











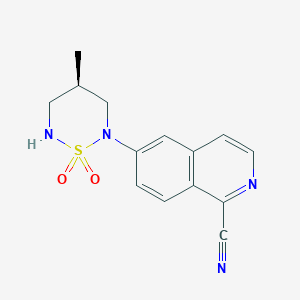
PF 14
| Molecular Formula: | C14H14N4O2S |
|---|---|
| Molecular Weight: | 302.35156 g/mol |
6-[(4R)-4-methyl-1,1-dioxo-1,2,6-thiadiazinan-2-yl]isoquinoline-1-carbonitrile
1612755-71-1 CAS
The androgen receptor (“AR”) is a ligand-activated transcriptional regulatory protein that mediates induction of male sexual development and function through its activity with endogenous androgens. Androgenic steroids play an important role in many physiologic processes, including the development and maintenance of male sexual characteristics such as muscle and bone mass, prostate growth, spermatogenesis, and the male hair pattern. The endogenous steroidal androgens include testosterone and dihydrotestosterone (“DHT”). Steroidal ligands which bind the AR and act as androgens (e.g. testosterone enanthate) or as antiandrogens (e.g. cyproterone acetate) have been known for many years and are used clinically.
PATENT
WO 2015173684
http://www.google.com/patents/WO2015173684A1?cl=en
The androgen receptor (“AR”) is a ligand-activated transcriptional regulatory protein that mediates induction of male sexual development and function through its activity with endogenous androgens. Androgenic steroids play an important role in many physiologic processes, including the development and maintenance of male sexual characteristics such as muscle and bone mass, prostate growth,
spermatogenesis, and the male hair pattern. The endogenous steroidal androgens include testosterone and dihydrotestosterone (“DHT”). Steroidal ligands which bind the AR and act as androgens (e.g. testosterone enanthate) or as antiandrogens (e.g.
cyproterone acetate) have been known for many years and are used clinically.
6-[(4f?)-4-Methyl-1 , 1-dioxido-1 ,2,6-thiadiazinan-2-yl]isoquinoline-1-carbonitrile (Formula I), in its free base form, has the chemical formula C14H14N4SO2 and the following structural formula:

Formula I
Synthesis of 6-[(4f?)-4-methyl-1 , 1-dioxido-1 ,2,6-thiadiazinan-2-yl]isoquinoline-1-carbonitrile is disclosed in co-pending international patent application,
PCT/IB2013/060381 , filed 25th November 2013, and published as WO 2014/087298 on 12th June 2014, assigned to the assignee of the present invention and which is incorporated herein by reference in its entirety. 6-[(4f?)-4-Methyl-1 , 1-dioxido-1 ,2,6-thiadiazinan-2-yl]isoquinoline-1-carbonitrile is known to be active as a selective androgen receptor modulator (SARM) and, as such, is useful for treating and/or preventing a variety of hormone-related conditions, for example, conditions associated with androgen decline, such as, inter alia, anaemia; anorexia; arthritis; bone disease; musculoskeletal impairment; cachexia; frailty; age-related functional decline in the elderly; growth hormone deficiency; hematopoietic disorders; hormone replacement; loss of muscle strength and/or function; muscular dystrophies; muscle loss following surgery; muscular atrophy; neurodegenerative disease; neuromuscular disease;
obesity; osteoporosis; and, muscle wasting.
Identification of new solid forms of a known pharmaceutical active ingredient provide a means of optimising either the physicochemical, stability, manufacturability and/or bioperformance characteristics of the active pharmaceutical ingredient without modifying its chemical structure. Based on a chemical structure, one cannot predict with any degree of certainty whether a compound will crystallise, under what conditions it will crystallise, or the solid state structure of any of those crystalline forms. The specific solid form chosen for drug development can have dramatic influence on the properties of the drug product. The selection of a suitable solid form is partially dictated by yield, rate and quantity of the crystalline structure. In addition, hygroscopicity, stability, solubility and the process profile of the solid form such as compressibility, powder flow and density are important considerations.
