirred at rt overnight. The white precipitate that forms is collected, washed with methanol and dried. The filtrate is evaporated and the residue is suspended in water (200 mL). The resulting mixture is heated to 60° C. Solid material is collected, washed with water and dried. The combined crops give 2-chloro-6-methoxy-isonicotinic acid (183 g) as a white solid; LC-MS: tR=0.80 min, [M+1]+=187.93.
b) To a suspension of 2-chloro-6-methoxy-isonicotinic acid (244 g, 1.30 mol) in methanol (2.5 L), H2SO4 (20 mL) is added. The mixture is stirred at reflux for 24 h before it is cooled to 0° C. The solid material is collected, washed with methanol (200 mL) and water (500 mL) and dried under HV to give 2-chloro-6-methoxy-isonicotinic acid methyl ester (165 g) as a white solid; LC-MS: tR=0.94 min, [M+1]+=201.89.
c) Under argon, Pd(dppf) (3.04 g, 4 mmol) is added to a solution of 2-chloro-6-methoxy-isonicotinic acid methyl ester (50 g, 0.248 mol) in THF (100 mL). A 0.5 M solution of 3-pentylzincbromide in THF (550 mL) is added via dropping funnel. Upon complete addition, the mixture is heated to 85° C. for 18 h before it is cooled to rt. Water (5 mL) is added and the mixture is concentrated. The crude product is purified by filtration over silica gel (350 g) using heptane:EA 7:3 to give 2-(1-ethyl-propyl)-6-methoxy-isonicotinic acid methyl ester (53 g) as a pale yellow oil; 1H NMR (CDCl3): δ0.79 (t, J=7.5 Hz, 6H), 1.63-1.81 (m, 4H), 2.47-2.56 (m, 1H), 3.94 (s, 3H), 3.96 (s, 3H), 7.12 (d, J=1.0 Hz, 1H), 7.23 (d, J=1.0 Hz, 1H).
d) A solution of 2-(1-ethyl-propyl)-6-methoxy-isonicotinic acid methyl ester (50 g, 0.211 mol) in ethanol (250 mL), water (50 mL) and 32% aq. NaOH (50 mL) is stirred at 80° C. for 1 h. The mixture is concentrated and the residue is dissolved in water (200 mL) and extracted with TBME. The org. phase is separated and washed once with water (200 mL). The TBME phase is discarded. The combined aq. phases are acidified by adding 25% aq. HCl and then extracted with EA (400+200 mL). The combined org. extracts are concentrated. Water (550 mL) is added to the remaining residue. The mixture is heated to 70° C., cooled to rt and the precipitate that forms is collected and dried to give the title compound (40.2 g) as a white solid; LC-MS: tR=0.95 min, [M+1]+=224.04; 1H NMR (D6-DMSO): δ 0.73 (t, J=7.3 Hz, 6H), 1.59-1.72 (m, 4H), 2.52-2.58 (m, 1H), 3.88 (s, 3H), 7.00 (d, J=1.0 Hz, 1H), 7.20 (d, J=1.0 Hz, 1H).
2-Methoxy-6-(3-methyl-butyl)-isonicotinic acid
The title compound is prepared in analogy to 2-(1-ethyl-propyl)-6-methoxy-isonicotinic acid; LC-MS: tR=0.94 min, [M+1]+=224.05; 1H NMR (D6-DMSO): δ 0.92 (d, J=5.8 Hz, 6H), 1.54-1.62 (m, 3H), 2.70-2.76 (m, 2H), 3.88 (s, 3H), 6.99 (s, 1H), 7.25 (s, 1H), 13.52 (s).
2-Cyclopentyl-6-methoxy-isonicotinic acid
The title compound is prepared in analogy to 2-(1-ethyl-propyl)-6-methoxy-isonicotinic acid; LC-MS: tR=0.93 min, [M+1]+=222.02; 1H NMR (CDCl3): δ 1.68-1.77 (m, 2H), 1.81-1.90 (m, 4H), 2.03-2.12 (m, 2H), 3.15-3.25 (m, 1H), 3.99 (s, 3H), 7.18 (d, J=1.0 Hz, 1H), 7.35 (d, J=0.8 Hz, 1H).
2-Cyclohexyl-6-methoxy-isonicotinic acid
The title compound is prepared in analogy to 2-(1-ethyl-propyl)-6-methoxy-isonicotinic acid; LC-MS: tR=0.98 min, [M+1]+=236.01; 1H NMR (D6-DMSO): δ 1.17-1.29 (m, 1H), 1.31-1.43 (m, 2H), 1.44-1.55 (m, 2H), 1.67-1.73 (m, 1H), 1.76-1.83 (m, 2H), 1.84-1.92 (m, 2H), 2.66 (tt, J=11.3, 3.3 Hz, 1H), 3.88 (s, 3H), 7.00 (d, J=1.0 Hz, 1H), 7.23 (d, J=1.0 Hz, 1H).
2-Cyclopentyl-N-hydroxy-6-methoxy-isonicotinamidine
a) A solution of 2-cyclopentyl-6-methoxy-isonicotinic acid methyl ester (3.19 g, 13.6 mmol) in 7 N NH3 in methanol (50 mL) is stirred at 60° C. for 18 h. The solvent is removed in vacuo and the residue is dried under HV to give crude 2-cyclopentyl-6-methoxy-isonicotinamide (3.35 g) as a pale yellow solid; LC-MS**: tR=0.57 min, [M+1]+=221.38.
b) Pyridine (8.86 g, 91.3 mmol) is added to a solution of 2-cyclopentyl-6-methoxy-isonicotinamide (3.35 g, 15.2 mmol) in DCM (100 mL). The mixture is cooled to 0° C. before trifluoroacetic acid anhydride (9.58 g, 45.6 mmol) is added portionwise. The mixture is stirred at 0° C. for 1 h before it is diluted with DCM (100 mL) and washed with sat. aq. NaHCO3 solution (100 mL) and brine (100 mL). The separated org. phase is dried over MgSO4, filtered and concentrated. The crude product is purified by CC on silica gel eluting with heptane:EA 9:1 to give 2-cyclopentyl-6-methoxy-isonicotinonitrile (2.09 g) as pale yellow oil; LC-MS**: tR=0.80 min, [M+1]+=not detectable; 1H NMR (D6-DMSO): δ 1.61-1.82 (m, 6H), 1.94-2.03 (m, 2H), 3.16 (quint, J=7.8 Hz, 1H), 3.89 (s, 3H), 7.15 (s, 1H), 7.28 (s, 1H).
c) To a solution of 2-cyclopentyl-6-methoxy-isonicotinonitrile (2.09 g, 10.3 mmol) in methanol (100 mL), hydroxylamine hydrochloride (2.15 g, 31.0 mmol) and NaHCO3 (3.04 g, 36.2 mmol) are added. The mixture is stirred at 60° C. for 18 h before it is filtered and the filtrate is concentrated. The residue is dissolved in EA (300 mL) and washed with water (30 mL). The washings are extracted back with EA (4×100 mL) and DCM (4×100 mL). The combined org. extracts are dried over MgSO4, filtered, concentrated and dried under HV to give the title compound (2.74 g) as a white solid; LC-MS**: tR=0.47 min, [M+1]+=236.24; 1H NMR (D6-DMSO): δ 1.61-1.82 (m, 6H), 1.92-2.01 (m, 2H), 3.04-3.13 (m, 1H), 3.84 (s, 3H), 5.90 (s, 2H), 6.86 (s, 1H), 7.13 (s, 1H), 9.91 (s, 1H).
2-Cyclopentyl-6-methoxy-isonicotinic acid hydrazide
a) To a solution of 2-cyclopentyl-6-methoxy-isonicotinic acid (2.00 g, 9.04 mmol), hydrazinecarboxylic acid benzyl ester (1.50 g, 9.04 mmol) and DIPEA (2.34 g, 18.1 mmol) in DCM (40 mL), TBTU (3.19 g, 9.94 mmol) is added. The mixture is stirred at rt for 2 h before it is diluted with EA (250 mL), washed twice with sat. aq. NaHCO3 solution (150 mL) followed by brine (100 mL), dried over MgSO4, filtered and concentrated. The crude product is purified by CC on silica gel eluting with heptane:EA 4:1 to give N′-(2-cyclopentyl-6-methoxy-pyridine-4-carbonyl)-hydrazinecarboxylic acid benzyl ester (2.74 g) as pale yellow oil; LC-MS**: tR=0.74 min, [M+1]+=369.69; 1H NMR (D6-DMSO): δ 1.62-1.83 (m, 6H), 1.95-2.05 (m, 2H), 3.10-3.21 (m, 1H), 3.88 (s, 3H), 5.13 (s, 2H), 6.97 (s, 1H), 7.23 (s, 1H), 7.28-7.40 (m, 5H), 9.45 (s, 1H), 10.52 (s, 1H).
b) Pd/C (500 mg, 10% Pd) is added to a solution of N′-(2-cyclopentyl-6-methoxy-pyridine-4-carbonyl)-hydrazinecarboxylic acid benzyl ester (2.74 g, 7.42 mmol) in THF (50 mL) and methanol (50 mL). The mixture is stirred at rt under 1 bar of H2 for 25 h. The catalyst is removed by filtration and the filtrate is concentrated and dried under HV to give the title compound (1.58 g) as an off-white solid; LC-MS**: tR=0.51 min, [M+1]+=236.20; 1H NMR (D6-DMSO): δ 1.60-1.82 (m, 6H), 1.94-2.03 (m, 2H), 3.08-3.19 (m, 1H), 3.86 (s, 3H), 4.56 (s br, 2H), 6.93 (d, J=1.0 Hz, 1H), 7.20 (d, J=1.0 Hz, 1H), 9.94 (s, 1H).
3-Ethyl-4-hydroxy-5-methyl-benzonitrile
The title compound is prepared from 3-ethyl-4-hydroxy-5-methyl-benzaldehyde following literature procedures (A. K. Chakraborti, G. Kaur, Tetrahedron 55 (1999) 13265-13268); LC-MS: tR=0.90 min; 1H NMR (CDCl3): δ1.24 (t, J=7.6 Hz, 3H), 2.26 (s, 3H), 2.63 (q, J=7.6 Hz, 2H), 5.19 (s, 1H), 7.30 (s, 2H).
3-Chloro-4-hydroxy-5-methyl-benzonitrile
The title compound is prepared from commercially available 2-chloro-6-methyl-phenol in analogy to literature procedures (see 3-ethyl-4-hydroxy-5-methyl-benzonitrile); LC-MS: tR=0.85 min. 1H NMR (CDCl3): δ2.33 (s, 3H), 6.10 (s, 1H), 7.38 (s, 1H), 7.53 (d, J=1.8 Hz, 1H).
3-Ethyl-4,N-dihydroxy-5-methyl-benzamidine
The title compound is prepared from 3-ethyl-4-hydroxy-5-methyl-benzonitrile or from commercially available 2-ethyl-6-methyl-phenol following literature procedures (G. Trapani, A. Latrofa, M. Franco, C. Altomare, E. Sanna, M. Usala, G. Biggio, G. Liso, J. Med. Chem. 41 (1998) 1846-1854; A. K. Chakraborti, G. Kaur, Tetrahedron 55 (1999) 13265-13268; E. Meyer, A. C. Joussef, H. Gallardo, Synthesis 2003, 899-905); LC-MS: tR=0.55 min; 1H NMR (D6-DMSO): δ 9.25 (s br, 1H), 7.21 (s, 2H), 5.56 (s, 2H), 2.55 (q, J=7.6 Hz, 2H), 2.15 (s, 3H), 1.10 (t, J=7.6 Hz, 3H).
3-Chloro-4,N-dihydroxy-5-methyl-benzamidine
The title compound is prepared from commercially available 2-chloro-6-methyl-phenol in analogy to literature procedures (e.g. B. Roth et al. J. Med. Chem. 31 (1988) 122-129; and literature cited for 3-ethyl-4,N-dihydroxy-5-methyl-benzamidine); 3-chloro-4-hydroxy-5-methyl-benzaldehyde: LC-MS: tR=0.49 min, [M+1]+=201.00; 1H NMR 82.24 (s, 2H), 2.35 (s, 4H), 5.98 (s br, 1H), 7.59 (d, J=1.8 Hz, 1H), 7.73 (d, J=1.8 Hz, 1H), 9.80 (s, 1H); 3-chloro-4,N-dihydroxy-5-methyl-benzamidine: 1H NMR (D6-DMSO): δ 2.21 (s, 3H), 5.72 (s br, 2H), 7.40 (s, 1H), 7.48 (s, 1H), 9.29 (s br, 1H), 9.48 (s br, 1H).
(R)-4-(2,2-Dimethyl-[1,3]dioxolan-4-ylmethoxy)-3-ethyl-N-hydroxy-5-methyl-benzamidine
a) To a solution of 3-ethyl-4-hydroxy-5-methyl-benzonitrile (2.89 g, 17.9 mmol) in THF (80 mL), (R)-(2,2-dimethyl-[1,3]dioxolan-4-yl)methanol (2.84 g, 21.5 mmol) followed by triphenylphosphine (5.81 g, 21.5 mmol) is added. The mixture is cooled with an ice-bath before DEAD (9.36 g, 21.5 mmol) is added dropwise. The mixture is stirred at rt for 1 h, the solvent is removed in vacuo and the residue is purified by CC on silica gel eluting with heptane:EA 85:15 to give (R)-4-(2,2-dimethyl-[1,3]dioxolan-4-ylmethoxy)-3-ethyl-5-methyl-benzonitrile (4.45 g) as a pale yellow oil; LC-MS**: tR=0.75 min, [M+1]+=not detected; 1H NMR (CDCl3): δ1.25 (t, J=7.5 Hz, 3H), 1.44 (s, 3H), 1.49 (s, 3H), 2.34 (s, 3H), 2.65-2.77 (m, 2H), 3.80-3.90 (m, 2H), 3.94-4.00 (m, 1H), 4.21 (t, J=7.3 Hz, 1H), 4.52 (quint, J=5.8 Hz, 1H), 7.35 (s, 1H), 7.38 (s, 1H).
b) To a mixture of (R)-4-(2,2-dimethyl-[1,3]dioxolan-4-ylmethoxy)-3-ethyl-5-methyl-benzonitrile (4.45 g, 16.2 mmol) and NaHCO3 (4.75 g, 56.6 mmol) in methanol (30 mL), hydroxylamine hydrochloride (3.37 g, 48.5 mmol) is added. The mixture is stirred at 60° C. for 18 h before it is filtered and the solvent of the filtrate is removed in vacuo. The residue is dissolved in EA and washed with a small amount of water and brine. The org. phase is separated, dried over MgSO4, filtered, concentrated and dried to give the title compound (5.38 g) as a white solid; LC-MS**: tR=0.46 min, [M+1]+=309.23; 1H NMR (D6-DMSO): δ 1.17 (t, J=7.5 Hz, 3H), 1.33 (s, 3H), 1.38 (s, 3H), 2.25 (s, 3H), 2.57-2.69 (m, 2H), 3.73-3.84 (m, 3H), 4.12 (t, J=7.0 Hz, 1H), 4.39-4.45 (m, 1H), 5.76 (s br, 2H), 7.34 (s, 1H), 7.36 (s, 1H), 9.47 (s, 1H).
(R)-3-Chloro-4-(2,2-dimethyl-[1,3]dioxolan-4-ylmethoxy)-N-hydroxy-5-methyl-benzamidine
The title compound is obtained as a colorless oil (1.39 g) in analogy to (R)-4-(2,2-dimethyl-[1,3]dioxolan-4-ylmethoxy)-3-ethyl-N-hydroxy-5-methyl-benzamidine starting from 3-chloro-4-hydroxy-5-methyl-benzonitrile and L-α,β-isopropyliden glycerol; LC-MS: tR=0.66 min, [M+H]+=314.96.
(S)-4-(3-Amino-2-hydroxypropoxy)-3-ethyl-5-methylbenzonitrile
a) To a solution of 3-ethyl-4-hydroxy-5-methyl-benzonitrile (5.06 g, 31.4 mmol) in THF (80 mL), PPh3 (9.06 g, 34.5 mmol) and (R)-glycidol (2.29 mL, 34.5 mmol) are added. The mixture is cooled to 0° C. before DEAD in toluene (15.8 mL, 34.5 mmol) is added. The mixture is stirred for 18 h while warming up to rt. The solvent is evaporated and the crude product is purified by CC on silica gel eluting with heptane:EA 7:3 to give 3-ethyl-5-methyl-4-oxiranylmethoxy-benzonitrile (5.85 g) as a yellow oil; LC-MS: tR=0.96 min; [M+42]+=259.08.
b) The above epoxide is dissolved in 7 N NH3 in methanol (250 mL) and the solution is stirred at 65° C. for 18 h. The solvent is evaporated to give crude (S)-4-(3-amino-2-hydroxypropoxy)-3-ethyl-5-methylbenzonitrile (6.23 g) as a yellow oil; LC-MS: tR=0.66 min; [M+1]+=235.11.
N—((S)-3-[2-Ethyl-4-(N-hydroxycarbamimidoyl)-6-methyl-phenoxy]-2-hydroxy-propyl)-2-hydroxy-acetamide
a) To a solution of (S)-4-(3-amino-2-hydroxypropoxy)-3-ethyl-5-methylbenzonitrile (6.23 g, 26.59 mmol) in THF (150 mL), glycolic acid (2.43 g, 31.9 mmol), HOBt (4.31 g, 31.9 mmol), and EDC hydrochloride (6.12 g, 31.9 mmol) are added. The mixture is stirred at rt for 18 h before it is diluted with sat. aq. NaHCO3 and extracted twice with EA. The combined org. extracts are dried over MgSO4, filtered and concentrated. The crude product is purified by CC with DCM containing 8% of methanol to give (S)—N-[3-(4-cyano-2-ethyl-6-methyl-phenoxy)-2-hydroxy-propyl]-2-hydroxy-acetamide (7.03 g) as a yellow oil; LC-MS: tR=0.74 min, [M+1]+=293.10; 1H NMR (CDCl3): δ 1.25 (t, J=7.5 Hz, 3H), 2.32 (s, 3H), 2.69 (q, J=7.5 Hz, 2H), 3.48-3.56 (m, 3H), 3.70-3.90 (m, 3H), 4.19 (s, br, 3H), 7.06 (m, 1H), 7.36 (s, 1H), 7.38 (s, 1H).
b) The above nitrile is converted to the N-hydroxy-benzamidine according to literature procedures (e.g. E. Meyer, A. C. Joussef, H. Gallardo, Synthesis 2003, 899-905); LC-MS: tR=0.51 min, [M+1]+=326.13; 1H NMR (D6-DMSO): δ 1.17 (t, J=7.4 Hz, 3H), 2.24 (s, 3H), 2.62 (q, J=7.4 Hz, 2H), 3.23 (m, 1H), 3.43 (m, 1H), 3.67 (m, 2H), 3.83 (s, 2H), 3.93 (m, 1H), 5.27 (s br, 1H), 5.58 (s br, 1H), 5.70 (s, 2H), 7.34 (s, 1H), 7.36 (s, 1H), 7.67 (m, 1H), 9.46 (s br, 1H).
(S)—N-(3-[2-Chloro-4-(N-hydroxycarbamimidoyl)-6-methyl-phenoxy]-2-hydroxy-propyl)-2-hydroxy-acetamide
The title compound is obtained as a beige wax (1.1 g) in analogy to N—((S)-3-[2-ethyl-4-(N-hydroxycarbamimidoyl)-6-methyl-phenoxy]-2-hydroxy-propyl)-2-hydroxy-acetamide starting from 3-chloro-4-hydroxy-5-methyl-benzonitrile; LC-MS: tR=0.48 min, [M+H]+=331.94.
3-Chloro-N-hydroxy-4-methanesulfonylamino-5-methyl-benzamidine
a) A mixture of 4-amino-3-chloro-5-methylbenzonitrile (155 mg, 930 μmol) and methanesulfonylchloride (2.13 g, 18.6 mmol, 1.44 mL) is heated under microwave conditions to 150° C. for 7 h. The mixture is cooled to rt, diluted with water and extracted with EA. The org. extract is dried over MgSO4, filtered and concentrated. The crude product is purified on prep. TLC using heptane:EA 1:1 to give N-(2-chloro-4-cyano-6-methyl-phenyl)-methanesulfonamide (105 mg) as an orange solid; LC-MS**: tR=0.48 min; 1H NMR (CDCl3): δ2.59 (s, 3H), 3.18 (s, 3H), 6.27 (s, 1H), 7.55 (d, J=1.3 Hz, 1H), 7.65 (d, J=1.5 Hz, 1H).
b) Hydroxylamine hydrochloride (60 mg, 858 μmol) and NaHCO3 (72 mg, 858 μmol) is added to a solution of N-(2-chloro-4-cyano-6-methyl-phenyl)-methanesulfonamide (105 mg, 429 μmol) in methanol (10 mL). The mixture is stirred at 65° C. for 18 h. The solvent is removed in vacuo and the residue is dissolved in a small volume of water (2 mL) and extracted three times with EA (15 mL). The combined org. extracts are dried over MgSO4, filtered, concentrated and dried to give the title compound (118 mg) as a white solid; LC-MS**: tR=0.19 min, [M+1]+=277.94; 1H NMR (CDCl3): δ2.57 (s, 3H), 3.13 (s, 3H), 6.21 (s, 1H), 7.49 (d, J=1.5 Hz, 1H), 7.63 (d, J=1.5 Hz).
3-Ethyl-N-hydroxy-4-methanesulfonylamino-5-methyl-benzamidine
a) In a 2.5 L three-necked round-bottom flask 2-ethyl-6-methyl aniline (250 g, 1.85 mol) is dissolved in DCM (900 mL) and cooled to 5-10° C. Bromine (310.3 g, 1.94 mol) is added over a period of 105 min such as to keep the temperature at 5-15° C. An aq. 32% NaOH solution (275 mL) is added over a period of 10 min to the greenish-grey suspension while keeping the temperature of the reaction mixture below 25° C. DCM (70 mL) and water (100 mL) are added and the phases are separated. The aq. phase is extracted with DCM (250 mL). The combined org. phases are washed with water (300 mL) and concentrated at 50° C. to afford the 4-bromo-2-ethyl-6-methyl-aniline (389 g) as a brown oil; 1H NMR (CDCl3): δ 1.27 (t, J=7.3 Hz, 3H), 2.18 (s, 3H), 2.51 (q, J=7.3 Hz, 2H), 3.61 (s br, 1H), 7.09 (s, 2H).
b) A double-jacketed 4 L-flask is charged with 4-bromo-2-ethyl-6-methyl-aniline (324 g, 1.51 mol), sodium cyanide (100.3 g, 1.97 mol), potassium iodide (50.2 g, 0.302 mol) and copper(I)iodide (28.7 g, 0.151 mol). The flask is evacuated three times and refilled with nitrogen. A solution of N,N′-dimethylethylenediamine (191.5 mL, 1.51 mol) in toluene (750 mL) is added. The mixture is heated to 118° C. and stirred at this temperature for 21 h. The mixture is cooled to 93° C. and water (1250 mL) is added to obtain a solution. Ethyl acetate (1250 mL) is added at 22-45° C. and the layers are separated. The org. phase is washed with 10% aq. citric acid (2×500 mL) and water (500 mL). The separated org. phase is evaporated to dryness to afford 4-amino-3-ethyl-5-methyl-benzonitrile (240 g) as a metallic black solid; 1H NMR (CDCl3): δ1.29 (t, J=7.5 Hz, 3H), 2.19 (s, 3H), 2.52 (q, J=7.3 Hz, 2H), 4.10 (s br, 1H), 7.25 (s, 2H).
c) The title compound is then prepared from the above 4-amino-3-ethyl-5-methyl-benzonitrile in analogy to 3-chloro-N-hydroxy-4-methanesulfonylamino-5-methyl-benzamidine; LC-MS**: tR=0.26 min, [M+1]+=272.32.
3-Chloro-4-ethanesulfonylamino N-hydroxy-5-methyl-benzamidine
The title compound is prepared in analogy to 3-chloro-N-hydroxy-4-methanesulfonylamino-5-methyl-benzamidine using ethanesulfonylchloride; LC-MS**: tR=0.27 min, [M+1]+=292.13; 1H NMR (D6-DMSO): δ 1.36 (t, J=7.5 Hz, 3H), 2.40 (s, 3H), 3.22 (q, J=7.5 Hz), 5.88 (s, 2H), 7.57 (d, J=1.5 Hz, 1H), 7.63 (d, J=1.5 Hz, 1H), 9.18 (s, 1H), 9.78 (s, 1H).
4-Benzyloxy-3-ethyl-5-methyl-benzoic acid
a) To a solution of 3-ethyl-4-hydroxy-5-methyl-benzaldehyde (34.9 g, 0.213 mol, prepared from 2-ethyl-6-methyl-phenol according to the literature cited for 3-ethyl-4,N-dihydroxy-5-methyl-benzamidine) in MeCN (350 mL), K2CO3 (58.7 g, 0.425 mol) and benzylbromide (36.4 g, 0.213 mol) are added. The mixture is stirred at 60° C. for 2 h before it is cooled to rt, diluted with water and extracted twice with EA. The org. extracts are washed with water and concentrated to give crude 4-benzyloxy-3-ethyl-5-methyl-benzaldehyde (45 g) as an orange oil. 1H NMR (CDCl3): δ1.29 (t, J=7.5 Hz, 3H), 2.40 (s, 3H), 2.77 (q, J=7.8 Hz, 2H), 4.90 (s, 2H), 7.31-7.52 (m, 5H), 7.62 (d, J=1.5 Hz, 1H), 7.66 (d, J=1.8 Hz, 1H), 9.94 (s, 1H).
b) To a mixture of 4-benzyloxy-3-ethyl-5-methyl-benzaldehyde (132 g, 0.519 mol) and 2-methyl-2-butene (364 g, 5.19 mol) in tert.-butanol (1500 mL), a solution of NaH2PO4 dihydrate (249 g, 2.08 mol) in water (1500 mL) is added. To this mixture, NaClO2 (187.8 g, 2.08 mol) is added in portions. The temperature of the reaction mixture is kept below 30° C., and evolution of gas is observed. Upon completion of the addition, the orange bi-phasic mixture is stirred well for 3 h before it is diluted with TBME (1500 mL). The org. layer is separated and washed with 20% aq. NaHS solution (1500 mL) and water (500 mL). The org. phase is then extracted three times with 0.5 N aq. NaOH (1000 mL), the aq. phase is acidified with 25% aq. HCl (500 mL) and extracted twice with TBME (1000 mL). These org. extracts are combined and evaporated to dryness to give the title compound; 1H NMR (D6-DMSO): δ 1.17 (t, J=7.5 Hz, 3H), 2.31 (s, 3H), 2.67 (q, J=7.5 Hz, 2H), 4.86 (s, 2H), 7.34-7.53 (m, 5H), 7.68 (s, 2H), 12.70 (s, 1H).
Example 1 (S)-3-(2-Ethyl-4-{5-[2-(1-ethyl-propyl)-6-methoxy-pyridin-4-yl]-[1,2,4]oxadiazol-3-yl}-6-methyl-phenoxy)-propane-1,2-diol
a) To a solution of 2-(1-ethyl-propyl)-6-methoxy-isonicotinic acid (190 mg, 732 μmol) in THF (10 mL) and DMF (2 mL), DIPEA (190 mg, 1.46 mmol) followed by TBTU (235 mg, 732 μmol) is added. The mixture is stirred at rt for 10 min before (R)-4-(2,2-dimethyl-[1,3]dioxolan-4-ylmethoxy)-3-ethyl-N-hydroxy-5-methyl-benzamidine 226 mg, 732 μmol) is added. The mixture is stirred at rt for 1 h before it is diluted with EA and washed with water. The org. phase is separated and concentrated. The remaining residue is dissolved in dioxane (10 mL) and heated to 105° C. for 18 h. The mixture is cooled to rt, concentrated and the crude product is purified on prep. TLC plates using DCM containing 10% of methanol to give 4-{3-[4-((R)-2,2-dimethyl-[1,3]dioxolan-4-ylmethoxy)-3-ethyl-5-methyl-phenyl]-[1,2,4]oxadiazol-5-yl}-2-(1-ethyl-propyl)-6-methoxy-pyridine (256 mg) as a yellow oil; LC-MS: tR=1.28 min, [M+H]+=496.23.
b) A solution of 4-{3-[4-((R)-2,2-dimethyl-[1,3]dioxolan-4-ylmethoxy)-3-ethyl-5-methyl-phenyl]-[1,2,4]oxadiazol-5-yl}-2-(1-ethyl-propyl)-6-methoxy-pyridine (250 mg, 504 μmol) in 4 M HCl in dioxane (10 mL) is stirred at rt for 90 min before it is concentrated. The crude product is purified on prep. TLC plates using DCM containing 10% of methanol to give the title compound (76 mg) as a pale brownish solid; LC-MS: tR=1.12 min, [M+H]+=456.12; 1H NMR (CDCl3): δ0.85 (t, J=7.0 Hz, 6H), 1.33 (t, J=7.0 Hz, 3H), 1.70-1.89 (m, 4H), 2.42 (s, 3H), 2.61-2.71 (m, 1H), 2.78 (q, J=7.3 Hz, 2H), 3.82-4.00 (m, 4H), 4.04 (s, 3H), 4.14-4.21 (m, 1H), 7.34 (s, 1H), 7.46 (s, 1H), 7.86-7.91 (m, 2H).
Example 2 (S)-3-{4-[5-(2-Cyclopentyl-6-methoxy-pyridin-4-yl)-[1,2,4]oxadiazol-3-yl]-2-ethyl-6-methyl-phenoxy}-propane-1,2-diol
The title compound is prepared in analogy to Example 1 starting from 2-cyclopentyl-6-methoxy-isonicotinic acid; LC-MS: tR=1.14 min, [M+H]+=454.16; 1H NMR (CDCl3): δ1.33 (t, J=7.5 Hz, 3H), 1.72-1.78 (m, 2H), 1.85-1.94 (m, 4H), 2.03-2.15 (m, 2H), 2.41 (s, 3H), 2.72 (d, J=5.3 Hz, 1H), 2.77 (q, J=7.5 Hz, 2H), 3.19-3.28 (m, 1H), 3.81-3.94 (m, 2 H), 3.95-3.98 (m, 2H), 4.02 (s, 3H), 4.14-4.21 (m, 1H), 7.31 (d, J=1.3 Hz, 1H), 7.51 (d, J=1.0 Hz, 1H), 7.88 (d, J=1.8 Hz), 7.89 (d, J=2.0 Hz, 1H).

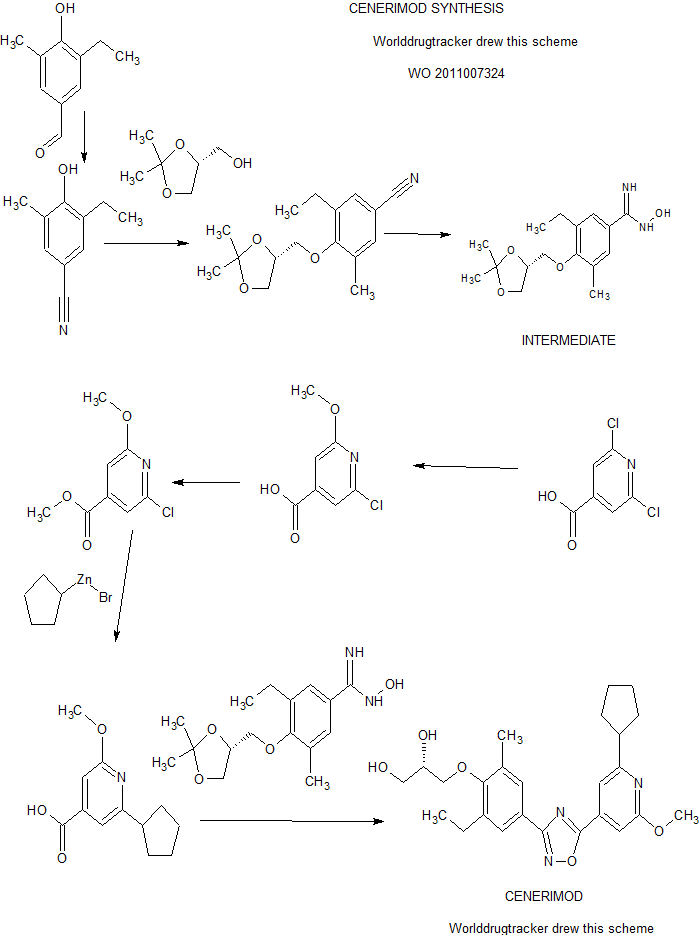
A practical synthesis of S1P receptor 1 agonist ACT-334441 (1) through late-stage convergent coupling of two key intermediates is described. The first intermediate is 2-cyclopentyl-6-methoxyisonicotinic acid whose skeleton was built from 1-cyclopentylethanone, ethyl oxalate, and cyanoacetate in a Guareschi–Thorpe reaction in 42% yield over five steps. The second, chiral intermediate, is a phenol ether derived from enantiomerically pure (R)-isopropylidene glycerol ((R)-solketal) and 3-ethyl-4-hydroxy-5-methylbenzonitrile in 71% yield in a one-pot reaction. The overall sequence entails 18 chemical steps with 10 isolated intermediates. All raw materials are cheap and readily available in bulk quantities, the reaction conditions match with standard pilot plant equipment, and the route reproducibly afforded 3–20 kg of 1 in excellent purity and yield for clinical studies.
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015133669 | 2015-05-14 | NEW PROCESS FOR THE PREPARATION OF 2-CYCLOPENTYL-6-METHOXY-ISONICOTINIC ACID |
| US8658675 | 2014-02-25 | Pyridin-4-yl derivatives |

OR
ULIXERTINIB
4-(5-chloro-2-isopropylaminopyridin-4-yl)-1H-pyrrole-2-carboxylic acid[1-(3-chlorophenyl)-2-hydroxyethyl]amide
| Molecular Formula: | C21H22Cl2N4O2 |
|---|---|
| Molecular Weight: | 433.33098 g/mol |
BVD-523; BVD-ERK; BVD-ERK/HM; BVD-ERK/ST; VRT-0752271; VRT-752271; VX-271, V

| Molecular Weight | 469.79 |
| Formula | C21H22Cl2N4O2●HCl |
| CAS | 1956366-10-1 |
| Chemical Name | 1H-Pyrrole-2-carboxamide, 4-[5-chloro-2-[(1-methylethyl)amino]-4-pyridinyl]-N-[(1S)-1-(3-chlorophenyl)-2-hydroxyethyl]-,hydrochloride(1:1) |
Ulixertinib malonate
4-(5-chloro-2-isopropylaminopyridin-4-yl)-1H-pyrrole-2-carboxylic acid[1-(3-chlorophenyl)-2-hydroxyethyl]amide (referred to as ulixertinib malonate)
![]()
INTRODUCTION
Ulixertinib is in phase I/II clinical trials for the treatment of acute myelogenous leukemia (AML), myelodysplasia and advanced solid tumors.
Members of the family of B-cell CLL/lymphoma 2 proteins (BCL-2) are apoptosis regulators. These proteins control mitochondrial outer
membrane permeabilization (MOMP). Expression of BCL-2 protein blocks cell death in response to various cellular injuries. A number of cancers, including melanoma, breast, prostate, chronic lymphocytic leukemia, and lung cancer, may be caused by damage to the BCL-2 gene. Mutations in BCL-2 may also be a cause of resistance to cancer treatments. Unfortunately, resistance can quickly develop using conventional BCL-2 inhibitor therapies to treat cancer.
Extracellular-signal-regulated kinases (ERKs) are protein kinases that are involved in cell cycle regulation, including the regulation of meiosis, mitosis, and postmitotic functions in differentiated cells. Disruption of the ERK pathway is common in cancers. However, to date, little progress has been made developing effective ERK inhibitors for the treatment of cancer.
As the understanding of the molecular basis of cancer grows, there is an increased emphasis on developing drugs that specifically target particular nodes in pathways that lead to cancer. In view of the deficiencies noted above, there is, inter alia, a need for effective molecularly targeted cancer treatments, including combination therapies. The present invention is directed to meeting these and other needs.

Mitogen-activated protein kinase (MAPK) pathways mediate signals which control diverse cellular processes including growth, differentiation, migration, proliferation and apoptosis. One MAPK pathway, the extracellular signal-regulated kinase (ERK) signaling pathway, is often found to be up-regulated in tumors. Pathway members, therefore, represent attractive blockade targets in the development of cancer therapies (Kohno and Pouyssegur, 2006). For example, U.S. Patent No. 7,354,939 B2 discloses, inter alia, compounds effective as inhibitors of ERK protein kinase. One of these compounds, 4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide, is a compound according to formula (I):

Pharmaceutical compositions are often formulated with a crystalline solid of the active pharmaceutical ingredient (API). The specific crystalline form of the API can have significant effects on properties such as stability and solubility / bioavailability. Instability and solubility characteristics can limit the ability to formulate a composition with an adequate shelf life or to effectively deliver a desired amount of a drug over a given time frame (Peterson et al., 2006).
Synergistic combination comprising an ERK1/2 inhibitor (such as ulixertinib) and a BCL-2 family inhibitor (such as navitoclax), assigned to BioMed Valley Discoveries (BVD), naming Decrescenzo and Welsch. BVD, presumably under license from Vertex, is developing ulixertinib (phase 2 trial), a small-molecule ERK 1/2 inhibitor for treating cancers including acute myelogenous leukemia and myelodysplastic syndrome. In June 2015, clinical data were presented at the 51st ASCO meeting in Chicago, IL.