As such, there is a need to identify solid forms of 6-[(4f?)-4-methyl-1 , 1-dioxido-1 ,2,6-thiadiazi
Example 1

Procedure:
Into a 2L 3-neck round bottom flask equipped with a mechanical stirrer, reflux condenser and thermocouple with heating mantle was placed 2-methyltetrahydrofuran (2-MeTHF) (10 mL/g; 8.15 moles; 817 ml_; 702 g) followed by racemic-2,2′-bis(diphenylphosphino)-1 ,1 ‘-binaphthyl (BINAP) (0.04 equiv (molar); 14.0 mmol; 8.74 g) and bis(dibenzylideneacetone)palladium (Pd2(dba)3) (0.04 equiv (molar); 14.0 mmol;
8.07 g). The mixture was degassed by pulling vacuum and refilling with nitrogen three times then heated to 75 °C for 15 minutes and cooled to ambient temperature. In a separate flask, (S)-3-amino-2-methylpropan-1-ol (1.60 equiv; 561 mmol; 50.0 g, prepared using literature methods, for example as disclosed in EP-A-0,089, 139 published on 21st September 1983) was dissolved in 2-methyltetrahydrofuran (5 ml_/g;
4.08 moles; 409 ml_; 351 g) and degassed by pulling vacuum and refilling with nitrogen three times. Into the pot containing the catalyst was added 6-(bromoisoquinoline-1- carbonitrile) (1.00 equiv; 351 mmol; 81.75 g) and cesium carbonate (1.6 equiv (molar); 561 mmol; 185 g) in single portions followed by the solution of the aminoalcohol via addition funnel. The reaction mixture was again degassed by pulling vacuum and refilling with nitrogen three times. The reaction was heated to 70 °C for 3 hours. The reaction was cooled to ambient temperature and filtered through a pad of Celite. The contents of the flask were rinsed out with three 100 mL portions of 2-methyltetrahydrofuran. The filtrate was transferred into a 2L round bottom flask equipped with a thermocouple and mechanical stirrer under nitrogen. Silica Gel (Silicylate SiliaMet® Thiol) (0.4 g/g-pure-LR; 544 mmol; 32.7 g) was charged and the flask was stirred at 40 °C overnight. The following morning, the reaction was cooled to < 30 °C and filtered again through Celite. The pad was washed with 100ml_ of 2-methyltetrahydrofuran (or until no yellow color persisted in the filtrate). The filtrate was placed into a 3L round bottom flask equipped with a magnetic stir bar, distillation head (with condenser and receiving flask), and thermocouple. The mixture was heated to 60 °C and placed under vacuum (-450-500 mbar) to distil out 1.3 L total of 2-methyltetrahydrofuran. 500 mL of toluene was added to precipitate the desired product. The heating mantle was removed and the reaction was allowed to reach ambient temperature. The mixture was stirred for 1 hour at ambient temperature and then the solids were collected by vacuum filtration on a sintered glass funnel. The cake was dried overnight on the funnel under vacuum. The following morning, the solids were transferred into an amber bottle and weighed (71.9 g; 298 mmol). The product was used in the next step without further purification.
Example 2

Procedure:
In a 1 L reactor equipped with a temperature probe and overhead stirring was added the product of Example 1 (20.0 g; 1.00 equiv; 82.9 mmol) and 2-methyltetrahydrofuran (2-MeTHF) (30 mL/g-pure-LR; 5.98 moles; 600 mL; 515 g). The reaction mixture was
gently warmed to 40°C to achieve partial solubility. The reaction was cooled to 0°C. Once the reaction reached 0°C methanesulfonyl chloride (MsCI) (1.4 equiv (molar); 1 16 mmol; 8.98 mL; 13.3 g) was added in a single portion followed immediately by triethylamine (TEA) (1.4 equiv (molar); 116 mmol; 16.2 mL; 11.7 g) dropwise via syringe over a period of 15 minutes. The reaction mixture was further stirred for 30 min at 0°C and then warmed to 23°C for 60 minutes. The product (26.47 g; 1.00 equiv; 82.88 mmol; 26.47 g; 100% assumed yield) was then used without purification for the sulfonylation reaction.