WO2005113541 PDT PATENT
I-9 COMPD
SEE BELOW
Novel crystalline forms of 4-(5-chloro-2-isopropylaminopyridin-4-yl)-1H-pyrrole-2-carboxylic acid[1-(3-chlorophenyl)-2-hydroxyethyl]amide (referred to as ulixertinib) can be prepared which exhibit improved properties, eg surprisingly improved stability and solubility characteristics. Also claimed is their use for treating cancer.
EXAMPLE 2
Preparation of Crystaline Free Base 4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide
4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide free base was prepared according to the following synthesis scheme.
Stepl

C5H2CIFIN
257.43 C8H10CIIN2
ASYM-11 1606 296.54
ASYM-1 12060

ASYM-111938 ASYM-112393

ASYM-1 11935
In Step 1 , a clean and dry 200 L glass-lined reactor was evacuated to <-0.08 MPa, and then filled with nitrogen to normal pressure three times. Anhydrous ethanol (49.90 kg) was charged into the 200 L glass-lined reactor. ASYM-1 1 1606 (Asymchem) (12.70 kg) and isopropylamine (29.00 kg) were added into the mixture in turn. The mixture was heated to 65-75°C for refluxing. The mixture reacted at 65-75°C. After 20 h, the reaction was sampled and analyzed by HPLC every 4-6 h until the content of ASYM-1 1 1606 was <1 %. The mixture was cooled to 40-45°C and was concentrated at <45°C under reduced pressure (<-0.08 MPa) until 13-26 Lremained. The organic phase was washed with a sodium chloride solution and was stirred for 20-30 min and settled for 20-30 min before separation. The organic phase was concentrated at <30°C under reduced pressure (<-0.06 MPa) until 13-20 L remained. Petroleum ether (8.55 kg) was added into the concentrated mixture. The mixture was transferred into a 20 L rotary evaporator and continued concentrating at <30°C under reduced pressure (<-0.06 MPa) until 13-20 L remained. Then petroleum ether (8.55 kg) was added into the concentrated mixture. The mixture was cooled to 0-5°C and stirred for crystallization. After 1 h, the mixture was sampled and analyzed by wt% every 1 -2 h until the wt% of the mother liquor was <1 1 % or the change of the wt% between consecutive samples was <1 %. The mixture was filtered with a 10 L filter flask. The filter cake was sampled and analyzed for purity by HPLC. 10.50 kg of product was recovered as a brownish yellow solid at 99.39% purity.
In Step 2, a clean and dry 300 L glass-lined reactor was evacuated to <-0.08 MPa, and then filled with nitrogen to normal pressure three times. Glycol dimethyl ether (73.10 kg) was charged into the 300 L glass-lined reactor at 20-30°C. ASYM-1 12060 (Asymchem) (10.46 kg) and ASYM-1 1 1938 (Asymchem) (12.34 kg, 1 1 .64 kg after corrected) were added into the mixture in turn under the protection of nitrogen. Maintaining the temperature at 20-30°C, purified water (10.50 kg) and anhydrous sodium carbonate (5.67 kg) were added into the mixture. Palladium acetate (0.239 kg) and tricyclohexylphosphonium tetrafluoroborate (0.522 kg) were added into the mixture under the protection of nitrogen. After addition, the mixture was evacuated to <-0.06 MPa, and then filled with nitrogen to normal pressure. This was repeated for ten times until residual oxygen was <300 ppm. The mixture was heated to 75-85°C for refluxing. The mixture reacted at 75-85°C. After 4 h, the mixture was sampled and analyzed by HPLC every 2-3 h for content of ASYM-
1 12060. The content of AS YM-1 12060 was 6.18%, so additional ASYM-1 1 1938 (0.72 kg) was added and continued reaction until the content of ASYM-1 12060 was <3%. The mixture was cooled to 25-35°C and filtered with a 30 L stainless steel vacuum filter. The filter cake was soaked and washed twice with THF (14.10kg). The filtrate and washing liquor were combined and concentrated at <50°C under reduced pressure (<-0.08 MPa) until 10-15 L remained. The mixture was cooled to 15-25°C. Methanol (1 1 .05 kg) was added into the concentrated mixture. Then the mixture was stirred for crystallization. After 2 h, the mixture was sampled and analyzed by HPLC every 2-4 h until the wt% of the mother liquor was <2%. The mixture was filtered with a 30 L stainless steel vacuum filter. The filter cake was soaked and washed twice with methanol (8.30 kg). The filter cake was transferred into a 50 L plastic drum. Then ethyl acetate (7.10 kg) and petroleum ether (46.30 kg) were added into the drum. The mixture was stirred for 1.5-2 h and then filtered with a nutsche filter. The filter cake was soaked and washed with petroleum ether (20.50 kg). The filter cake was dried in the nutsche filter under nitrogen at 30-40°C. After 8 h, the solid was sampled and Karl Fischer (KF) analysis was performed in intervals of 4-8 h to monitor the drying process. Drying was completed when the KF result was <1 .0% water. During drying, the solid was turned over and mixed every 4-6 h. 12.15 kg of product was recovered as a brownish yellow solid at 98.32% purity.
In Step 3, a clean and dry 300 L glass-lined reactor was evacuated to <-0.08 MPa, and then filled with nitrogen to normal pressure three times. THF (62.58 kg) was charged into the 300 L glass-lined reactor at 15-30°C. Then the stirrer was started. ASYM-1 12393 (12.00 kg, 1 1 .70 kg after corrected) was added into the mixture. The mixture was stirred until the solid dissolved completely. Maintaining the temperature at 15-30°C, a lithium hydroxide solution which was
prepared with lithium hydroxide monohydrate (5.50 kg) in purified water (70.28 kg) was added into the mixture. Then diethylamine (3.86 kg) was added. The mixture was heated to 60-70°C for refluxing. The mixture reacted at 60-70°C. After 30 h, the reaction was sampled and analyzed by HPLC every 4-6 h until the content of intermediate at relative retention time (RRT)=1 .39-1 .44 was <1 % and the content of ASYM-1 12393 was <1 %. HPLC conditions for this analysis are set forth in Table 1 .
Table 1 : HPLC Parameters

The mixture was cooled to 25-35°C and MTBE (25.97 kg) was added into the mixture. The mixture was stirred for 20-30 min and filtered via an in-line fluid filter. The filtrate was transferred into a 300 L glass-lined reactor and settled for 20-30 min before separation. The pH of the obtained aqueous phase was adjusted with a 6 N hydrochloric acid solution which was prepared from concentrated hydrochloric acid (14.86 kg) in purified water (10.88 kg) at the rate of 5-8 kg/h at 15-25°C until the pH was 1 -2. The pH of the mixture was adjusted again with a saturated sodium carbonate solution which was prepared from sodium carbonate (5.03 kg) in purified water (23.56 kg) at the rate of 3-5 kg/h at 15-25°C until the pH was 6.4-6.7. Then the pH of the mixture was adjusted with a hydrochloric acid solution which was prepared from concentrated hydrochloric acid (1 .09 kg) in purified water (0.80 kg) until the pH was 6.2-6.4. The mixture was filtered with a nutsche filter. The filter cake was transferred into a 300 L glass-lined reactor and purified water (1 17.00 kg) was added. The mixture was stirred and sampled and analyzed by HPLC until the p-toluenesulfonic acid residue of the filter cake was <0.5%. Then the mixture was filtered. The filter cake was dried in the tray drier under nitrogen at 55-65°C until KF<10%. The solid and MTBE (8.81 kg) were charged into a 50 L stainless steel drum. The mixture was stirred for 1 -2 h. The mixture was filtered with a 30 L stainless steel vacuum filter. The filter cake was dried in the nutsche filter at 50-60°C. After 8 h, the solid was sampled and analyzed by KF every 4-8 h until KF<5%. During drying, the solid was turned over and mixed every 4-6 h. 6.3 kg of product was recovered as an off-white solid at 98.07% purity.
In Step 4, a dry and clean 50 L flask was purged with nitrogen for 20 min. DMF (30.20 kg) was charged into the 50 L flask reactor. Then the stirrer was started. Maintaining the temperature at 15-25°C, ASYM-1 12394 (3.22 kg, 2.76 kg after corrected) was added into the mixture. The mixture was stirred until the solid dissolved completely. The mixture was cooled to -10 to -20°C and 1 -hydroxybenzotriazole hydrate (2.10 kg) was added into the mixture at -10 to -20°C. Then EDCI (2.41 kg) was added into the mixture in five portions at an interval of about 5-10 min. The mixture was cooled to -20 to -30°C and ASYM-1 1 1888 (Asymchem) (1 .96 kg) was added into the mixture at -20 to -30°C. Then DIEA (1 .77 kg) was added into the mixture at the rate of 3-4 kg/h. The mixture was heated to 15-25°C at the rate of 5-10°C/h. The mixture was reacted at 15-25°C. After 6-8 h, the mixture was sampled and analyzed by HPLC every 2-4 h until the content of ASYM-1 12394 was <2%. The mixture was cooled to 0-10°C and the reaction mixture was quenched with a solution which was prepared from ethyl acetate (28.80 kg) in purified water (12.80 kg) at 0-10°C. The mixture was extracted three times with ethyl acetate (28.80 kg). For each extraction the mixture was stirred for 20-30 min and settled for 20-30 min before separation. The organic phases were combined and washed twice with purified water (12.80 kg). The mixture was stirred for 20-30 min and settled for 20-30 min before separation for each time. Then the obtained organic phase was filtered through an in-line fluid filter. The filtrate was transferred into a 300 L glass-lined reactor. The mixture was washed twice with a 5% acetic acid solution, which was prepared from acetic acid (2.24 kg) in purified water (42.50 kg). The solution was added at the rate of 10-20 kg/h. The organic phase was washed twice with a sodium carbonate solution, which was prepared from sodium carbonate (9.41 kg) in purified water (48.00 kg). The organic phase was washed twice with a sodium chloride solution, which was prepared from sodium chloride (16.00 kg) in purified water (44.80 kg). The organic phase was transferred into a 300 L glass-lined reactor. Anhydrous sodium sulfate (9.70 kg) was added into the mixture and the mixture was stirred for 2-4 h at 15-30°C. The mixture was filtered with a nutsche filter, which was pre-loaded with about 1 cm thick silica gel (7.50 kg). The filter cake was soaked and washed with ethyl acetate (14.40 kg) before filtration. The filtrates were combined and the combined filtrate was added into a 72 L flask through an in-line fluid filter. The mixture was concentrated at T≤40°C under reduced pressure (P<-0.08 MPa) until 3-4 L remained. Then MTBE (4.78 kg) was added into the mixture. The mixture was cooled to 0-10°C for crystallization with stirring. After 1 h, the mixture was sampled and analyzed by wt% every 1-2 h until the wt% of the mother liquor was <5% or the change of wt% between consecutive samples was <1%. The mixture was filtered with a vacuum filter flask and the filter cake was dried in the tray drier under nitrogen at 30-40°C until KF<0.5%. 3.55 kg of product was recovered as an off-white solid at 100% purity.
EXAMPLE 3A
Preparation of 4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide Form C

4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide Form C was prepared from 4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide free base as follows. ASYM-1 1 1935 (10.4 kg) was added to a stirred mixture of anhydrous ethanol (73.9 kg), methanol (4.1 kg) and isopropanol (4.1 kg). The mixture was heated to 70-75°C and stirred until all the solids dissolved. Anhydrous HCI (37 wt%, 1 .1 eq) in a mixture of ethanol/methanol/isopropanol (90:5:5) was added and the mixture maintained at 70-75°C for 2 hours after the addition was completed. The mixture was then cooled to 15-25°C at a rate of 5-15°C per hour and stirred at this temperature until the desired polymorphic purity was reached. The end point of the crystallization/polymorph conversion was
determined by the absence of an XRPD peak at about 10.5° 2Θ in three successive samples.
The mixture was then filtered and washed successively with a pre-prepared solution of anhydrous ethanol (14.8 kg), methanol (0.8 kg) and isopropanol (0.8 kg), followed by MTBE (2 x 21 kg). Avoidance of delay in the washing of the filter cake is preferable because the polymorph may be unstable in the wet filter cake in the presence of reagent alcohol and improved stability was observed after the MTBE wash has been performed. The wet filter cake was then dried in a heated filter funnel or a tray drier at 40-50°C until dry. Typical yields were about 85-90%.
EXAMPLE 3B
Alternative Preparation of 4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide Form C

ASYM-1 15985
4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide Form C was also prepared from 4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide free base as follows. A dry and clean 72 L flask was purged with nitrogen for 20 min. Anhydrous ethanol (21 .35 kg) methanol (1 .17 kg) and isopropanol (1 .19 kg) were charged into the 72 L flask at 15-25°C and the mixture was stirred for 20-30 min. ASYM-1 1 1935 (3.01 kg) was added into the mixture and heated to 70-75°C at the rate of 15-25°C/h and stirred until the solid dissolved completely.
An alcohol / HCI solution was prepared as follows. Anhydrous ethanol (1.500 kg) methanol (0.088 kg) and isopropanol (0.087 kg) were charged into a 5 L flask at 15-25°C and the mixture was stirred for 20-30 min. The mixture was bubbled with hydrogen chloride through a dip tube under stirring at 10-25°C. After 2 h, the mixture was sampled and analyzed every 2-4 h until the wt% of hydrogen chloride was > 35%.
The alcohol / HCI solution (0.519 kg) prepared above was added dropwise into the mixture at the rate of 0.5-1.0 kg/h at 70-75°C. Seed crystal (0.009 kg) was added into the mixture and the alcohol / HCI solution (0.173 kg) prepared above was added into the mixture at the rate of 0.5-1 .0 kg/h at 70-75°C. After addition, the mixture was stirred for 1 -2 h at 70-75°C. The mixture was cooled to 15-25°C at the rate of 5-15°C/h and stirred for 4-6 h. The mixture was heated to 70-75°C at the rate of 15-25°C/h and stirred for 8-10 h at 70-75°C. The mixture was cooled to 15-25°C at the rate of 5-15°C/h and stirred for 4-6 h. The mixture was filtered with a vacuum filter flask. The filter cake was soaked and rinsed with a solution which was prepared from anhydrous ethanol (4.25 kg) and methanol (0.24 kg) and isopropanol (0.24 kg) before filtration. The filter cake was dried in a drying room under nitrogen at 40-50°C until the ethanol residue was <0.5% and methanol residue was <0.3% and isopropanol residue was <0.3%. 2.89 kg of product was recovered as a white solid at 99.97% purity.
WO-2016123581
Novel crystalline malonate salt forms of 4-(5-chloro-2-isopropylaminopyridin-4-yl)-1H-pyrrole-2-carboxylic acid[1-(3-chlorophenyl)-2-hydroxyethyl]amide (referred to as ulixertinib malonate) and composition comprising them. Also claimed is their use for treating cancer.
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016123581&redirectedID=true
EXAMPLE 6
Aqueous Disolution of 4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1-(3-chlorophenyl)-2-hydroxyethyl]amide Malonate Form A
Samples of 4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide Form C and 4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2 -carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide malonate Form A were each shaken at ambient temperature in fasting state simulated gastric fluid (FaSSGF) pH 1.6 for 30 minutes. Concentration of 4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide was measured at 5, 15 and 30 minutes.
After 30 minutes, the samples were removed from FaSSGF, placed in fasting state simulated intestinal fluid (FaSSIF) pH 6.5, with shaking, for an additional 5 hours. Concentration of 4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide was measured at 10, 30, 60 90, 120, 180, 270, and 300 minutes. Results are summarized in Table 13 and shown in FIG. 10A (FaSSGF) and FIG. 10B (FaSSIF).
Table 13: Solubility of 4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide Form C and Malonate Form A.

WO2015095834
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015095834&redirectedID=true
![]()
Example 1 Compound 1-9 was prepared as follows:

1-9
2,2,2-TrichIoro-l-(4-iodo-lH-pyrrol-2-yl)ethanone: To a stirred solution of 50 g (235 mmol, 1.0 equiv.) of 2,2,2-trichloro-l-(lH-pyrrol-2-yl)-ethanone in dry dichloromethane (400 mL) under nitrogen, a solution of iodine monochloride (39 g, 240 mmol, 1.02 equivalents) in of dichloromethane (200 mL) was added dropwise. The resulting mixture was stirred at room temperature for 2 hours. The solution was washed with 10% potassium carbonate, water, 1.0 M sodium thiosulfate, saturated sodium chloride, dried over sodium sulfate, filtered, and concentrated under reduced pressure. The solid was recrystallized from hexanes/methyl acetate to afford the title compound (68.5g, 86%) as a colorless solid (86%). MS FIA: 335.8, 337.8 ES-.
4-Iodo-lH-pyrrole-2-carboxyIic acid methyl ester: To a stirred solution of 2,2,2- trichloro-l-(4-iodo-lH-pyrrol-2-yl)ethanone (68g, 201 mmol, 1.0 equivalent) in dry methanol (400 mL) under nitrogen, was added a solution of sodium methoxide in methanol (4.37 M, 54 mL, 235 mmol, 1.2 equivalents) over 10 minutes. The resulting mixture was stirred at room temperature for 1 hour. The volatiles were removed under reduced pressure and the crude was then partitioned between water and tert- butylmethyl ether. The organic phase was separated, washed two times with water, saturated sodium chloride, dried over sodium sulfate, filtered and concentrated under vacuum to afford the title compound (48g, 96%) as a colorless solid, that was used directly without further purification.
4-Iodo-l-(toluene-4-sulfonyl)-lH-pyrrole-2-carboxylic acid methyl ester: 4-Iodo- lH-pyrrole-2-carboxylic acid methyl ester (24.6 g, 98 mmol, 1.0 equivalent) was dissolved in dichloromethane (150 mL) and triethylamine (30 mL, 215.6 mmol, 2.2 equivalents). 4-(Dimethylamino)pyridine (1.2 g, 9.8 mmol, 0.1 equivalent) and p- toluenesulfonylchloride (20.6 g, 107.8 mmol, 1.1 equivalents) were added and the reaction mixture was stirred for 16 hours at room temperature. The reaction was quenched with 1 M ΗC1 and the organic layer was washed with aqueous sodium bicarbonate and brine. After drying over magnesium sulfate, the solvent was removed under reduced pressure and the residue was crystallized from tert-butylmethyl ether, yielding the title compound as a pale yellow solid (30 g, 75%). Rt(min) 8.259 minutes.
4-(4,4,5,5-Tetramethyl-[l,3,2]dioxaborolan-2-yI)-l-(toluene-4-sulfonyl)-lH- pyrrole-2-carboxylic acid methyl ester: To a degassed solution of 4-iodo-l- (toluene-4-sulfonyl)-lH-pyrrole-2-carboxylic acid methyl ester (20 g, 49.4 mmol, 1.0 equivalent) and bis(pinacolato)diborane (15 g, 65 mmol, 1.3 equivalents) in DMF (200 mL) under nitrogen, was added dichloro[l,l ‘- bis(diphenylphosphino)ferrocene]palladium (II) dichloromethane adduct (3.6 g, 4.9 mmol, 0.1 equivalent). The reaction mixture was then stirred at 80 °C for 18 hours. After removing the DMF under reduced pressure, the resulting thick oil residue was suspended in diethyl ether (500 mL) and a solid precipitated immediately. This solid was removed by filtration and the filtrate was washed with IM HCl, water, brine and dried over MgS0 . Concentration afforded the title compound as a white solid and used without further purification (10 g, 50%). LC/MS: Rt(min) 4.6; 406.4 ES+. MS FIA: 406.2 ES+. ‘pfNMR δ 1.2 (s, 12H), 2.35 (s, 3H), 3.8 (s, 3H), 7.2 (m, 3H), 7.8 (d, 2H), 8.0 (s, IH).
N,N’-2-(5-Chloro-4-iodo-pyridyI)-isopropyIarnine:
Method A. (Microwave)
In a 10 mL microwave tube, 5-chloro-2-fluoro-4-iodopyridine (1.0 g, 3.9 mmol, 1.0 equivalent) was dissolved in DMSO (4.0 mL) and then ispropylamine (0.99 mL, 11.7 mmol, 3.0 equivalents) was added. The tube was sealed and placed under microwave irradiation for 600 sec at 150 °C. This reaction was repeated six times. The reaction mixtures were combined, then diluted in ethyl acetate and washed with water. After drying over sodium sulfate, the solvent was evaporated to afford the title compound as a thick brown oil (5.6 g, 80% ) which was used directly without further purification. Rt(min) 4.614; MS FIA: 296.9 ES+. ‘pfNMRsssssss δ 1.25 (d, 6H), 3.65 (m, IH), 7.15 (s, IH), 7.75 (s, IH).
Method B: (Thennal)
5-Chloro-2-fluoro-4-iodopyridine (400 mg, 1.55 mmol, 1.0 equivalent) was dissolved in ethanol (5.0 mL) and then isopropylamine (0.66 mL, 7.8 mmol, 5.0 equivalents) was added. The resulting solution was stirred at 80 °C for 48 hours. The reaction mixture was then diluted in ethyl acetate and washed with water. After drying over sodium sulfate, the solvent was evaporated and a thick brown oil was obtained, which was then purified by flash chromatography on silica gel eluting with mixtures of hexanes/ethyl acetate (from 99:1 to 80:20) to afford the title compound as a pale yellow solid (96 mg, 21%).
4-(5-Chloro-2-isopropylaminopyridin-4-yl)-l-(toluene-4-suIfonyl)-lH-pyrrole-2- carboxylic acid methyl ester: To a solution of N,N’-2-(5-chloro-4-iodo-pyridyl)- isopropylamine (0.53 g, 1.8 mmol, 1.0 equivalent) and 4-(4,4,5,5-tetramethyl- [l,3,2]dioxaborolan-2-yl)-l-(toluene-4-sulfonyl)-lH-pyrrole-2-carboxylic acid methyl ester (0.78 g, 1.8 mmol, 1.0 equivalent) in DME (4.0 mL) was added a solution of aqueous 2 M sodium carbonate (1.0 mL) followed by Pd(PPh3)4 (0.21 mg, 0.18 mmol, 0.1 equivalent). The microwave tube was sealed and the reaction mixture was irradiated by microwave for 1800 sec. at 170 °C. The cmde of six reactions were combined and diluted in ethyl acetate and washed with water. After drying the organic layer with sodium sulfate, the solvent was removed and the resulting thick oil was adsorbed on silica gel. The crude was then purified by flash chromatography on silica, eluting with hexanes/ethyl acetate mixtures (from 99:1 to 70:30) to afford the title compound as a yellow solid (3.1 g, 61% over two steps). Rt(min) 6.556. MS FIA: 448.1 ES+. ‘HNMR δ 1.45 (d, 6H), 2.5 (s, 3H), 3.81 (s, 3H), 6.8 (s, IH), 7.35 (s, IH),
7.4 (d, 2H), 8.0 (m ,3H), 8.3 (s, IH).
4-(5-Chloro-2-isopropylaminopyridin-4-yl)-l-(2,4,6-trimethylbenzenesulfonyl)- lH-pyrrole-2-carboxylic acid methyl ester: To a solution of N,N’-2-(5-chloro-4- iodo-pyridyl)-isopropylamine (96 mg, 0.32 mmol, 1.0 equivalent) and 4-(4,4,5,5- tetramethyl-[ 1 ,3,2]dioxaborolan-2-yl)- 1 -(2,4,6-trimethylbenzenesulfonyl)- lH-pyrrole- 2-carboxylic acid methyl ester (152 mg, 0.35 mmol, 1.1 equivalents) in DME (2 mL), was added a solution of aqueous 2 M sodium carbonate (0.2 mL) followed by Pd(PPh ) (37 mg, 0.032 mmol, 0.1 equivalent). The reaction mixture was stirred at 80 °C for 16 hours. The crude was diluted in ethyl acetate and washed with water. After drying the organic layer with sodium sulfate, the solvent was removed and the resulting thick oil was adsorbed on silica gel. The cmde was then purified by flash chromatography on silica, eluting with hexanes/ethyl acetate mixtures (from 99:1 to 80:20) to afford the title compound as a yellow solid (65 mg, 43%). Rt(min) 7.290. MS FIA:476.1 ES+.
4-(5-Chloro-2-isopropylaminopyridin-4-yl)-lH-pyrrole-2-carboxyIic acid:
Method A. (Microwave)
A solution of 4-(5-chloro-2-isopropylaminopyridin-4-yl)-l-(toluene-4-sulfonyl)-lH- pyrrole-2-carboxylic acid methyl ester (3.1 g, 6.9 mmol, 1.0 equivalent) in TΗF (2.0 mL) was added to a solution of lithium hydroxide monohydrated (710 mg, 17.3 mmol,
2.5 equivalents) in water (3.0 mL). The microwave tube was sealed and the reaction mixture was irradiated by microwave for 1200 sec. at 150 °C. The cmde solution was acidified with aqueous 6Ν ΗC1. The solvent was evaporated off to afford the title compound which was used directly without further purification. Rt(min): 3.574. FIA MS: 279.9 ES+; 278.2 ES-.
Method B: (Thermal)
A solution of 4-(5-chloro-2-isopropylaminoρyridin-4-yl)-l-(2,4,6- trimethylbenzenesulfonyl)-lH-pyrrole-2-carboxylic acid methyl ester (0.69 g, 1.4 mmol, 1.0 equivalent) in TΗF (3.0 mL) was added to a solution of lithium hydroxide monohydrated (1.19 g, 29 mmol, 20.0 equivalents) in water (3.0 mL). The mixture was then refluxed for 8 hours. The cmde solution was acidified with aqueous 6N ΗC1 until cloudy, the organic solvent was partially removed and the product precipitated. The title compound was isolated by filtration and washed with water and diethyl ether, yielding a white solid (0.38 g, 96%).
4-(5-Chloro-2-isopropylaminopyridin-4-yl)-lH-pyrrole-2-carboxyIic acid [l-(3- ch!orophenyl)-2-hydroxyethyl] amide: To a suspension of 4-(5-chloro-2- isopropylaminopyridin-4-yl)-lH-pyrrole-2-carboxylic acid (1.93 g, 6.9 mmol, 1.0 equivalent) in DMF (5.0 mL) was added EDCI (1.45 g, 7.6 mmol, 1.1 equivalents), ΗOBt (0.94 g, 6.9 mmol, 1.0 equivalent) and (5)-3-chlorophenylglycynol (1.58 g, 7.6 mmol, 1.1 equivalents). Dusopropylethylamme (2.7 mL) was then added and the resulting mixture was stirred a room temperature overnight. The mixture was then poured into water and extracted with ethyl acetate. After drying over sodium sulfate, the solvent was removed and the crude was adsorbed on silica gel. Purification was effected by flash chromatography on silica, eluting with mixtures of hexanes/acetone (from 80:20 to 60:40) to afford the title compound as white solid (1.9 g, 64%). Rt(min) 4.981s. FIA MS: 433.1 ES+; 431.2 ES-. 1ΗNMR (CD3OD) δ 1.31 (d, 6H), 3.85 (m, 3H), 5.15 (t, IH), 7.01 (s, IH), 7.25 (m, 3H), 7.4 (s, IH), 7.45 (s, IH), 7.7 (s, IH), 7.95 (s, IH).
Example 2 Compound 1-9 was also prepared according to following alternate method:

2,5-DichIoro-4-nitropyridine N-oxide: To a suspension of 2-chloro-5-chloropyridine (10 g, 0.067 mol) in acetic anhydride (25 mL) was added hydrogen peroxide 30% (25 mL) in small portions. This mixture was stirred at room temperature for 24 hours and then heated at 60 °C for 30 hours. After removing the excess of acetic acid under reduced pressure, the residue was added in small portions to concentrated sulfuric acid (15 mL). The resulting solution was added to a mixture of concentrated sulfuric acid (15 mL) and fuming nitric acid (25 mL) and then heated at 100 °C for 90 minutes. The reaction mixture was poured on ice, neutralized with solid ammonium carbonate and finally with aqueous ammonia until a basic pH was obtained and. A precipitate formed. The precipitate was collected by filtration to afford the title compound as a pale yellow solid (3.1 g), Rt(min) 3.75. MS FIA shows no peak. ‘pfΝMR (DMSO-de) δ 8.78 (s, IH), 9.15 (s, IH).
4-Bromo-2-chloro-5-N-isopropylpyridin-2-amine N-oxide: To 2,5-dichloro-4- nitropyridine Ν-oxide (400 mg, 1.9 mmol) was added acetyl bromide (2 mL) very slowly. The reaction mixture was then heated at 80 °C for 10 minutes. The solvent was removed under a stream of nitrogen and the cmde product was dried under high vacuum. The cmde material (165 mg, 0.62 mmol) was dissolved in ethanol (2 mL), z‘so-propylamine (0.53 mL) added and the resulting mixture was heated at 80 °C for 2 hours. The cmde solution was then purified by reversed phase HPLC (acetonitrile/water/TFA 1%) to afford the title compound as a pale yellow solid (60 mg, 36.6%). Rt(min) 5.275. MS FIA264.8, 266.9 ES+.
4-(5-chloro-2-isopropylaminopyridin-4-yl)-lH-pyrrole-2-carboxylic acid [l-(3- chlorophenyl)-2-hydroxyethyl] amide (1-9): 4-Bromo-2-chloro-5-N- isopropylpyridin-2-amine N-oxide (25 mg, 0.094 mmol, 1.0 equivalent) and 4- (4,4,5, 5-tetramethyl-[l,3,2]dioxaborolan-2-yl)-l-(2,4,6-trimethylbenzensulfonyl)-lH- pyrrole-2-carboxylic acid methyl ester (39 mg, 0.094 mmol, 1.0 equivalent) were dissolved in benzene (5 mL) then aqueous 2M Νa2C03 (1 mL) and Pd(PPh3)4 (115.6 mg, 0.1 mmol, 0.2 equivalent) were added and the resulting suspension was heated at reflux at 80 °C for 16 hours. The reaction mixture was diluted in ethyl acetate, washed with water and dried over anhydrous sodium sulfate to afford 4-(5-chloro-2- isopropylamino-pyridin-4-yl)- 1 -(2,4,6-trimethyl-benzenesulfonyl)- lH-pyrrole-2- carboxylic acid methyl ester N-oxide (R (min) 6.859. MS FIA: 492.0 ES+) which was then treated with a 2 M solution of PC13 in dichloromethane (1 mL) at room temperature. After 10 minutes, the solvent was removed under a stream of nitrogen and the cmde oil was dissolved in methanol (1 mL) and aqueous 1 M ΝaOΗ (1 mL). The resulting mixture was heated at reflux for 16 hours then the cmde solution was acidified using aqueous 1 M ΗC1 and the solvent was removed. The resulting 4-(5- chloro-2-isopropylamino-pyridin-4-yl)-lΗ-pyrrole-2-carboxylic acid (R (min) 3.527. MS FIA: 279.4 ES+; 278.2 Es-) was suspended in DMF (3 mL) together with EDCI (36 mg, 0.19 mmol, 2 equivalents), HOBt (26 mg, 0.19 mmol, 2 equivalents), (S)-3- chlorophenylglycinol HCl salt (59 mg, 0.28 mmol, 3 equivalents) and DIEA (0.12 mL, 0.75 mmol, 8 equivalents). The resulting mixture was stirred at room temperature for 16 hours. The reaction mixture was diluted in ethyl acetate, washed with water and dried over sodium sulfate. After removing the solvent under reduced pressure, the cmde product was purified by reversed phase HPLC (acetonitrile/water/TFA 1%) to afford the title compound as a white solid (4.8 mg, 8.1%).
US20150512092015-02-19COMPOUNDS AND COMPOSITIONS AS INHIBITORS OF MEK
US73549392008-04-08Pyrrole inhibitors of ERK protein kinase, synthesis thereof and intermediates thereto
Research scientist Tony Huang works in a laboratory at Vertex Pharmaceuticals Inc. in San Diego
REFERENCES
1 . Kohno M, Pouyssegur J (2006) Targeting the ERK signaling pathway in cancer therapy. Ann Med 38: 200-21 1 .
2. Kuby, J., Immunology, 3rd Ed., W.H. Freeman & Co., New York.
3. Lee DC, Webb ML(2003) Pharmaceutical Analysis. John Wiley & Sons, Inc., New York: 255-257.
4. Peterson ML, Hickey MB, Zaworotko MJ and Almarsson O (2006) Expanding the Scope of Crystal Form Evaluation in Pharmaceutical Science. J Pharm Pharmaceut Sci 9(3):317-326.
5. Pierce Catalog and Handbook, 1994-1995; Pierce Chemical Co., Rockford, III.
6. Remington, The Science and Practice of Pharmacy (21 st Edition, Lippincott Williams and Wilkins, Philadelphia, PA.
7. The United States Pharmacopeia-National Formulary, The United States Pharmacopeial Convention, Rockville, MD.

Director, Chemistry at Sage Therapeutics

July 2012 – Present (4 years 2 months)

March 2008 – July 2012 (4 years 5 months)

2002 – 2008 (6 years)
PIC NOT AVAILABLE
///////////ULIXERTINIB, BVD-523; BVD-ERK, BVD-ERK/HM, BVD-ERK/ST, VRT-0752271, VRT-752271, VX-271, уликсертиниб ,أوليكسيرتينيب ,优立替尼 , PHASE 2, Vertex Pharmaceuticals, BioMed Valley Discoveries, UNII:16ZDH50O1U, 869886-67-9
CC(C)NC1=NC=C(C(=C1)C2=CNC(=C2)C(=O)NC(CO)C3=CC(=CC=C3)Cl)Cl

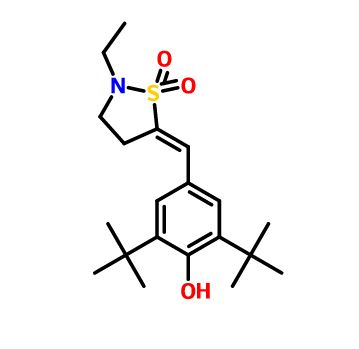

S-2474
(E)-(5)-(3,5-Di-tert-butyl-4-hydroxybenzylidene)-2-ethyl-1,2-isothiazolidine-1,1-dioxide
Shionogi Research Laboratories
cyclooxygenase-2 (COX-2) and 5-lipoxygenase (5-LO)
S-2474,158089-95-3, 158089-96-4 ((Z)-isomer),C20-H31-N-O3-S,
E)-5-(3,5-Di-tert-butyl-4-hydroxybenzylidene)-2-ethylisothiazolidine 1,1-dioxide
(E)-(5)-(3,5-Di-tert-butyl-4-hydroxybenzylidene)-2-ethyl-1,2-isothiazolidine-1,1-dioxide (S-2474, ), which was discovered at Shionogi Research Laboratories, shows potent inhibitory effects on both cyclooxygenase-2 (COX-2) and 5-lipoxygenase (5-LO) and is anticipated to be promising as an antiarthritic drug
![]()
synthesis of novel γ-sultam derivatives containing the di-tert-butylphenol antioxidant moiety. Several compounds with lower alkyl groups at the 2-position of the γ-sultam skeleton showed potent inhibitory activities against PGE2 production via the COX pathway and LTB4 production via the 5-LO pathway, as well as production of IL-1 in in vitro assays. Extensive pharmacological characterizations revealed that 2-ethyl-γ-sultam derivative 10b displays multiple inhibition of COX, 5-LO, and IL-1 production similar to tenidap and also good selective COX-2 inhibition like NS-398 and celecoxib. It exerted excellent antiinflammatory activity without any ulcerogenic effects and was designated as S-2474 an agent having both NSAID and cytokine modulating properties. S-2474 is now being developed as a promising alternative antiarthritic drug candidate
SYNTHESIS
17th Symp Med Chem (Nov 19 1997 , Tsukuba), EP 0595546; JP 1994211819; US 5418230
The intermediate gamma-sultam (III) was prepared by condensation of 3-chloropropylsulfonyl chloride (I) with ethylamine, followed by cyclization of the resulting chloro sulfonamide (II) under basic conditions. Condensation of 3,5-di- tert-butyl-4- (methoxymethoxy) benzaldehyde (IV) with sultam (III) in the presence of LDA produced the aldol addition compound (V). Then, acid-promoted dehydration and simultaneous methoxymethyl group deprotection gave rise to a mixture of the desired E-benzylidene sultam and the corresponding Z-isomer (VII), which were separated by column chromatography.

PAPER

Various 1,2-isothiazolidine-1,1-dioxide (γ-sultam) derivatives containing an antioxidant moiety, 2,6-di-tert-butylphenol substituent, were prepared. Some compounds, which have a lower alkyl group at the 2-position of the γ-sultam skeleton, showed potent inhibitory effects on both cyclooxygenase (COX)-2 and 5-lipoxygenase (5-LO), as well as production of interleukin (IL)-1 in in vitro assays. They also proved to be effective in several animal arthritic models without any ulcerogenic activities. Among these compounds, (E)-(5)-(3,5-di-tert-butyl-4-hydroxybenzylidene)-2-ethyl-1,2-isothiazolidine-1,1-dioxide (S-2474) was selected as an antiarthritic drug candidate and is now under clinical trials. The structure−activity relationships (SAR) examined and some pharmacological evaluations are described.
http://pubs.acs.org/doi/abs/10.1021/jm9906015
PAPER


PAPER
* To whom correspondence should be addressed. Telephone: +81-6-6401-8198 . Fax: +81-6-6401-1371. E-mail:takemasa.hida@shionogi.co.jp., †
Chemical Development Department, CMC Development Laboratories.
, ‡Shionogi Research Laboratories.

A one-pot synthesis of S-2474 was developed to overcome the problems of a large number of steps, low stereoselectivity, low yield, a large amount of waste, and severe reaction conditions. Aldol-type condensation of 3,5-di-tert-butyl-4-hydroxybenzaldehyde and N-ethyl-γ-sultam was carried out with LDA and then quenched with water. Dehydration proceeded under basic conditions, providing S-2474 directly as a single isomer on the benzylidene double bond. The reaction mechanism appears to involve a quinone methide intermediate. Environmental assessment of the development of this compound is also discussed in this paper.
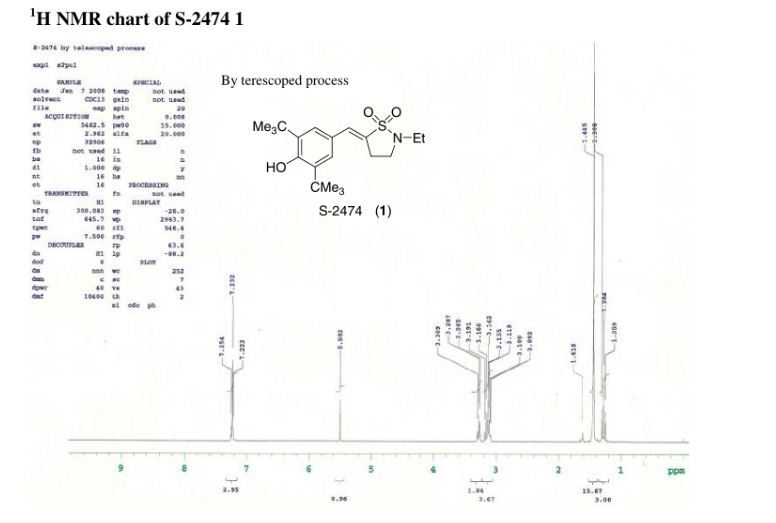
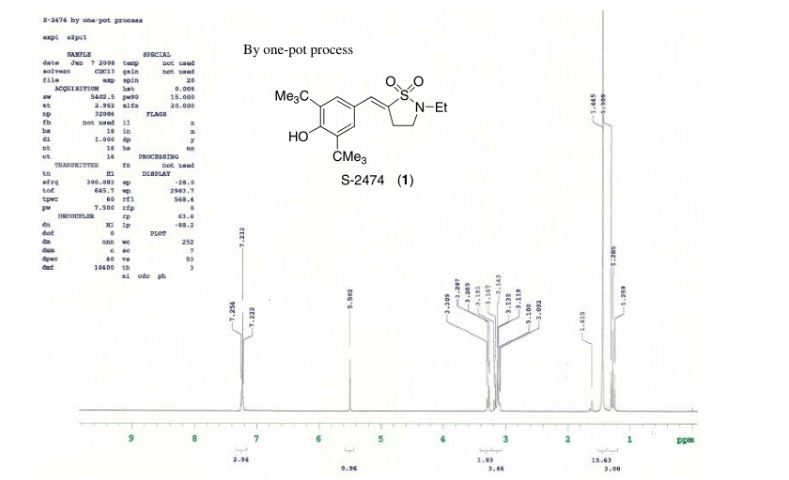
///////New, Antiarthritic , Drug Candidate, S-2474, Shionogi Research Laboratories, cyclooxygenase-2, (COX-2), 5-lipoxygenase , (5-LO), PHASE 2, 158089-95-3, 158089-96-4, S2474, S 2474
CCN2CC\C(=C/c1cc(c(O)c(c1)C(C)(C)C)C(C)(C)C)S2(=O)=O

 as CH3COOH salt
as CH3COOH salt
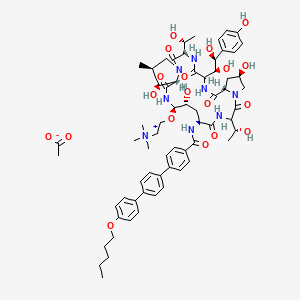
Several structural representations above
Biafungin™; CD 101 IV; CD 101 Topical; CD101; SP 3025, Biafungin acetate, Echinocandin B
UNII-G013B5478J FRE FORM,
CAS 1396640-59-7 FREE FORM
MF, C63-H85-N8-O17, MW, 1226.4035
| Treat and prevent invasive fungal infections; Treat and prevent systemic Candida infections; Treat candidemia |
Biafungin acetate
CAS 1631754-41-0 ACETATE, Molecular Formula, C63-H85-N8-O17.C2-H3-O2, Molecular Weight, 1285.4472,
UNII: W1U1TMN677
CD101 – A novel echinocandin antifungal C. albicans (n=351) MIC90 = 0.06 µg/mL C. glabrata (n=200) MIC90 = 0.06 µg/mL Echinocandins have potent fungicidal activity against Candida species
U.S. – Fast Track (Treat candidemia);
U.S. – Fast Track (Treat and prevent invasive fungal infections);
U.S. – Orphan Drug (Treat and prevent invasive fungal infections);
U.S. – Orphan Drug (Treat candidemia);
U.S. – Qualified Infectious Disease Program (Treat candidemia);
U.S. – Qualified Infectious Disease Program (Treat and prevent invasive fungal infections)
Fungal infections have emerged as major causes of human disease, especially among the immunocompromised patients and those hospitalized with serious underlying disease. As a consequence, the frequency of use of systemic antifungal agents has increased significantly and there is a growing concern about a shortage of effective antifungal agents. Although resistance rates to the clinically available antifungal agents remains low, reports of breakthrough infections and the increasing prevalence of uncommon fungal species that display elevated MIC values for existing agents is worrisome. Biafungin (CD101, previously SP 3025) is a novel echinocandin that displays chemical stability and long-acting pharmacokinetics that is being developed for once-weekly or other intermittent administration (see posters #A-693 and A- 694 for further information). In this study, we test biafungin and comparator agents against a collection of common Candida and Aspergillus species, including isolates resistant to azoles and echinocandins.
The echinocandins are an important class of antifungal agents, but are administered once daily by intravenous (IV) infusion. An echinocandin that could be administered once weekly could facilitate earlier hospital discharges and could expand usage to indications where daily infusions are impractical. Biafungin is a highly stable echinocandin for once-weekly IV administration. The compound was found to have a spectrum of activity and potency comparable to other echinocandins. In chimpanzees single dose pharmacokinetics of IV and orally administered biafungin were compared to IV anidulafungin, which has the longest half-life (T1/2 ) of the approved echinocandins.
Background Vulvovaginal candidiasis (VVC) is a highly prevalent mucosal infection VVC is caused by Candida albicans (~85%) and non-albicans (~15%) 5-8% of women have recurrent VVC (RVVC) which is associated with a negative impact on work/social life Oral fluconazole prescribed despite relapse, potential DDIs and increased risk to pregnant women No FDA-approved therapy for RVVC and no novel agent in >20 years