Example 3
t-BuOH, 2-MeTHF
o 0 °C to 23 °C o
CI-S-N=C=0 CI-S-NHBoc
0 O
Procedure:
To a solution of t-butyl alcohol (t-BuOH) (1 equiv (molar); 116 mmol; 1 1.0 mL; 8.60 g) in 2-methyltetrahydrofuran (2-MeTHF) (1 M; 1.16 moles; 116 mL; 99.6 g) at 0°C was added chlorosulfonyl isocyanate (116 mmol; 1.00 equiv; 10.1 mL; 16.4 g) dropwise. The homogeneous solution was stirred for 30 minutes at ambient temperature and then used directly in the sulfonylation reaction.
Example 4

Sulfonylation Reaction Procedure:
A previously prepared solution of the product of Example 3 (1.4 equiv (molar); 1 16 mmol; 116 g) in 2-methyltetrahydrofuran was added to a suspension of the product of Example 2 (1.00 equiv; 82.89 mmol; 26.5 g) at 0°C. The mixture was warmed to ambient temperature over 30 minutes. HPLC analysis revealed the reaction was complete. The reaction was quenched with a 10% sodium carbonate solution (2 equiv
(molar); 165 mmol; 101 mL; 1 17 g) and water (to dissolve salts) (5 L/kg; 7.35 moles; 132 mL; 132 g). The top organic layer was removed and passed through a plug of Carbon (Darco G60) (0.5 g/g) on a filter. A significant improvement in color (dark orange to yellow) was observed. The solution was concentrated to 10 total volumes and used in the next step without purification.
Example 5

Procedure:
A solution of the product of Example 4 (1.OOequiv; 82.9 mmol; 41.3 g) in 2-methyltetrahydrofuran (2-MeTHF) (10ml_/g; 4.12 moles; 413 mL; 355 g) was placed into a 1 L reactor equipped with an overhead stirrer and temperature probe. Next, potassium carbonate (K2CO3) (325 mesh) (6 equiv (molar); 497 mmol; 69.4 g) and water (0.0 L/100-g-bulk-LR; 459 mmol; 8.26 mL; 8.26 g) were added and the mixture heated to 40°C (jacket temperature) and stirred overnight. The reaction was cooled to ambient temperature and water (4L/kg-pure-LR; 9.17 moles; 165 mL; 165 g]) was added. The biphasic reaction was stirred for 1 hour at 23 °C. The aqueous layer was extracted and removed. The organic layer was passed through a plug of Carbon (Darco G60) (0.5 g/g-pure-LR; 20.7g) in a disposable filter. The 2-methyltetrahydrofuran solution was switched to a 10 volume solution of toluene via a constant strip-and-replace distillation to no more than 1 % 2-methyltetrahydrofuran. The toluene solution of the reaction product (1.00 equiv; 82.9 mmol; 33.4 g; 100% assumed yield) was used as-is in the next step without further purification.
Example 6

Procedure:
To a 1 L reactor under nitrogen and equipped with overhead stirring and a temperature probe was added the product of Example 5 (1.00 equiv; 78.7 mmol; 33.4 g) as a solution in toluene (10 mL/g-pure-LR; 3.00 moles; 317 ml_; 276 g). Next, trifluoroacetic acid (TFA) (10 equiv (molar); 787 mmol; 59.5 ml_; 89.8 g) was added to the reaction over a period of 1 hour keeping the internal temperature below 30°C. The dark red mixture was stirred for 1 hour. The reaction was quenched at 23 °C by the addition of sodium carbonate (5 equiv (molar); 394 mmol; 240 ml_; 278 g). The reaction was quenched slowly, over a period of 1 hour to form the TFA salt of the product. Once the charge was complete, the mixture was cooled to 0°C, held for 1 hour and filtered. The next morning, the solid product (6-[(4R)-4-methyl-1 , 1-dioxido-1 ,2,6-thiadiazinan-2-yl]isoquinoline-1-carbonitrile in its free base form) was weighed (0.89 equiv; 70.0 mmol; 21.2 g; 89.0% yield) and used in the next step without further purification.
Example 7
Crystalline 6-[(4f?)-4-methyl-1 , 1-dioxido-1 ,2,6-thiadiazinan-2-yl]isoquinoline-1-carbonitrile free base (Form (1)) was prepared as follows.