Cidara Therapeutics 6310 Nancy Ridge Drive, Suite 101 San Diego, CA 92121
The incidence of invasive fungal infections, especially those due to Aspergillus spp. and Candida spp., continues to increase. Despite advances in medical practice, the associated mortality from these infections continues to be substantial. The echinocandin antifungals provide clinicians with another treatment option for serious fungal infections. These agents possess a completely novel mechanism of action, are relatively well-tolerated, and have a low potential for serious drug–drug interactions. At the present time, the echinocandins are an option for the treatment of infections due Candida spp (such as esophageal candidiasis, invasive candidiasis, and candidemia). In addition, caspofungin is a viable option for the treatment of refractory aspergillosis. Although micafungin is not Food and Drug Administration-approved for this indication, recent data suggests that it may also be effective. Finally, caspofungin- or micafungin-containing combination therapy should be a consideration for the treatment of severe infections due to Aspergillus spp. Although the echinocandins share many common properties, data regarding their differences are emerging at a rapid pace. Anidulafungin exhibits a unique pharmacokinetic profile, and limited cases have shown a potential far activity in isolates with increased minimum inhibitory concentrations to caspofungin and micafungin. Caspofungin appears to have a slightly higher incidence of side effects and potential for drug–drug interactions. This, combined with some evidence of decreasing susceptibility among some strains ofCandida, may lessen its future utility. However, one must take these findings in the context of substantially more data and use with caspofungin compared with the other agents. Micafungin appears to be very similar to caspofungin, with very few obvious differences between the two agents.
Echinocandins are a new class of antifungal drugs[1] that inhibit the synthesis of glucan in the cell wall, via noncompetitive inhibition of the enzyme 1,3-β glucan synthase[2][3] and are thus called “penicillin of antifungals”[4] (a property shared with papulacandins) as penicillin has a similar mechanism against bacteria but not fungi. Beta glucans are carbohydrate polymers that are cross-linked with other fungal cell wall components (The bacterial equivalent is peptidoglycan). Caspofungin, micafungin, and anidulafungin are semisynthetic echinocandin derivatives with clinical use due to their solubility, antifungal spectrum, and pharmacokinetic properties.[5]


List of echinocandins:[17]
Discovery of echinocandins stemmed from studies on papulacandins isolated from a strain of Papularia sphaerosperma (Pers.), which were liposaccharide – i.e., fatty acid derivatives of a disaccharide that also blocked the same target, 1,3-β glucan synthase – and had action only on Candida spp. (narrow spectrum). Screening of natural products of fungal fermentation in the 1970s led to the discovery of echinocandins, a new group of antifungals with broad-range activity against Candida spp. One of the first echinocandins of the pneumocandin type, discovered in 1974, echinocandin B, could not be used clinically due to risk of high degree of hemolysis. Screening semisynthetic analogs of the echinocandins gave rise to cilofungin, the first echinofungin analog to enter clinical trials, in 1980, which, it is presumed, was later withdrawn for a toxicity due to the solvent system needed for systemic administration. The semisynthetic pneumocandin analogs of echinocandins were later found to have the same kind of antifungal activity, but low toxicity. The first approved of these newer echinocandins was caspofungin, and later micafungin and anidulafungin were also approved. All these preparations so far have low oral bioavailability, so must be given intravenously only. Echinocandins have now become one of the first-line treatments for Candida before the species are identified, and even as antifungal prophylaxis in hematopoietic stem cell transplant patients.
CIDARA THERAPEUTICS DOSES FIRST PATIENT IN PHASE 2 TRIAL OF CD101 TOPICAL TO TREAT VULVOVAGINAL CANDIDIASIS
SAN DIEGO–(BUSINESS WIRE)–Jun. 9, 2016– Cidara Therapeutics, Inc. (Nasdaq:CDTX), a biotechnology company developing novel anti-infectives and immunotherapies to treat fungal and other infections, today announced that the first patient has been dosed in RADIANT, a Phase 2 clinical trial comparing the safety and tolerability of the novel echinocandin, CD101, to standard-of-care fluconazole for the treatment of acute vulvovaginal candidiasis (VVC). RADIANT will evaluate two topical formulations of CD101, which is Cidara’s lead antifungal drug candidate.
“There have been no novel VVC therapies introduced for more than two decades, so advancing CD101 topical into Phase 2 is a critical step for women with VVC and for Cidara,” said Jeffrey Stein, Ph.D., president and chief executive officer of Cidara. “Because of their excellent safety record and potency against Candida, echinocandin antifungals are recommended as first line therapy to fight systemic Candida infections. CD101 topical will be the first echinocandin tested clinically in VVC and we expect to demonstrate safe and improved eradication of Candida with rapid symptom relief for women seeking a better option over the existing azole class of antifungals.”
RADIANT is a Phase 2, multicenter, randomized, open-label, active-controlled, dose-ranging trial designed to evaluate the safety and tolerability of CD101 in women with moderate to severe episodes of VVC. The study will enroll up to 125 patients who will be randomized into three treatment cohorts. The first cohort will involve the treatment of 50 patients with CD101 Ointment while a second cohort of 50 patients will receive CD101 Gel. The third cohort will include 25 patients who will be treated with oral fluconazole.
The primary endpoints of RADIANT will be the safety and tolerability of a single dose of CD101 Ointment and multiple doses of CD101 Gel in patients with acute VVC. Secondary endpoints include therapeutic efficacy in acute VVC patients treated with CD101. Treatment evaluations and assessments will occur on trial days 7, 14 and 28.
The RADIANT trial will be conducted at clinical trial centers across the United States. More information about the trial is available at www.clinicaltrials.gov, identifier NCT02733432.
About VVC and RVVC
Seventy-five percent of women worldwide suffer from VVC in their lifetime, and four to five million women in the United Statesalone have the recurrent form of the infection, which is caused by Candida. Many women will experience recurrence after the completion of treatment with existing therapies. Most VVC occurs in women of childbearing potential (the infection is common in pregnant women), but it affects women of all ages. In a recent safety communication, the U.S. Food and Drug Administration(FDA) advised caution in the prescribing of oral fluconazole for yeast infections during pregnancy based on a published study concluding there is an increased risk of miscarriage. The Centers for Disease Control and Prevention (CDC) guidelines recommend using only topical antifungal products to treat pregnant women with vulvovaginal yeast infections. Vaginal infections are associated with a substantial negative impact on day-to-day functioning and adverse pregnancy outcomes including preterm delivery, low birth weight, and increased infant mortality in addition to predisposition to HIV/AIDS. According to the CDC, certain species of Candida are becoming increasingly resistant to existing antifungal medications. This emerging resistance intensifies the need for new antifungal agents.
About CD101 Topical
CD101 topical is the first topical agent in the echinocandin class of antifungals and exhibits a broad spectrum of fungicidal activity against Candida species. In May 2016, the FDA granted Qualified Infectious Disease Product (QIDP) and Fast Track Designation to CD101 topical for the treatment of VVC and the prevention of RVVC.
About Cidara Therapeutics
Cidara is a clinical-stage biotechnology company focused on the discovery, development and commercialization of novel anti-infectives for the treatment of diseases that are inadequately addressed by current standard-of-care therapies. Cidara’s initial product portfolio comprises two formulations of the company’s novel echinocandin, CD101. CD101 IV is being developed as a once-weekly, high-exposure therapy for the treatment and prevention of serious, invasive fungal infections. CD101 topical is being developed for the treatment of vulvovaginal candidiasis (VVC) and the prevention of recurrent VVC (RVVC), a prevalent mucosal infection. In addition, Cidara has developed a proprietary immunotherapy platform, Cloudbreak™, designed to create compounds that direct a patient’s immune cells to attack and eliminate pathogens that cause infectious disease. Cidara is headquartered inSan Diego, California. For more information, please visit www.cidara.com.
REF http://ir.cidara.com/phoenix.zhtml?c=253962&p=irol-newsArticle&ID=2176474
CLIP
Cidara Therapeutics (formerly K2 Therapeutics) grabbed $42 million in a private Series B funding round Wednesday to continue developing its once-weekly anti-fungal therapy. Just in June 2014, the company completed a $32 million Series A financing led by 5AM Ventures, Aisling Capital, Frazier Healthcare and InterWest Partners, which was the fourth largest A round in 2014 for innovative startups[1]. FierceBiotech named the company as one of 2014 Fierce 15 biotech startups.
Cidara has an impressive executive team. The company was co-founded by Kevin Forrest, former CEO of Achaogen (NASDAQ: AKAO), and Shaw Warren. Jeffrey Stein, former CEO of Trius Therapeutics (NASDAQ: TSRX) and Dirk Thye, former president of Cerexa, have joined Cidara as CEO and CMO, respectively. Trius successfully developed antibiotic tedizolid and was acquired in 2013 by Cubist Pharmaceuticals (NASDAQ: CBST) for $818 million.
Cidara’s lead candidate, biafungin (SP3025), was acquired from Seachaid Pharmaceuticals for $6 million. Biafungin’s half-life is much longer than that of similar drugs known as echinocandins (e.g., caspofungin, micafungin, anidulafungin), which may allow it to be developed as a once-weekly therapy, instead of once daily. The company is also developing a topical formulation of biafungin, namely topifungin. Cidara intends to file an IND and initiate a Phase I clinical trial in the second half of 2015.
Merck’s Cancidas (caspofungin), launched in 2001, was the first of approved enchinocandins. The drug generated annual sales of $596 million in 2008. The approved echinocandins must be administered daily by intravenous infusion. Biafungin with improved pharmacokinetic characteristics has the potential to bring in hundreds of millions of dollars per year.
[1] Nat Biotechnol. 2015, 33(1), 18.
CLIP
Biafungin is a potent and broad-spectrum antifungal agent with excellent activity against wild-type and troublesome azole- and echinocandin-resistant strains of Candida spp. The activity of biafungin is comparable to anidulafungin. • Biafungin was active against both wild-type and itraconazole-resistant strains of Aspergillus spp. from four different species. • In vitro susceptibility testing of biafungin against isolates of Candida and Aspergillus may be accomplished by either CLSI or EUCAST broth microdilution methods each providing comparable results. • The use of long-acting intravenous antifungal agents that could safely be given once a week to select patients is desirable and might decrease costs with long-term hospitalizations. Background: A novel echinocandin, biafungin, displaying long-acting pharmacokinetics and chemical stability is being developed for once-weekly administration. The activities of biafungin and comparator agents were tested against 173 fungal isolates of the most clinically common species. Methods: 106 CAN and 67 ASP were tested using CLSI and EUCAST reference broth microdilution methods against biafungin (50% inhibition) and comparators. Isolates included 27 echinocandin-resistant CAN (4 species) with identified fks hotspot (HS) mutations and 20 azole nonsusceptible ASP (4 species). Results: Against C. albicans, C. glabrata and C. tropicalis, the activity of biafungin (MIC50, 0.06, 0.12 and 0.03 μg/ml, respectively by CLSI method) was comparable to anidulafungin (AND; MIC50, 0.03, 0.12 and 0.03 μg/ml, respectively) and caspofungin (CSP; MIC50, 0.12, 0.25 and 0.12 μg/ml, respectively; Table). C. krusei strains were very susceptible to biafungin, showing MIC90 values of 0.06 μg/ml by both methods. Biafungin (MIC50/90, 1/2 μg/ml) was comparable to AND and less potent than CSP against C. parapsilosis using CLSI methodology. CLSI and EUCAST methods displayed similar results for most species, but biafungin (MIC50, 0.06 μg/ml) was eight-fold more active than CSP (MIC50, 0.5 μg/ml) against C. glabrata using the EUCAST method. Overall, biafungin was two- to four-fold more active against fks HS mutants than CSP and results were comparable to AND. Biafungin was active against A. fumigatus (MEC50/90, ≤0.008/0.015 μg/ml), A. terreus (MEC50/90, 0.015/0.015 μg/ml), A. niger (MEC50/90, ≤0.008/0.03 μg/ml) and A. flavus (MEC50/90, ≤0.008/≤0.008 μg/ml) using CLSI method. EUCAST results for ASP were also low for all echinocandins and comparable to CLSI results. Conclusions: Biafungin displayed comparable in vitro activity with other echinocandins against common wild-type CAN and ASP and resistant subsets that in combination with the long-acting profile warrants further development of this compound. 1. Arendrup MC, Cuenca-Estrella M, Lass-Florl C, Hope WW (2013). Breakpoints for antifungal agents: An update from EUCAST focussing on echinocandins against Candida spp. and triazoles against Aspergillus spp. Drug Resist Updat 16: 81-95. 2. Castanheira M, Woosley LN, Messer SA, Diekema DJ, Jones RN, Pfaller MA (2014). Frequency of fks mutations among Candida glabrata isolates from a 10-year global collection of bloodstream infection isolates. Antimicrob Agents Chemother 58: 577-580. 3. Clinical and Laboratory Standards Institute (2008). M27-A3. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts: third edition. Wayne, PA: CLSI. 4. Clinical and Laboratory Standards Institute (2008). M38-A2. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Filamentous Fungi: Second Edition. Wayne, PA: CLSI. 5. Clinical and Laboratory Standards Institute (2012). M27-S4. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts: 4th Informational Supplement. Wayne, PA: CLSI. 6. European Committee on Antimicrobial Susceptibility Testing (2014). Breakpoint tables for interpretation of MICs and zone diameters. Version 4.0, January 2014. Available at: http://www.eucast.org/clinical_breakpoints/. Accessed January 1, 2014. 7. Pfaller MA, Diekema DJ (2010). Epidemiology of invasive mycoses in North America. Crit Rev Microbiol 36: 1-53. 8. Pfaller MA, Diekema DJ, Andes D, Arendrup MC, Brown SD, Lockhart SR, Motyl M, Perlin DS (2011). Clinical breakpoints for the echinocandins and Candida revisited: Integration of molecular, clinical, and microbiological data to arrive at species-specific interpretive criteria. Drug Resist Updat 14: 164-176. ABSTRACT Activity of a Novel Echinocandin Biafungin (CD101) Tested against Most Common Candida and Aspergillus Species, Including Echinocandin- and Azole-resistant Strains M CASTANHEIRA, SA MESSER, PR RHOMBERG, RN JONES, MA PFALLER JMI Laboratories, North Liberty, Iowa, USA C
https://www.google.com/patents/WO2015035102A2?cl=en
BIAFUNGIN ACETATE IS USED AS STARTING MATERIAL
Example 30b: Synthesis of Compound 31


Step a. Nitration of Biafungin Acetate

To a stirring solution of biafungin (1 00 mg, 0.078 mmol) in glacial acetic acid(1 .5 ml_) was added sodium nitrite (1 1 mg, 0.159 mmol) and the reaction was stirred at ambient temperature for 20 hours. The mixture was applied directly to reversed phase H PLC (Isco CombiFlash Rf; 50g RediSep C1 8 column, 5 to 95% acetonitrile in Dl water containing 0.1 % formic acid: 15 minute gradient). The pure fractions were pooled and lyophilized to yield 85 mg of the desired product as a light yellow solid, formate salt. 1 H-NMR (300 M Hz, Methanol-d4) δ 8.58 (d, 1 H, J = 1 1 .7 Hz), 8.47 (t, 2H, J = 8.7Hz), 8.05 (d, 1 H, J = 2.1 Hz), 7.99 (d, 2H, J = 9.3 Hz), 7.82 (d, 2H, J = 8.7 Hz), 7.79-7.60 (m, 12H), 7.1 7 (d, 1 H, J = 8.7 Hz), 7.03 (d, 2H, J = 9 Hz), 5.48 (d, 1 H, J = 6 Hz), 5.08 (dd, 1 H, J = 1 .2, 5.7 Hz), 4.95-4.73 (m, 5H), 4.68-4.56 (m, 2H), 4.53 (d, 1 H, J = 5.7 Hz), 4.48-4.39 (m, 2H), 4.31 -3.79 (m, 6H), 4.04 (t, 2H, J = 5.7 Hz), 3.72-3.44 (m,3H), 3.1 8 (s, 9H), 2.60-1 .99 (m, 5H), 1 .83 (m, 2H, J = 8.7 Hz), 1 .56-1 .35 (m, 5H), 1 .28 (d, 6H, J = 4.2 Hz), 1 .09 (d, 3H, J = 1 0.2 Hz), 0.99 (t, 3H, J = 8.7 Hz) ; LC/MS, [M/2+H]+: 635.79, 635.80 calculated.
Step b. Reduction of Nitro-Biafungin To Amino-Biafungin

To a stirring solution of Nitro-Biafungin (1 00 mg, 0.075 mmol) in glacial acetic acid(1 .5 ml_) was added zinc powder (50 mg, 0.77 mmol) and the reaction was stirred at ambient temperature for 1 hour. The mixture was filtered and applied directly to reversed phase HPLC (Isco CombiFlash Rf, 50g Redisep C18 column; 5 to 95% acetonitrile in Dl water containing 0.1 % formic acid: 15 minute gradient). The pure fractions were pooled and lyophilized to yield 55 mg of the desired product as a white solid, formate salt. 1 H-NMR (300 MHz, Methanol-d4) 5 8.47 (bs, 1 H), 7.99 (d, 2H, J = 1 0.8Hz), 7.82 (d, 2H, J = 7.5 Hz), 7.80-7.67 (m, 6H), 7.62 (d, 2H, J = 8.7 Hz), 7.03 (d, 2H, J = 7.5 Hz), 6.77 (d, 1 H, J = 1 .9 Hz), 6.68 (d, 1 H, J = 8.2 Hz), 6.55 (dd, 2H, J = 8.2, 1 .9 Hz), 5.43 (d, 1 H, J = 2.5 Hz), 5.05 (d, 1 H, J = 3 Hz), 4.83-4.73 (m, 2H), 4.64- 4.56 (m, 2H), 4.43-4.34 (m, 2H), 4.31 -4.15 (m, 4H), 4.03-4.08 (m, 1 H), 4.1 1 -3.89 (m, 8H), 3.83 (d, 1 H, J = 1 0.8 Hz), 3.68-3.47 (m, 3H), 3.1 7 (s, 9H), 2.57-2.42 (m, 2H), 2.35-2.27 (m, 1 H), 2.14-1 .98 (m, 2H), 1 .83 (m, 2H, J = 6 Hz), 1 .56-1 .38 (m, 4H), 1 .28 (dd, 6H, J = 6.5, 2 Hz), 1 .09 (d, 3H, J = 7 Hz), 0.986 (t, 3H, J = 7 Hz); High Res LC/MS: [M+H]+ 1241 .61 63; 1241 .6136 calculated.
Step c. Reaction of Amino-Biafungin with lnt-2 to Produce Compound 31