In a 1 L 3-neck round bottom flask was added 6-[(4R)-4-methyl-1 , 1-dioxido-1 ,2,6-thiadiazinan-2-yl]isoquinoline-1-carbonitrile free base (1.00 equiv; 70.0 mmol; 21.2 g) a magnetic stir bar and acetone (40ml_/g; 1 1.5 moles; 847 ml_; 669 g). The mixture was heated to reflux (approximately 57°C) and stirred for 1 hour. The mixture was concentrated by atmospheric distillation (heating mantle set at 65°C) and 40ml_ of acetone was collected into a graduated cylinder. Next, water (25 mL/g; 29.4 moles; 530 ml_; 530 g) was charged over a period of one hour. The mixture was stirred at ambient temperature for 60min before being cooled to 0°C at 1 °C /min for 1 hour. The solids were collected by filtration in a disposable funnel. Crystalline 6-[(4f?)-4-methyl-1 , 1-dioxido-1 ,2,6-thiadiazinan-2-yl]isoquinoline-1-carbonitrile (Form (1), 0.88 equiv; 61.9 mmol; 18.7 g; 88.3% yield) was dried under vacuum overnight at 40 °C. Typical purity after crystallization is 98%.
PATENT
US 20140155390
http://www.google.com/patents/US20140155390
PATENT
WO 2015181676
http://www.google.com/patents/WO2015181676A1?cl=en
xample 9
6-[(3S)-3-methyl-1 , 1 -dioxido-1 ,2,5-thiadiazolidin-2-yl1naphthalene-1 -carbonitrile
(stereochemistry is arbitrarily assigned)
LCMS m/z = 286.0 (M – H). 1 H NMR (400 MHz, cf6-DMSO): δ 1 .31 (d, J = 6.2 Hz, 3H), 3.13 – 3.25 (m, 1 H), 3.71 (dt, J = 12.5, 6.8 Hz, 1 H), 4.49 – 4.62 (m, 1 H), 7.62 – 7.70 (m, 1 H), 7.75 – 7.83 (m, 2H), 7.99 (t, J = 7.8 Hz, 1 H), 8.07 (d, J = 6.6 Hz, 1 H), 8.14 (d, J = 8.9 Hz, 1 H), 8.28 (d, J = 8.4 Hz, 1 H). Chiral HPLC purity: 99.1 % (retention time 17.12 minutes)

Step 1. Synthesis of aminoester (#D1). Thionylchlride (8.5 ml_, 1 16.5 mmol) was added to the solution of amino acid (4.0 g, 38.8 mmol) in MeOH (170 ml_) at 0 °C, and the reaction mixture was stirred for 6 h at room temperature. The reaction was monitored by TLC, and after disappearance of the starting material it was cooled to room temperature and solid NaHC03 was added. The reaction mixture was filtered, concentrated in vacuo and the resulting residue was triturated with diethyl ether to obtain crude #D1 (4 g, 90%) as a white solid. Rf: 0.4 (f-BuOH: AcOH: H20 (4:0.5:0.5)).
GCMS m/z = 1 17.1 (M). 1H NMR (400 MHz, cf6-DMSO): δ 1.17 (d, J = 6.8 Hz, 3H), 2.83 – 2.88 (m, 2H), 3.03 – 3.05 (m, 1 H), 3.65 (s, 3H), 8.02 – 8.30 (br s, 3H).
Step 2. Synthesis of aminoalcohol (#D2). #D1 (2.0 g, 13.0 mmol) was added
portionwise to a suspension of LiAIH4 (1.4 g, 39.2 mmol) in THF (75 ml_) under nitrogen atmosphere at 0 °C. The reaction mixture was stirred for 30 minutes and then allowed to stir at room temperature for another 30 minutes. The reaction mixture was refluxed for 2 h, and then it was cooled to -10 °C and quenched carefully with ice cold water (1.4 ml_). 10% NaOH solution (2.8 ml_) and ice cold water (4.2 ml_) were added, and the mixture was stirred for 15 minutes. It was filtered, and the filtrate washed with EtOAc (3 x 100 ml_), dried over anhydrous Na2S04 and concentrated under vacuum to obtain #D2 (1.2 g, 86%) as a pale yellow liquid. Rf: 0.2 (20% MeOH in DCM).
1H NMR (400 MHz, cf6-DMSO): δ 0.78 (d, J = 6.8 Hz, 3H), 1.46 – 1.54 (m, 1 H), 2.41 -2.45 (m, 2H), 2.50 – 2.54 (m, 1 H), 3.22 – 3.34 (m, 4H).