To a stirring solution of Amino-Biafungin (50 mg, 0.04 mmol) in DM F (1 ml_) was added formyl-Met-Leu-Phe- -Ala-OSu (lnt-2) (36 mg, 0.06 mmol) and DI PEA (7 uL, 0.04 mmol). The reaction was stirred at ambient temperature for 1 8 hours. The mixture was applied directly to reversed phase HPLC (Isco CombiFlash Rf; 50g Redisep C1 8 column; 5 to 95% acetonitrile in Dl water containing 0.1 % formic acid: 15 minute gradient). The pure fractions were pooled and lyophilized to yield 26 mg of a white solid as a formate salt. 1 H-NMR (300 M Hz, Methanol-d4) 5 8.55 (bs, 1 H), 8.44 (t, 1 H, J = 10 Hz), 8.1 8 (d, 1 H, J = 6 Hz), 8.1 1 (s, 1 H), 7.99 (d, 2H, J = 1 0 Hz), 7.84-7.70 (m, 6H), 7.63 (d, 2H, J = 7.8 Hz), 7.32-7.1 9 (m, 6H), 7.03 (d, 4H, J = 9 Hz), 6.87 (d, 1 H, J = 8.1 Hz), 5.44 (d, 1 H, J = 1 0.5 Hz), 5.05 (d, 1 H, J = 4.5 Hz), 4.83-4.74 (m, 2H), 4.66-4.50 (m, 6H), 4.45-4.29 (m, 10H), 4.1 9-3.82 (m, 1 0H), 3.67-3.57 (m, 6H), 3.1 7 (s, 9H), 2.64-2.46 (m, 6 H), 2.14-1 .92 (m, 6H), 1 .84 (m, 4H, J = 6 Hz), 1 .62-1 .40 (m, 8H), 1 .32-1 .22 (m, 6H), 1 .09 (d, 3H, J = 9 Hz), 0.99 (t, 3H, J = 7.5 Hz), 0.88 (m, 6H, J = 6.8 Hz) ; High Res LC/MS, [M/2+H]+ 865.4143, 865.4147 calculated.
17 Eschenauer, G; Depestel, DD; Carver, PL (March 2007). “Comparison of echinocandin antifungals.”. Therapeutics and clinical risk management 3 (1): 71–97. PMC 1936290.PMID 18360617.
///////////Biafungin™, CD 101 IV, CD 101 Topical, CD101, SP 3025, PHASE 2, CIDARA, Orphan Drug, Fast Track Designation, Seachaid Pharmaceuticals, Qualified Infectious Disease Product, QIDP, UNII-G013B5478J, 1396640-59-7, 1631754-41-0, Vulvovaginal candidiasis, Echinocandin B, FUNGIN
FREE FORM
CCCCCOc1ccc(cc1)c2ccc(cc2)c3ccc(cc3)C(=O)N[C@H]4C[C@@H](O)[C@H](NC(=O)[C@@H]5[C@@H](O)[C@@H](C)CN5C(=O)[C@@H](NC(=O)C(NC(=O)[C@@H]6C[C@@H](O)CN6C(=O)C(NC4=O)[C@@H](C)O)[C@H](O)[C@@H](O)c7ccc(O)cc7)[C@@H](C)O)OCC[N+](C)(C)C
AND OF ACETATE
CCCCCOc1ccc(cc1)c2ccc(cc2)c3ccc(cc3)C(=O)N[C@H]4C[C@@H](O)[C@H](NC(=O)[C@@H]5[C@@H](O)[C@@H](C)CN5C(=O)[C@@H](NC(=O)C(NC(=O)[C@@H]6C[C@@H](O)CN6C(=O)[C@@H](NC4=O)[C@@H](C)O)[C@H](O)[C@@H](O)c7ccc(O)cc7)[C@@H](C)O)OCC[N+](C)(C)C.CC(=O)[O-]
Three antifungal drugs approved by the United States Food and Drug Administration, caspofungin, anidulafungin, and micafungin, are known to inhibit β-1 ,3-glucan synthase which have the structures shown below.

caspofungin

Anidulafungin

Other exemplary p-1 ,3-glucan synthase inhibitors include,

echinocandin B

cilofungin

pneumocandin A0

pneumocandin B0

L-705589

L-733560

A-174591 

 or a salt thereof,
or a salt thereof,
Biafungin

or a salt thereof,
Amino-biafungin

or a salt thereof,

Amino-AF-053 


ASP9726
Yet other exemplary p-1 ,3-glucan synthase inhibitors include, without limitation:

Papulacandin B


Ergokonin
//////////////
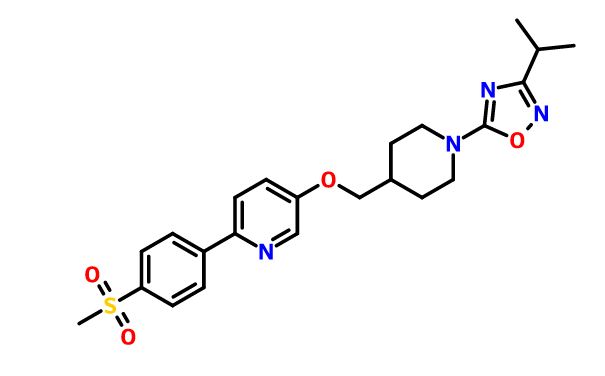
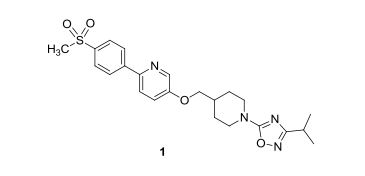
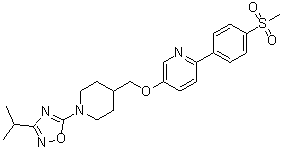
GSK-1292263
CAS 1032823-75-8
3-isopropyl-5-(4-(((6-(4-(methylsulfonyl)phenyl)pyridin-3-yl)oxy)methyl)piperidin-1-yl)-1,2,4-oxadiazole
5-[1-(3-Isopropyl-1,2,4-oxadiazol-5-yl)piperidin-4-ylmethoxy]-2-[4-(methylsulfonyl)phenyl]pyridine
5-[({1-[3-(1-Methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}methyl)oxy]-2-[4-(methylsulfonyl)phenyl]pyridine
MF C23H28N4O4S
MW: 456.18313
1292263
GSK-1292263
GSK-1292263A
GSK-263A
Smithkine Beecham Corp, INNOVATOR
GSK-1292263 is a novel GPR119 receptor agonist that is currently under development for the treatment of type 2 diabetes. Treatment of male Sprague-Dawley rats with a single dose of GSK-1292263 (3-30 mg/kg) in the absence of nutrients correlated with increased levels of circulating gastrointestinal peptides; glucagon-like peptide 1 (GLP-1), gastric inhibitory polypeptide (GIP), peptide YY (PYY) and glucagon.
GSK-1292263 had been evaluated in phase II clinical studies at GlaxoSmithKline for the oral treatment of type 2 diabetes and as monotherapy or in combination with sitagliptin for the treatment of dyslipidemia; however no recent development has been reported for this research.
Following administration of glucose in the oral glucose tolerance test (OGTT), greater increases in total GLP-1, GIP and PYY were seen in GSK-1292263-treated rats than in control animals. Despite significant decreases in the glucose AUC, no statistically significant differences in insulin responses and insulin AUC were observed between rats administered GSK-1292263 and those receiving vehicle control.
In the intravenous glucose tolerance test, significant increases in the peak insulin response and insulin AUC(0-15 min) of 30-60% were reported in the GSK-1292263 treatment group, compared with values in the vehicle control cohort. This insulin upregulation correlated with a significant increase in the glucose disposal rate (Brown, K.K. et al. Diabetes [70th Annu Meet Sci Sess Am Diabetes Assoc (ADA) (June 25-29, Orlando) 2010] 2010, 59(Suppl. 1): Abst 407).
The safety, tolerability, pharmacokinetics and pharmacodynamics of single and multiple oral doses of GSK-1292263 were evaluated in a recently completed randomized, placebo-controlled clinical trial in healthy volunteers (ClinicalTrials.gov Identifier NCT00783549).
A total of 69 subjects received single escalating doses of GSK-1292263 (10-400 mg) prior to administration of a 250-mg dose given once daily for 2 and 5 days, which was also evaluated in combination with sitagliptin (100 mg). Treatment with GSK-1292263 at all doses was described as well tolerated, with the most common drug-related effects being mild headache, dizziness, hyperhidrosis, flushing and post-OGTT hypoglycemia.
NMR
1H NMR (400 MHz, DMSO-d6) δ 8.44 (d, J = 3.0 Hz, 1H), 8.28 (d, J = 8.8 Hz, 2H), 8.06 (d, J = 8.8 Hz, 1H), 7.99 (bd, J = 8.5 Hz, 2H), 7.54 (dd, J = 8.8, 3.0 Hz, 1H), 4.03 (d, J = 6.3 Hz, 2H), 4.03–3.97 (m, 2H), 3.25 (s, 3H), 3.20–3.09 (m, 2H), 2.81 (q, J = 6.7 Hz, 1H), 2.13–2.00 (m, 1H), 1.88 (bd, J = 12.8 H, 2H), 1.42–1.29 (m, 2H), 1.18 (d, J = 7.0 Hz, 6H).
13C NMR (100.6 MHz, DMSO-d6) 175.3, 170.9, 155.5, 147.0, 143.5, 140.5, 138.6, 127.9, 127.0, 122.4, 122.3, 72.5, 45.7, 44.1, 35.0, 28.0, 26.7, 20.8.
HRMS calcd for C23H29N4O4S (M + H)+ 457.1904, found, 457.1900.
Anal. Calcd for C23H28N4O4S: C, 60.51; H, 6.18; N, 12.27. Found: C, 60.64; H, 6.16; N, 12.24.
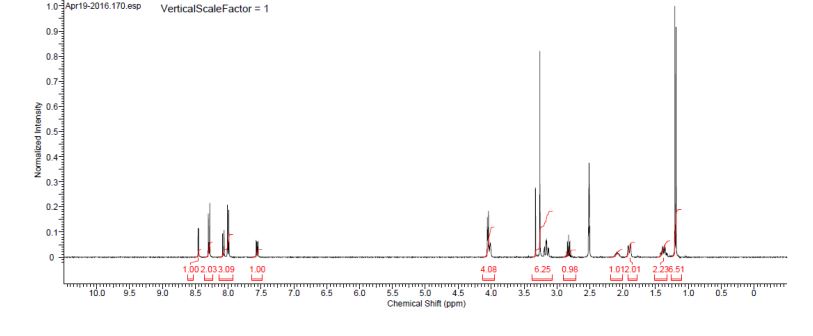
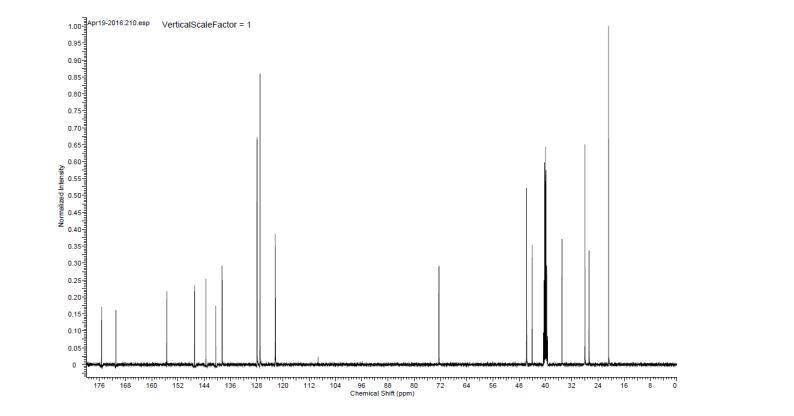
Hypoglycemia was not reported with the 5-day dosing schedule. Pharmacokinetic profiling revealed dose-proportional AUC and Cmax at single lower doses, but not at single higher ones. Following repeated once-daily dosing (5 days), drug accumulation was observed consistent with a mean half-life of 12-18 hours. A dose-dependent increase in glucose AUC(0-3 h) during OGTT was seen in GSK-1292263-treated subjects. The treatment was also associated with an increase in PYY during the prandial periods.
Coadministration with sitagliptin led to increases in the plasma concentrations of active GLP-1 but reduced the levels of total GLP-1, GIP and PYY. Sitagliptin affected the exposure to GSK-1292263 (50% increase) but GSK-1292263 did not affect sitagliptin exposure. The data support further evaluation of GSK-1292263 for the treatment of type 2 diabetes (Source: Nunez, D.J. et al. Diabetes [70th Annu Meet Sci Sess Am Diabetes Assoc (ADA) (June 25-29, Orlando) 2010] 2010, 59(Suppl. 1): Abst 80-OR).
WO 2008070692
http://www.google.com.au/patents/WO2008070692A2?cl=en
Example 169: 5-[({1 -[3-(1 -Methylethyl)-1,2,4-oxadiazol-5-yl]-4- piperidinyl}methyl)oxy]-2-[4-(methylsulfonyl)phenyl]pyridine hydrochloride

Step 1 : A mixture of 6-bromo-3-pyridinol (7 g, 40 mmol), [4-(methylsulfonyl)phenyl]boronic acid (8 g, 40 mmol), 2M Na2CO3 (30 ml_), PdCI2(PPh3)2 (1 g) and DME (60 ml.) under N2 was heated at 80 0C overnight. The reaction was allowed to cool to room temperature and was diluted with EtOAc and water. The resulting precipitate was filtered off and the aqueous layer was extracted with EtOAc. The combined organic extracts were dried over MgSO4, filtered and concentrated. The aqueous phase was also concentrated. Each of the residues was recrystallized from MeOH. The solid material from the organic phase recrystallization and the mother liquors from both aqueous and organic recrystallizations were combined, concentrated and purified by chromatography on a silica gel column using 0 to 10% MeOH/CH2CI2 to give 6-[4-(methylsulfonyl)phenyl]-3-pyridinol (2.9 g, 29%) as a tan solid. Step 2: Diisopropyl azodicarboxylate (0.175 ml_, 0.89 mmol) was added dropwise to a solution of 6-[4-(methylsulfonyl)phenyl]-3-pyridinol (150 mg, 0.59 mmol), {1-[3-(1- methylethyl)-1 ,2,4-oxadiazol-5-yl]-4-piperidinyl}methanol (prepared as in Example 20, Steps 1-3, 200 mg, 0.89 mmol), PPh3 (233 mg, 0.89 mmol), and THF (10 ml.) at ambient temperature. The mixture was stirred at ambient temperature for 4 h. The mixture was concentrated, and the resulting crude was purified by reverse-phase preparative HPLC using a CH3CN:H2O gradient (10:90 to 100:0) with 0.05% TFA as a modifier, then taken up in CH2CI2 and free-based with saturated NaHCO3 (aq) to give 5-[({1-[3-(1-methylethyl)-1 ,2,4-oxadiazol-5-yl]-4-piperidinyl}methyl)oxy]-2-[4- (methylsulfonyl)phenyl]pyridine (220 mg) as a white solid. Step 3: A mixture of the resulting white solid (50 mg, 0.1 1 mmol) in THF (3 ml.) was stirred at ambient temperature as 4Λ/ HCI in dioxane (28 μl_) was added dropwise. The resulting white precipitate was filtered, air-dried, then triturated with diethyl ether to give 35 mg (65%) of the title compound as a white solid. 1H NMR (400 MHz, CDCI3): δ 8.46 (d, 1 H, J = 0.7 Hz), 8.18 (bs, 2H), 8.05 (bs, 2H), 7.83 (bs, 1 H), 7.61- 7.45 (m, 1 H), 4.24 (d, 2H, J = 10.4 Hz), 4.00 (d, 2H, J = 0.6 Hz), 3.21-3.03 (m, 5H), 2.89 (m, 1 H), 2.15 (d, 1 H, J = 1.1 Hz), 1.96 (bs, 2H), 1.50 (bs, 2H), 1.28 (d, 6H, J = 6.9 Hz); LRMS (ESI), m/z 457 (M+H).
PATENT
http://www.google.co.ug/patents/US20120077812
Example 100
5-[({1-[3-(1-Methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}methyl)oxy]-2-[4-(methylsulfonyl)phenyl]pyridine[0480]
Step 1: A mixture of 2-methylpropanenitrile (100 g, 1.45 mol), hydroxylamine hydrochloride (111 g, 1.59 mol) and NaOH (64 g, 1.59 mol) in EtOH (2 L) and water (500 mL) was stirred at reflux overnight. The mixture was evaporated to dryness and extracted with dichloromethane. The organic extract was dried over Na2SO4 and concentrated to afford the desired N-hydroxy-2-methylpropanimidamide (50 g, 34%).
Step 2: A solution of 4-piperidinemethanol (140 g, 1.22 mol) in CH2Cl2 (1 L) was treated with a slurry of NaHCO3(205 g, 2.44 mol) in water (1.4 L) at 0° C. The mixture was stirred at 0° C. for 15 min, and then charged with a solution of cyanogen bromide in CH2Cl2, (1.34 mol) at 0° C. The reaction mixture was stirred and allowed to warm to ambient temperature, and stirred overnight. The aqueous layer was separated and extracted with CH2Cl2. The combined organic extracts were dried over Na2SO4, filtered, and the filtrate was concentrated. The crude product was combined with other batches made similarly and purified by chromatography on a silica gel column to give 300 g of 4-(hydroxymethyl)-1-piperidinecarbonitrile. Step 3: A solution of 1N ZnCl2 in Et2O (182 mL, 182 mmol) was added to a solution of 4-(hydroxymethyl)-1-piperidinecarbonitrile (21.3 g, 152 mmol) and N-hydroxy-2-methylpropanimidamide (18.6 g, 182 mmol) in EtOAc (50 mL) at ambient temperature. The reaction mixture was left at ambient temperature for 30 min, decanted, and was treated with concentrated HCl (45 mL) and ethanol 20 mL). The mixture was heated at reflux for 2 h. The mixture was evaporated to dryness, and the resulting residue was charged with water and the pH was adjusted to basic with K2CO3. The mixture was extracted with EtOAc and the material obtained was combined with 9 other batches prepared similarly and purified by silica gel chromatography to give 150 g of {1-[3-(1-methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}methanol.
Step 4: A solution of {1-[3-(1-methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}methanol (prepared as in Step 3, 174 g, 0.77 mol) and triethylamine (140 mL, 1.0 mol) in dichloromethane (1 L) at 5° C. was treated with a solution of methanesulfonyl chloride (69 mL, 0.89 mol) in dichloromethane (150 mL) over a 1 h period. The mixture was stirred at 5° C. for 30 min, and then was quenched by the addition of water (400 mL). The mixture was stirred for 30 min, and then the organic extract was washed with water (2×400 mL), dried (MgSO4) and concentrated. The residue was treated with heptane (1 L), stirred for 3 h, and the resulting solid was collected by filtration (heptane wash) and air-dried to afford {1-[3-(1-methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}methyl methanesulfonate (219.7 g, 94%) as an off-white solid. 1NMR (400 MHz, CDCl3): δ 4.21-4.15 (m, 2H), 4.08 (d, 2H, J=6.6 Hz), 3.04 (m, 2H), 3.01 (s, 3H), 2.86 (septet, 1H, J=6.9 Hz), 2.05-1.93 (m, 1H), 1.88-1.81 (m, 2H), 1.43-1.31 (m, 2H), 1.26 (d, 6H, J=6.8 Hz); LRMS (ESI), m/z 304 (M+H).
Step 5: A mixture of 6-bromo-3-pyridinol (36 g, 207 mmol), [4-(methylsulfonyl)phenyl]boronic acid (50 g, 250 mmol), 2M Na2CO3 (315 mL) and DME (500 mL) was degassed with N2 for 30 min, and then Pd(PPh3)4 (12 g, 10 mmol) was added and the mixture was heated at 80° C. for 18 h. The reaction was allowed to cool to room temperature and was diluted with dichloromethane (500 mL) and water (500 mL) and stirred for 30 min. The reaction was filtered and the solids were rinsed with dichloromethane and the aqueous layer was extracted with dichloromethane. The combined organic extracts were extracted with 1N NaOH (2×600 mL), and then cooled to 5° C. and the pH was adjusted to ˜8 with 6N HCl. The resulting precipitate was collected by filtration (water wash) and air-dried to afford a yellow solid. This procedure was repeated and the solids were combined to provide (71.2 g, 68%) of 6-[4-(methylsulfonyl)phenyl]-3-pyridinol. 1H NMR (400 MHz, DMSO-d6): δ 10.27 (s, 1H), 8.25 (d, 1H, J=2.7 Hz), 8.21 (d, 2H, J=8.5 Hz), 8.00-7.90 (m, 3H), 7.27 (dd, 1H, Ja=8.7 Hz, Jb=2.8 Hz), 3.21 (s, 3H); LRMS (ESI), m/z 250 (M+H).
Step 6: A mixture of {1-[3-(1-methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}methyl methanesulfonate (82.3 g, 271 mmol), 6-[4-(methylsulfonyl)phenyl]-3-pyridinol (71.0 g, 285 mmol), powdered potassium carbonate (118 g, 855 mmol) and N,N-dimethylformamide (750 mL) was mechanically stirred and heated at 80° C. under nitrogen for 20 h. The reaction was cooled to ambient temperature, poured onto ice water (3 L) and allowed to stand for 1 h. The resulting solid was filtered, rinsed with water (2×500 mL) and air-dried. The solid was taken up in dichloromethane (300 mL) and methanol (500 mL). The dichloromethane was slowly removed via rotovap at 55° C. The methanol solution was allowed to stand at ambient temperature for 16 h. The resulting crystalline solid was filtered, rinsed with cold methanol and dried under vacuum at 60° C. for 18 h to afford the desired product (105.7 g, 84%) as a light tan solid. 1H NMR (400 MHz, CDCl3): δ 8.41 (d, 1H, J=2.8 Hz), 8.13 (d, 2H, J=8.6 Hz), 8.01 (d, 2H, J=8.6 Hz), 7.74 (d, 1H, J=8.7 Hz), 7.29 (dd, 1H, Ja=8.7 Hz, Jb=3.0 Hz), 4.24 (d, 2H, J=13.1 Hz), 3.95 (d, 2H, J=6.2 Hz), 3.17-3.04 (m, 5H), 2.94-2.84 (m, 1H), 2.11 (bs, 1H), 1.97 (d, 2H, J=12.6 Hz), 1.54-1.42 (m, 2H), 1.29 (d, 6H, J=7.0 Hz); LRMS (ESI), m/z 457 (M+H).
Alternative preparation: Step 1: 2-Bromo-5-[({1-[3-(1-methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}methyl)oxy]pyridine (220 mg, 29%) was prepared as a white solid from {1-[3-(1-methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}methanol (prepared as in Example 20, Steps 1-3, 348 mg, 2.0 mmol), 6-bromo-3-pyridinol (348 mg, 2.0 mmol) and Ph3P (629 mg, 2.4 mmol) in THF (5 mL) followed by diisopropyl azodicarboxylate (0.51 mL, 2.6 mmol) in a manner similar to Example 1, Step 2. 1H NMR (400 MHz, CDCl3): δ 8.04 (s, 1H), 7.37 (d, 1H, J=8.8 Hz), 7.08 (d, 1H, J=8.8 Hz), 4.26-4.16 (m, 2H), 3.85 (d, 2H, J=6.2 Hz), 3.14-3.04 (m, 2H), 2.95-2.76 (m, 1H), 2.11-1.96 (m, 1H), 1.98-1.88 (m, 2H), 1.52-1.36 (m, 2H), 1.28 (d, 6H, J=6.9 Hz); LRMS (ESI), m/z 381/383 (M+H).
Step 2: 5-[({1-[3-(1-Methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}methyl)oxy]-2-[4-(methylsulfonyl)phenyl]pyridine (51 mg, 21%) was prepared from 2-bromo-5-[({1-[3-(1-methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}methyl)oxy]pyridine (220 mg, 0.52 mmol), [4-(methylsulfonyl)phenyl]boronic acid (105 mg, 0.52 mmol), 2M Na2CO3 (5 mL), Pd(PPh3)4 (50 mg, 0.04 mmol) and DME (5 mL) in a manner similar to Example 21, Step 3.
Paper