Step 3. Synthesis of coupling product (#D3). K3P04 (6.1 g, 28.8 mmol), BINAP (0.44 g, 0.72 mmol) and Pd2(dba)3 (0.32.0 g, 0.36 mmol) was added to the degassed
suspension of 6-bromo-1 -cyanoisoquinoline #A3 (1.7 g, 7.2 mmol), #D2 (1.2 g, 14.5 mmol) in DMSO at room temperature. The reaction mixture was heated at 105 °C for 2 h. The reaction was cooled to room temperature, water (500 ml_) followed by EtOAc (100 ml_) were added, and the mixture was stirred for 10 minutes. The biphasic mixture was filtered through a Celite™ pad and washed with EtOAc (100 ml_). The organic layer was separated, and the aqueous layer was extracted with EtOAc (3 x 100 ml_). The combined organic layers were dried over anhydrous Na2S04, concentrated under reduced pressure to get a crude material. This was purified by column chromatography on 100 – 200 mesh silica gel, using 50 – 70% EtOAc in petroleum ether as the eluent to obtain #D3 (0.5 g, 48.5%) as a yellow solid. Rf: 0.4 (60% EtOAC in petroleum ether).
LCMS m/z = 242.0 (M + H). 1 H NMR (400 MHz, cf6-DMSO): δ 0.97 (d, J = 6.4 Hz, 3H), 1.87 – 1.99 (m, 1 H), 2.92 – 2.99 (m, 1 H), 3.20 – 3.27 (m, 1 H), 3.38 – 3.42 (m, 2H), 4.59 (t, J = 5.2 Hz, 1 H), 6.77 (d, J = 2.0, 1 H), 7.01 (t, J = 5.6 Hz, 1 H), 7.34 (dd, J = 9.2 Hz, J = 2.0 Hz, 1 H), 7.73 (d, J = 6.0 Hz, 1 H), 7.88 (d, J = 8.8 Hz, 1 H), 8.312 (d, J = 6.0 Hz, 1 H).
Step 4. Methanesulfonated coupling product (#D4). Triethylamine (0.44 mL, 3.1 mmol) was added to a solution of #D3 (0.50 g, 2.0 mmol) in DCM at 0 °C.
Methanesulfonylchloride (0.25 mL, 3.1 mmol) was added over 10 minutes, and the reaction mixture was stirred for 1 h at room temperature. After disappearance of the starting material by TLC, it was diluted with DCM and washed with water. The organic layer was separated, dried over Na2S04, concentrated under reduced pressure to obtain crude #D4 (0.6 g, crude) as yellow solid. This was used for next step without any purification. Rf: 0.6 (50% EtOAc in petroleum ether).
LCMS m/z = 320.0 (M + H). 1 H NMR (400 MHz, CDCI3): δ 1.17 (d, J = 6.8 Hz, 3H), 2.32 – 2.37 (m, 1 H), 3.06 (s, 3H), 3.26 – 3.41 (m, 2H), 4.16 – 4.20 (m, 1 H), 4.33 – 4.37 (m, 1 H), 4.75 (br s, 1 H), 6.70 (d, J = 2.4, 1 H), 7.09 (dd, J = 9.2 Hz, 2.4 Hz, 1 H), 7.57 (d, J = 6.0 Hz, 1 H), 8.05 (d, J = 9.2 Hz, 1 H), 8.39 (d, J = 5.6 Hz, 1 H).
Step 5. Cyclized and uncyclized intermediates (#D5, #D6). Chlorosulfonylisocyanate (1.2 mL, 13.1 mmol) was added dropwise to a solution f-BuOH (1.4 mL, 13.1 mmol) in toluene (4.0 mL) at -5 °C. The reaction mixture was stirred at room temperature for 20 minutes, and then THF (1 mL) was added to the resulting suspension to obtain clear solution. In another flask, DIPEA (2.3 mL, 13.1 mmol) was added to a solution of #D4 (0.6 g, crude 2.6 mmol) in dry THF (3 mL). The above prepared reagent (CIS02NH-Soc) was added to this reaction mixture dropwise at room temperature over a period of 20 minutes. The resulting reaction mixture was then stirred for 16 h at room temperature. The mixture was diluted with EtOAc (100 mL) and washed with water (100 mL). The aqueous layer was washed with EtOAc (2 x 100 mL), combined all the organic layers, dried over Na2S04, concentrated under reduced pressure to obtain the crude product (LCMS shows desired #D6 and uncyclized #D5. This crude was purified by column chromatography on 100 – 200 mesh silica gel, using 10 – 30% EtOAc in petroleum ether as an eluent to obtain desired #D6 (0.35 g, 47.8%), and uncyclized #D5 (0.22 g, crude).