Practical and scalable syntheses were developed that were used to prepare multikilogram batches of GSK1292263A (1) and GSK2041706A (15), two potent G protein-coupled receptor 119 (GPR119) agonists. Both syntheses employed relatively cheap and readily available starting materials, and both took advantage of an SNAr synthetic strategy.
///////////1292263, GSK-1292263, GSK-1292263A, GSK-263A, GSK1292263, GSK1292263A, GSK 1292263, GSK 1292263A, GSK 263A, GSK263A, 1032823-75-8
O=S(C1=CC=C(C2=CC=C(OCC3CCN(C4=NC(C(C)C)=NO4)CC3)C=N2)C=C1)(C)=O

Sreeni Labs Private Limited, Hyderabad, India is ready to take up challenging synthesis projects from your preclinical and clinical development and supply from few grams to multi-kilo quantities. Sreeni Labs has proven route scouting ability to design and develop innovative, cost effective, scalable routes by using readily available and inexpensive starting materials. The selected route will be further developed into a robust process and demonstrate on kilo gram scale and produce 100’s of kilos of in a relatively short time.
Accelerate your early development at competitive price by taking your route selection, process development and material supply challenges (gram scale to kilogram scale) to Sreeni Labs…………
WEBSITE………. https://sreenilabs.com/
Sreeni Labs based in Hyderabad, India is working with various global customers and solving variety of challenging synthesis problems. Their customer base ranges from USA, Canada, India and Europe. Sreeni labs Managing Director, Dr. Sreenivasa Reddy Mundla has worked at Procter & Gamble Pharmaceuticals and Eli Lilly based in USA.
The main strength of Sreeni Labs is in the design, development of innovative and highly economical synthetic routes and development of a selected route into a robust process followed by production of quality product from 100 grams to 100s of kg scale. Sreeni Labs main motto is adding value in everything they do.
They have helped number of customers from virtual biotech, big pharma, specialty chemicals, catalog companies, and academic researchers and drug developers, solar energy researchers at universities and institutions by successfully developing highly economical and simple chemistry routes to number of products that were made either by very lengthy synthetic routes or by using highly dangerous reagents and Suzuki coupling steps. They are able to supply materials from gram scale to multi kilo scale in a relatively short time by developing very short and efficient synthetic routes to a number of advanced intermediates, specialty chemicals, APIs and reference compounds. They also helped customers by drastically reducing number of steps, telescoping few steps into a single pot. For some projects, Sreeni Labs was able to develop simple chemistry and avoided use of palladium & expensive ligands. They always begin the project with end in the mind and design simple chemistry and also use readily available or easy to prepare starting materials in their design of synthetic routes
Over the years, Sreeni labs has successfully made a variety of products ranging from few mg to several kilogram scale. Sreeni labs has plenty of experience in making small select libraries of compounds, carbocyclic compounds like complex terpenoids, retinal derivatives, alkaloids, and heterocyclic compounds like multi substituted beta carbolines, pyridines, quinolines, quinolones, imidazoles, aminoimidazoles, quinoxalines, indoles, benzimidazoles, thiazoles, oxazoles, isoxazoles, carbazoles, benzothiazoles, azapines, benzazpines, natural and unnatural aminoacids, tetrapeptides, substituted oligomers of thiophenes and fused thiophenes, RAFT reagents, isocyanates, variety of ligands, heteroaryl, biaryl, triaryl compounds, process impurities and metabolites.
Sreeni Labs is Looking for any potential opportunities where people need development of cost effective scalable routes followed by quick scale up to produce quality products in the pharmaceutical & specialty chemicals area. They can also take up custom synthesis and scale up of medchem analogues and building blocks. They have flexible business model that will be in sink with customers. One can test their abilities & capabilities by giving couple of PO based (fee for service) projects.
Some of the compounds prepared by Sreeni labs;
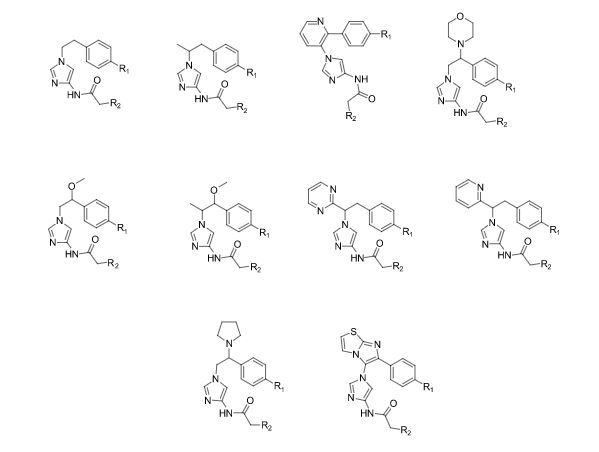
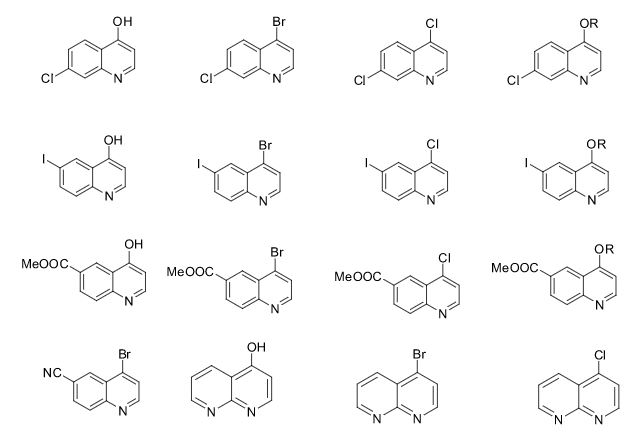
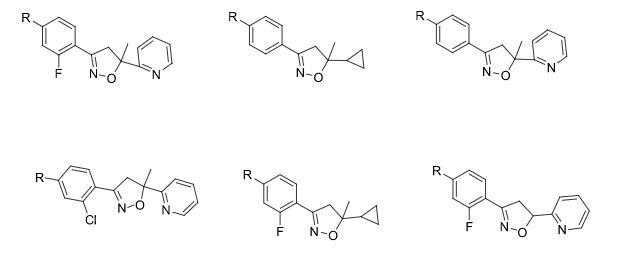
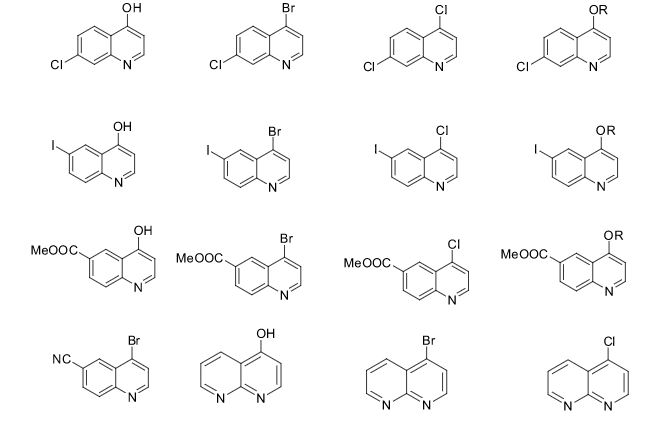
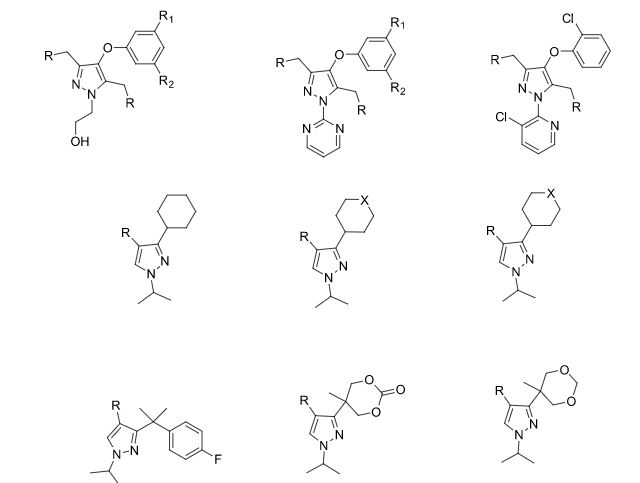
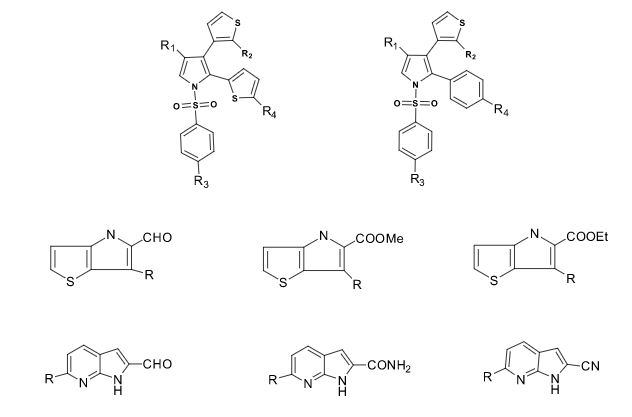
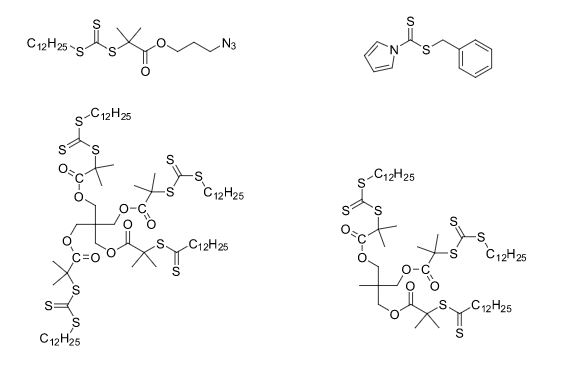
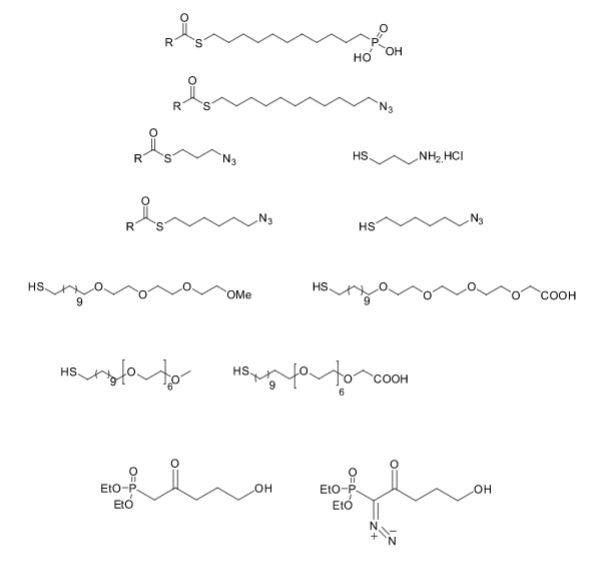



See presentation below
LINK ON SLIDESHARE
Managing Director at Sreeni Labs Private Limited
QUOTE………….
One virtual biotech company customer from USA, through a common friend approached Sreeni Labs and told that they are buying a tetrapeptide from Bachem on mg scale at a very high price and requested us to see if we can make 5g. We accepted the challenge and developed solution phase chemistry and delivered 6g and also the process procedures in 10 weeks time. The customer told that they are using same procedures with very minor modifications and produced the tetrapeptide ip to 100kg scale as the molecule is in Phase III.
One East coast customer in our first meeting told that they are working with 4 CROs of which two are in India and two are in China and politely asked why they should work with Sreeni Labs. We told that give us a project where your CROs failed to deliver and we will give a quote and work on it. You pay us only if we deliver and you satisfy with the data. They immediately gave us a project to make 1.5g and we delivered 2g product in 9 weeks. After receiving product and the data, the customer was extremely happy as their previous CRO couldn’t deliver even a milligram in four months with 3 FTEs.
One Midwest biotech company was struggling to remove palladium from final API as they were doing a Suzuki coupling with a very expensive aryl pinacol borane and bromo pyridine derivative with an expensive ligand and relatively large amount of palldium acetate. The cost of final step catalyst, ligand and the palladium scavenging resin were making the project not viable even though the product is generating excellent data in the clinic. At this point we signed an FTE agreement with them and in four months time, we were able to design and develop a non suzuki route based on acid base chemistry and made 15g of API and compared the analytical data and purity with the Suzuki route API. This solved all three problems and the customer was very pleased with the outcome.
One big pharma customer from east coast, wrote a structure of chemical intermediate on a paper napkin in our first meeting and asked us to see if we can make it. We told that we can make it and in less than 3 weeks time we made a gram sample and shared the analytical data. The customer was very pleased and asked us to make 500g. We delivered in 4 weeks and in the next three months we supplied 25kg of the same product.
Through a common friend reference, a European customer from a an academic institute, sent us an email requesting us to quote for 20mg of a compound with compound number mentioned in J. med. chem. paper. It is a polycyclic compound with four contiguous stereogenic centers. We gave a quote and delivered 35 mg of product with full analytical data which was more pure than the published in literature. Later on we made 8g and 6g of the same product.
One West coast customer approached us through a common friend’s reference and told that they need to improve the chemistry of an advanced intermediate for their next campaign. At that time they are planning to make 15kg of that intermediate and purchased 50kg of starting raw material for $250,000. They also put five FTEs at a CRO for 5 months to optimize the remaining 5 steps wherein they are using LAH, Sodium azide, palladium catalyst and a column chromatography. We requested the customer not to purchase the 50kg raw material, and offered that we will make the 15kg for the price of raw material through a new route in less than three months time. You pay us only after we deliver 15 kg material. The customer didn’t want to take a chance with their timeline as they didn’t work with us before but requested us to develop the chemistry. In 7 weeks time, we developed a very simple four step route for their advanced intermediate and made 50g. We used very inexpensive and readily available starting material. Our route gave three solid intermediates and completely eliminated chromatographic purifications.
One of my former colleague introduced an academic group in midwest and brought us a medchem project requiring synthesis of 65 challenging polyene compounds on 100mg scale. We designed synthetic routes and successfully prepared 60 compounds in a 15 month time.
UNQUOTE…………
The man behind Seeni labs is Dr.Sreenivasa Reddy Mundla

Managing Director at Sreeni Labs Private Limited
Sreeni Labs Private Limited
Road No:12, Plot No:24,25,26
Links
LINKEDIN https://in.linkedin.com/in/sreenivasa-reddy-10b5876
FACEBOOK https://www.facebook.com/sreenivasa.mundla
RESEARCHGATE https://www.researchgate.net/profile/Sreenivasa_Mundla/info
EMAIL mundlasr@hotmail.com, Info@sreenilabs.com, Sreeni@sreenilabs.com
Dr. Sreenivasa Mundla Reddy

Dr. M. Sreenivasa Reddy obtained Ph.D from University of Hyderabad under the direction Prof Professor Goverdhan Mehta in 1992. From 1992-1994, he was a post doctoral fellow at University of Wisconsin in Professor Jame Cook’s lab. From 1994 to 2000, worked at Chemical process R&D at Procter & Gamble Pharmaceuticals (P&G). From 2001 to 2007 worked at Global Chemical Process R&D at Eli Lilly and Company in Indianapolis.
In 2007 resigned to his job and founded Sreeni Labs based in Hyderabad, Telangana, India and started working with various global customers and solving various challenging synthesis problems.
The main strength of Sreeni Labs is in the design, development of a novel chemical route and its development into a robust process followed by production of quality product from 100 grams to 100’s of kg scale.
They have helped number of customers by successfully developing highly economical simple chemistry routes to number of products that were made by Suzuki coupling. they are able to shorten the route by drastically reducing number of steps, avoiding use of palladium & expensive ligands. they always use readily available or easy to prepare starting materials in their design of synthetic routes.
Sreeni Labs is Looking for any potential opportunities where people need development of cost effective scalable routes followed by quick scale up to produce quality products in the pharmaceutical & specialty chemicals area. They have flexible business model that will be in sink with customers. One can test their abilities & capabilities by giving PO based projects
August 2007 – Present (8 years 11 months)
Sreeni Labs Profile

March 2001 – August 2007 (6 years 6 months)

July 1994 – February 2001 (6 years 8 months)


With Sreenivasa Mundla, Narahara sastry, Ram Kishan Rao, Jagadeesh Bharatam, Jagadish Gunjur and Jagadish Bharatham.
PUBLICATIONS
Jianye Zhang · Zhiqian Dong · Sreenivasa Reddy Mundla · X Eric Hu · William Seibel ·Ruben Papoian · Krzysztof Palczewski · Marcin Golczak
Article: ChemInform Abstract: Regioselective Synthesis of 4Halo ortho-Dinitrobenzene Derivative
Aug 2010 · ChemInform
Hong-yu Li · William T. McMillen · Charles R. Heap · Denis J. McCann · Lei Yan · Robert M. Campbell · Sreenivasa R. Mundla · Chi-Hsin R. King · Elizabeth A. Dierks · Bryan D. Anderson · Karen S. Britt · Karen L. Huss
Apr 2008 · Journal of Medicinal Chemistry
Hong-yu Li · Yan Wang · William T. McMillen · Arindam Chatterjee · John E. Toth ·Sreenivasa R. Mundla · Matthew Voss · Robert D. Boyer · J. Scott Sawyer
Feb 2008 · ChemInform
Hong-yu Li · Yan Wang · William T. McMillen · Arindam Chatterjee · John E. Toth ·Sreenivasa R. Mundla · Matthew Voss · Robert D. Boyer · J. Scott Sawyer
Nov 2007 · Tetrahedron
Hong-yu Li · Yan Wang · Charles R Heap · Chi-Hsin R King · Sreenivasa R Mundla · Matthew Voss · David K Clawson · Lei Yan · Robert M Campbell · Bryan D Anderson · Jill R Wagner ·Karen Britt · Ku X Lu · William T McMillen · Jonathan M Yingling
Apr 2006 · Journal of Medicinal Chemistry

Hui Cao · Sreenivasa R. Mundla · James M. Cook
Aug 2003 · Tetrahedron Letters
Article: ChemInform Abstract: A New Method for the Synthesis of 2,6-Dinitro and 2Halo6-nitrostyrenes
Nov 2000 · ChemInform
Article: ChemInform Abstract: A Novel Method for the Efficient Synthesis of 2-Arylamino-2-imidazolines


TGF-β inhibitors
The present invention provides 2-(6-methyl-pyridin-2-yl)-3-[6-amido-quinolin-4-yl) -5,6-dihydro-4H-pyrrolo[1,2-b]pyrazole monohydrate, i.e., Formula I.

EXAMPLE 1 Preparation of 2-(6-methyl-pyridin-2-yl)-3-[6-amido-quinolin-4-yl-5,6-dihydro-4H -pyrrolo[1,2-b]pyrazole monohydrate

Galunisertib
1H NMR (CDCl3): δ=9.0 ppm (d, 4.4 Hz, 1H); 8.23-8.19 ppm (m, 2H); 8.315 ppm (dd, 1.9 Hz, 8.9 Hz, 1H); 7.455 ppm (d, 4.4 Hz, 1H); 7.364 ppm (t, 7.7 Hz, 1H); 7.086 ppm (d, 8.0 Hz, 1H); 6.969 ppm (d, 7.7 Hz, 1H); 6.022 ppm (m, 1H); 5.497 ppm (m, 1H); 4.419 ppm (t, 7.3 Hz, 2H); 2.999 ppm (m, 2H); 2.770 ppm (p, 7.2 Hz, 7.4 Hz, 2H); 2.306 ppm (s, 3H); 1.817 ppm (m, 2H). MS ES+: 370.2; Exact: 369.16
ABOVE MOLECULE IS
https://newdrugapprovals.org/2016/05/04/galunisertib/
Galunisertib
Phase III
A TGF-beta receptor type-1 inhibitor potentially for the treatment of myelodysplastic syndrome (MDS) and solid tumours.
LY-2157299
CAS No.700874-72-2
READ MY PRESENTATION ON
KEYWORDS Sreenivasa Mundla Reddy, Managing Director, Sreeni Labs Private Limited, Hyderabad, Telangana, India, new, economical, scalable routes, early clinical drug development stages, Custom synthesis, custom manufacturing, drug discovery, PHASE 1, PHASE 2, PHASE 3, API, drugs, medicines

Motolimod
VTX-2337, 莫托莫德 , мотолимод , موتوليمود ,
(1E,4E)-2-amino-N,N-dipropyl-8-(4-(pyrrolidine-1-carbonyl)phenyl)-3H-benzo[b]azepine-4-carboxamide,
Cancer; Lymphoma
George A. Doherty, C. Todd Eary, Robert D. Groneberg, Zachary Jones
| Originator: | Array BioPharma |
| Developer: | VentiRx Pharmaceuticals |
| Class: | Antineoplastics, immunomodulator |
| Mechanism of Action: | Toll-like receptor 8 (TLR8) agonist |
| WHO ATC code: | L03A-X |
| EPhMRA code: | L3A9 |
Useful for treating a toll-like receptor (TLR)-associated diseases eg cancer. VentiRx, under license from Array BioPharma, and collaborator Celgene are developing Motolimod
A TLR-8 agonist, for treating cancer. In June 2016, Motolimod was reported to be in phase 2 clinical development.
Clinical Trials:
| Conditions | Phases | Interventions | Recruitment |
| Epithelial Ovarian Cancer|Fallopian Tube Cancer|Primary Peritoneal Cancer | Phase 2 | Combination | Active, not recruiting |
| Carcinoma, Squamous Cell of Head and Neck | Phase 2 | Combination | Active, not recruiting |
| Ovarian Cancer | Phase 1|Phase 2 | Combination | Not yet recruiting |
| Low Grade B Cell Lymphoma | Phase 1|Phase 2 | Combination | Terminated |
| Locally Advanced, Recurrent, or Metastatic Squamous Cell Cancer of Head and Neck | Phase 1 | Combination | Completed |
| Recurrent or Persistent Ovarian Epithelial, Fallopian Tube, or Peritoneal Cavity Cancer | Phase 1 | Combination | Completed |
| Squamous Cell Carcinoma of the Head and Neck | Phase 1 | Combination | Recruiting |
| Advanced Solid Tumors|Lymphoma | Phase 1 | Alone | Completed |
| Description | Motolimod (VTX-2337) is a selective and potent Toll-like receptor (TLR) 8 agonist with EC50 of 100 nM, > 50-fold selectivity over TLR7. Phase 2. | |||||
|---|---|---|---|---|---|---|
| Targets | TLR8 [1] | |||||
| IC50 | 100 nM(EC50) | |||||
| In vitro | VTX-2337 stimulates the production of both TNFα with EC50 of 140 nM and IL-12 with EC50 of 120 nM in PBMCs. In monocytes and mDCs, VTX-2337 selectively induces the production of TNFα and IL-12 via NF-κB activation. VTX-2337 also stimulates IFNγ production from NK cells, augments the lytic function of NK cells and enhances ADCC. [1] | |||||
| In vivo | In an ovarian cancer mouse model, TX-2337 enhances the effect of pegylated liposomal doxorubicin (PLD). [2] | |||||
| Features | ||||||
| Activity assay | The activity of specific TLR agonists is assessed using the secretory embryonic alkaline phosphatase (SEAP) reporter gene that is linked to NF-κB activation in response to TLR stimulation. Measurement of SEAP activity using the Quanti-blue substrate (InvivoGen) after TLR agonist treatment is carried out. |
|---|
| Cell lines | PBMCs or purified NK cells |
|---|---|
| Concentrations | ~500 nM |
| Incubation Time | 48 h |
| Method | PBMCs or purified NK cells are prepared as previously described, and the purity of NK cells was approximately 99%. NK cell–mediated cytotoxicity is assessed by Calcein AM release from labeled target cells. In brief, PBMCs or purified NK cells are cultured for 48 hours in RPMI medium in the presence of VTX-2337 (167 or 500 nmol/L) before incubation with target cells. |
| Species | Mouse | Rat | Rabbit | Guinea pig | Hamster | Dog |
| Weight (kg) | 0.02 | 0.15 | 1.8 | 0.4 | 0.08 | 10 |
| Body Surface Area (m2) | 0.007 | 0.025 | 0.15 | 0.05 | 0.02 | 0.5 |
| Km factor | 3 | 6 | 12 | 8 | 5 | 20 |
| Animal A (mg/kg) = Animal B (mg/kg) multiplied by | Animal B Km |
| Animal A Km |
For example, to modify the dose of resveratrol used for a mouse (22.4 mg/kg) to a dose based on the BSA for a rat, multiply 22.4 mg/kg by the Km factor for a mouse and then divide by the Km factor for a rat. This calculation results in a rat equivalent dose for resveratrol of 11.2 mg/kg.
| Rat dose (mg/kg) = mouse dose (22.4 mg/kg) × | mouse Km(3) | = 11.2 mg/kg |
| rat Km(6) |
[1] Lu H, et al. Clin Cancer Res. 2012, 18(2), 499-509.
[2] Monk BJ, et al. J Clin Oncol 31, 2013 (suppl; abstr 3077).
| NCT Number | Recruitment | Conditions | Sponsor /Collaborators |
Start Date | Phases |
|---|---|---|---|---|---|
| NCT02650635 | Recruiting | Colorectal Adenocarcinoma|Metastatic Pancreatic Adenocarcinoma|Recurrent Breast Carcinoma|Recurrent Colorectal Carcinoma|Recurrent Melanoma of the …more | Mayo Clinic|National Cancer Institute (NCI) | February 2016 | Phase 1 |
| NCT02431559 | Recruiting | Ovarian Cancer | Ludwig Institute for Cancer Research|MedImmune LLC|VentiR …more | November 2015 | Phase 1|Phase 2 |
| NCT02124850 | Recruiting | Squamous Cell Carcinoma of the Head and Neck | VentiRx Pharmaceuticals Inc. | September 2014 | Phase 1 |
| NCT01836029 | Active, not recruiting | Carcinoma, Squamous Cell of Head and Neck | VentiRx Pharmaceuticals Inc. | July 2013 | Phase 2 |
| NCT01666444 | Active, not recruiting | Epithelial Ovarian Cancer|Fallopian Tube Cancer|Primary Peritoneal Cancer | VentiRx Pharmaceuticals Inc.|Gynecologic Oncology Group | October 2012 | Phase 2 |
Download Motolimod (VTX-2337) SDF
| Molecular Weight (MW) | 458.6 |
|---|---|
| Formula | C28H34N4O2 |
| CAS No. | 926927-61-9 |
| Storage | 3 years -20℃powder |
|---|---|
| 6 months-80℃in solvent | |
| Synonyms | N/A |
| Solubility (25°C) * | In vitro | DMSO | 55 mg/mL warming (119.93 mM) |
|---|---|---|---|
| Ethanol | 15 mg/mL (32.7 mM) | ||
| Water | <1 mg/mL (<1 mM) | ||
| In vivo | |||
| * <1 mg/ml means slightly soluble or insoluble. * Please note that Selleck tests the solubility of all compounds in-house, and the actual solubility may differ slightly from published values. This is normal and is due to slight batch-to-batch variations. |
|||
PATENT
formula (I).

((IE, 4E)-2-amino-N,N-dipropyl-8-(4-(pyrrolidine-l-carbonyl)phenyl)-3H-benzo[b]azepine-4-carboxamide (“Compound A”)). The crystalline form can be an unsolvated or solvated crystalline form of the compound of formula (I).
Also provided herein are compositions including the crystalline forms of the compound of formula (I) described herein, methods of making the crystalline forms, and methods of using the crystalline forms for the treatment of diseases, including, for example, cancer.
Further provided herein are methods of agonizing a Toll-like receptor using the crystalline forms of the compound of formula (I) described herein. In one aspect the method includes agonizing a Toll-like receptor (TLR8) by contacting TLR8 with an effective amount of a crystalline form of the compound formula (I) described herein, wherein the effective amount agonizes the TLR8.
PATENT
WO2007024612
https://www.google.com/patents/WO2007024612A2?cl=en
Example 10
Synthesis of ClE, 4E)-2-ammo-N,N-dipropyl-8-(4-rpyrrolidine-l-carbonyl)phenyl)-3H- benzorbiazepine-4-carboxamide C27)

Compound (27) was prepared from compound (24) by a method similar to that described in Example 2 to provide 49 mg (43%) of the desired compound. 1H NMR (CDCl3) δ 0.93 (t, 6H), 1.63-1.71 (m, 4H), 1.89 (m, 2H), 1.98 (m, 2H), 2.83 (s, 2H), 3.40-3.51 (m, 6H), 3.67 (t, 2H), 6.83 (s, IH), 7.3 (dd, IH), 7.35 (d, IH), 7.49 (d, IH)5 7.64 (q, 4H).
EXAMPLE 2 CLIP, QUANTITIES MAY VARY USE YOUR DISCRETION
Trimethylaluminum (0.34 mL of a 2.0 M solution in toluene) was added to bis(2- methoxyethyl)amine (92 mg, 0.69 mmol) in DCE (3 mL). After 10 minutes solid COMPD 24, 0.23 mmol) was added and the vessel was sealed and heated to 75 0C for 16-20 hours. Upon cooling the reaction was quenched with saturated Rochelle’s salt (2 mL) and after 20 minutes the mixture was partitioned between CH2Cl2 (50 mL) and brine (50 mL). The phases were separated and the aqueous was extracted with CH2Cl2 (2 x 20 mL). The combined organics were dried and concentrated. The crude material was purified via preparative TLC (2, 0.5 mm plates, eluting with 5-10% MeOH/CH2Cl2 with 4-6 drops of NH4OH)
Synthesis of (IE, 4E)-ethyl 2-ammo-8-(pyrrolidine-l-carbonyl)-3H-benzorb]azepine-4- carboxylate (24)

The reaction scheme for the synthesis of compound (24) is shown in Figure 4. Step A: Preparation of (E)-2-(4-bromo-2-nitrophenyl)-N,N-dimethylethenamine (18):
To a solution of l-methyl-2-nitro-4-bromobenzene (17) (29.86 g, 138.2 mmol) in toluene (200 niL) was added dimethylformamide dimethylacetal (17.52 g, 138.2 mmol). The mixture was heated to reflux for 14 hours. After cooling to room temperature the mixture was concentrated under vacuum and the resulting oil was immediately used in the next reaction. Step B: Preparation of 4-bromo-2-nitrobenzaldehyde (19): To a solution of crude (E)-
2-(4-bromo-2-nitrophenyl)-N,N-dimethylethenamine (35.5 g, 131 mmol) in THF (300 mL) and pH 7.2 phosphate buffer (300 mL) was added NaIO4 (56.0 g, 262 mmol). The solids were removed and the filter cake was washed with EtOAc (200 mL). The filtrate was washed with brine (2 X 100 mL), dried and concentrated. The concentrate was purified via flash chromatography (5% EtOAc/hexanes to 10% EtOAc/hexanes) to provide 4-bromo-2- nitrobenzaldehyde (8.41 g, 28% yield).
Step C: Preparation of (E)-ethyl 3-(4-bromo-2-nitrophenyl)-2-(cyanomethyl)acrylate (20): To a solution of 4-bromo-2-nitrobenzaldehyde (3.45 g, 15.0 mmol) in toluene (15 mL) was added α-cyanomethylcarboethoxyethylidene triphenylphosphorane (6.1O g, 15.7 mmol). The mixture was heated to 75 °C for 16 hours. The reaction was allowed to cool and the solvent was removed under vacuum. The concentrate was purified via flash chromatography (100% hexanes to 20% EtOAc) to yield (E)-ethyl 3-(4-bromo-2-nitrophenyl)-2- (cyanomethyl)acrylate (2.25 g, 44% yield) as an off white solid.
Step D: Preparation of (IE, 4E)-ethyl 2-ammo-8-bromo-3H-benzo|“b1azepine-4- carboxylate (21): To a solution of (E)-ethyl 3-(4-bromo-2-nitrophenyl)-2- (cyanomethyl)acrylate (1.00 g, 2.9 mmol) in acetic acid (25 mL) was added iron powder (1.10 g, 19.0 mmol). The mixture was heated to 90 °C for 5 hours. Upon cooling the acetic acid was removed under vacuum and the resulting semisolid was dissolved in 50% K2CO3 (100 mL) and EtOAc (100 mL). The mixture was filtered to remove insoluble material and the phases were separated. The aqueous phase was extracted with EtOAc (2 x 100 mL). The combined organics were dried and concentrated. The concentrate was purified via flash chromatography (Biotage 40m, 5% MeOH/CH2Cl2) to yield (lE,4E)-ethyl 2-amino-8-bromo- 3H-benzo[b] azepine-4-carboxylate (0.52 g, 57%).
Step E: Preparation of (IE. 4E)-ethyl-8-bromo-2-(tert-butoxycarbonyl)-3H- benzo FbI azepine-4-carboxylate (22) : To a CH2Cl2 (5 mL) solution containing (IE, 4E)-ethyl 2-amino-8-bromo-3H-benzo[b]azepine-4-carboxylate (198 mg, 0.640 mmol) was added Boc anhydride (140 mg, 0.640 mmol). The solution was stirred at room temperature for 72 hours. The reaction was concentrated to dryness and purified by column chromatography (Biotage 12m, 4:1 hexanes :EtO Ac) to provide (IE, 4E)-ethyl-8-bromo-2-(tert-butoxycarbonyl)-3H- benzo[b] azepine-4-carboxylate (245 mg, 94% yield) as a white solid. Step F: Preparation of (IE, 4E)-ethyl-2-(tert-butoxycarbonyl)-8-(pyrrolidine-l- carbonyl)-3H-benzo Fb] azepme-4-carboxylate (23) : To an ethanol solution (15 mL) containing K3PO4 (938 mg, 4.42 mmol), 4-(pyrrolidine-l-carbonyl)phenylboronic acid (785 mg, 3.58 mmol), and (IE, 4E)-ethyl-8-bromo-2-(tert-butoxycarbonyl)-3H-benzo[b]azepine-4- carboxylate (489 mg, 1.19 mmol), was added palladium acetate (80.5 mg, 0.358 mmol). The reaction was heated to 60 °C for 2 hours, then cooled to room temperature and concentrated to dryness. The brown oil was purified by preparative LC plate (100% EtOAc) to provide (lE,4E)-ethyl-2-(tert-butoxycarbonyl)-8-(pyrrolidine-l-carbonyl)-3H-benzo[b]azepine-4- carboxylate (277 mg, 46% yield) as a tan oil.
Step G: Preparation of (IE, 4E)-ethyl 2-amino-8-(pyrrolidine-l-carbonyl)-3H- benzoFbl azepine-4-carboxylate (24V (IE, 4E)-ethyl-2-(tert-butoxycarbonyl)-8-(pyrrolidine-l- carbonyl)-3H-benzo[b]azepine-4-carboxylate (110 mg, 0.218 mmol) was diluted with a 1:4 TFA:CH2C12 solution (4 mL). The reaction was stirred at room temperature for 1 hour, and then diluted with CH2Cl2. The organic phase was washed with 10% K2CO3 and brine (30 mL). The CH2Cl2 solution was dried over Na2SO4, filtered, and concentrated to provide (IE, 4E)-ethyl 2-amino-8-(pyrrolidine-l-carbonyl)-3H-benzo[b]azepine-4-carboxylate (88 mg, 81% yield) as a yellow solid. 1H NMR (CDCl3) δ 1.39 (t, 3H), 1.88-1.99 (m, 4H), 2.98 (s, 2H), 3.49-3.52 (m, 2H), 3.66-3.69 (m, 2H), 4.30-4.35 (m, 2H), 7.32 (d, IH), 7.46-7.49 (m, 2H), 7.60 (d, 2H) 7.67 (d, 2H), 7.84 (s, IH).
PATENT
WO2012045090
(assigned to VentiRx), claiming an aqueous composition comprising a TLR-8 agonist (ie motolimod) and an anti-cancer agent (eg doxorubicin, gemcitabine or cyclophosphamide), useful for treating cancer.
| Patent ID | Date | Patent Title |
|---|---|---|
| US2016045502 | 2016-02-18 | THERAPEUTIC BENEFIT OF SUBOPTIMALLY ADMINISTERED CHEMICAL COMPOUNDS |
| US2015182490 | 2015-07-02 | METHODS FOR TREATING TYROSINE-KINASE-INHIBITOR-RESISTANT MALIGNANCIES IN PATIENTS WITH GENETIC POLYMORPHISMS OR AHI1 DYSREGULATIONS OR MUTATIONS EMPLOYING DIANHYDROGALACTITOL, DIACETYLDIANHYDROGALACTITOL, DIBROMODULCITOL, OR ANALOGS OR DERIVATIVES THEREOF |
| US2014066432 | 2014-03-06 | Substituted Benzoazepines As Toll-Like Receptor Modulators |
| US2013236449 | 2013-09-12 | METHODS OF ENHANCING ANTIBODY-DEPENDENT CELLULAR CYTOTOXICITY |
| US2013018042 | 2013-01-17 | Toll-Like Receptor Agonist Formulations and Their Use |
| US8304407 | 2012-11-06 | 8-substituted benzoazepines as toll-like receptor modulators |
| US2012219615 | 2012-08-30 | Therapeutic Use of a TLR Agonist and Combination Therapy |
| US8242106 | 2012-08-14 | TOLL-LIKE RECEPTOR AGONIST FORMULATIONS AND THEIR USE |
| US8153622 | 2012-04-10 | 8-Substituted Benzoazepines as Toll-Like Receptor Modulators |
| US2012082658 | 2012-04-05 | Methods for the Treatment of Allergic Diseases |
//////Motolimod, VTX-2337, 莫托莫德 , мотолимод , موتوليمود , VTX 2337, VTX-378, 926927-61-9, phase 2, TLR-8 agonist
CCCN(CCC)C(=O)C1=CC2=C(C=C(C=C2)C3=CC=C(C=C3)C(=O)N4CCCC4)N=C(C1)N