The uncyclized #D5 (0.22 g, crude) was dissolved in THF (1 mL) and DIPEA (0.6 mL) was added to the solution. The reaction mixture was stirred for another 12 h at room temperature. After which time, it was diluted with EtOAc (100 mL) and washed with water (100 mL). The aqueous layer was washed with EtOAc (2 x 100 mL), combined all the organic layers, dried over Na2S04, concentrated under reduced pressure to obtain crude product. This crude was purified by column chromatography on 100 – 200 mesh silica gel, using 10 – 30% EtOAc in petroleum ether as an eluent to obtain desired #D6 (1 .1 g, 13.2%). Total amount of #D6 was (0.5 g, 60% for two steps, 82% LCMS purity). Rf: 0.8 (60% EtOAc in petroleum ether).
LCMS m/z = 403.1 (M + H). 1 H NMR (400 MHz, CDCI3): δ 1 .04 (d, J = 6.8 Hz, 3H), 1 .50 (s, 9H), 2.38 – 2.48 (m, 1 H), 3.65 – 3.82 (m, 2H), 3.92 – 4.02 (m, 1 H), 4.30 – 4.38 (m, 1 H), 7.79 – 7.81 (m, 1 H), 7.86 – 7.88 (m, 2H), 8.34 – 8.37 (d, J = 9.2 Hz, 1 H), 8.67 (d, J = 6.0 Hz, 1 H).
Step 6. Racemate #D7 and final products (#10, #11 ). TFA (5 mL) was added to a solution of #D6 (0.15 g, 0.37 mmol) in DCM (100 mL) at 0 °C. The reaction mixture was stirred for 1 h at 0 °C. The solution was neutralized with saturated aqueous NaHC03 solution at 0 °C. The mixture was diluted with water, extracted with DCM (3 x 100 mL). The combined organic layers were dried over anhydrous Na2S04 and concentrated under reduced pressure to obtain racemic #D7 (0.10 mg, 73%).
LCMS m/z = 303.0 (M + H). Rf: 0.3 (60% EtOAc in petroleum ether).
Enantiomeric separation: #D7 was submitted for chiral separation to obtain final compounds #10 (0.015 mg) and #11 (0.016 mg).
Column: CHIRALPAK IA, 4.6 χ 250 mm, 5 m; Mobile phase: n-Hexane/ /-PrOH/DCM (60%/15%/15%); Flow rate: 0.8 mL/min.
Example 10
6-[(4R)-4-methyl-1 , 1 -dioxido-1 ,2,6-thiadiazinan-2-yl1isoquinoline-1 -carbonitrile (#10; R = (R)-CH3)
LCMS m/z = 303.0 (M + 1 ). 1 H NMR (400 MHz, cf6-DMSO): δ 0.98 (d, J = 6.4 Hz, 3H), 2.22 – 2.26 (m, 1 H), 3.16 – 3.22 (m, 1 H), 3.34 – 3.39 (m, 1 H), 3.59 – 3.65 (m, 1 H), 3.77 – 3.81 (m, 1 H), 7.75 – 7.79 (m, 1 H, disappeared in D20 exchange), 7.95 (dd, J = 8.8 Hz, J = 2.0 Hz, 1 H), 8.06 (d, J = 1 .6 Hz, 1 H), 8.23 – 8.27 (m, 2H), 8.703 (d, J = 5.2 Hz, 1 H). Rf: 0.3 (60% EtOAc in petroleum ether). Chiral HPLC purity: 98.2% (retention time 1 1 .43 minutes).
| Patent ID | Date | Patent Title |
|---|---|---|
| US2014155390 | 2014-06-05 | NOVEL SELECTIVE ANDROGEN RECEPTOR MODULATORS |
//////////pf 14,
C[C@H]3CN(c1cc2ccnc(C#N)c2cc1)S(=O)(=O)NC3