Tout sur les médicaments הכל על תרופות كل شيئ عن الأدوية Все о наркотиках 关于药品的一切 డ్రగ్స్ గురించి అన్ని 마약에 관한 모든 것 Όλα για τα Ναρκωτικά Complete Tracking of Drugs Across the World by Dr Anthony Melvin Crasto, Worldpeacepeaker, worlddrugtracker, PH.D (ICT), MUMBAI, INDIA, Worlddrugtracker, Helping millions, 9 million hits on google on all websites, 2.5 lakh connections on all networks, “ALL FOR DRUGS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This is a compilation for educational purposes only. P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
D4A (Anti-Pruritics, Including Topical Antihistamines, Anaesthetics, etc), N2A (Narcotics)
Indication
Pain, Osteoarthritis, Pruritus
A kappa opioid receptor agonist potentially for treatment of post-operative pain and uremic pruritus.
Difelikefalin, also known CR845, is a novel and potent kappa opioid receptor agonist. CR845 exhibit low P450 CYP inhibition and low penetration into the brain. CR845 may be useful in the prophylaxis and treatment of pain and inflammation associated with a variety of diseases and conditions .
κ opioid receptor agonists are known to be useful as therapeutic agents for various pain. Among, kappa opioid receptor agonist with high selectivity for peripheral kappa opioid receptors, are expected as a medicament which does not cause the central side effects. Such as peripherally selective κ opioid receptor agonist, a synthetic pentapeptide has been reported (Patent Documents 1 and 2).
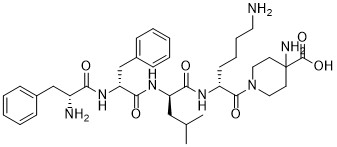
The following formula among the synthetic pentapeptide (A)
[Formula 1] Being Represented By Compounds Are Useful As Pain Therapeutics. The Preparation Of This Compound, Solid Phase Peptide Synthesis Methods In Patent Documents 1 And 2 Have Been Described.
Compound (1) or a salt thereof and compound (A), for example as shown in the following reaction formula, 4-aminopiperidine-4-carboxylic acid, D- lysine (D-Lys), D- leucine (D-Leu) , it can be prepared by D- phenylalanine (D-Phe) and D- phenylalanine (D-Phe) sequentially solution phase peptide synthesis methods condensation.
[Of 4]
The present invention will next to examples will be described in further detail.
Example
1 (1) Synthesis of Cbz-D-Lys (Boc) -α-Boc-Pic-OMe (3)
to the four-necked flask of 2L, α-Boc-Pic- OMe · HCl [α-Boc-4 – aminopiperidine-4-carboxylic acid methyl hydrochloride] were charged (2) 43.7g (148mmol), was suspended in EtOAc 656mL (15v / w). To the suspension of 1-hydroxybenzotriazole (HOBt) 27.2g (178mmol), while cooling with Cbz-D-Lys (Boc) -OH 59.2g (156mmol) was added an ice-bath 1-ethyl -3 – (3-dimethylcarbamoyl amino propyl) was added to the carbodiimide · HCl (EDC · HCl) 34.1g (178mmol). After 20 minutes, stirring was heated 12 hours at room temperature. After completion of the reaction, it was added and the organic layer was 1 N HCl 218 mL of (5.0v / w). NaHCO to the resulting organic layer 3 Aq. 218ML (5.0V / W), Et 3 N 33.0 g of (326Mmol) was stirred for 30 minutes, and the mixture was separated. The organic layer HCl 218ML 1N (5.0V / W), NaHCO 3 Aq. 218mL (5.0v / w), NaClaq . Was washed successively with 218ML (5.0V / W), Na 2 SO 4 dried addition of 8.74g (0.2w / w). Subjected to vacuum filtration, was concentrated under reduced pressure resulting filtrate by an evaporator, and pump up in the vacuum pump, the Cbz-D-Lys (Boc) -α-Boc-Pic-OMe (3) 88.9g as a white solid obtained (96.5% yield, HPLC purity 96.5%).
[0033]
(2) D-Lys (Boc) Synthesis Of -Arufa-Boc-Pic-OMe (4)
In An Eggplant-Shaped Flask Of 2L, Cbz-D-Lys (Boc) -Arufa-Boc-Pic-OMe (3) 88.3g (142mmol) were charged, it was added and dissolved 441mL (5.0v / w) the EtOAc. The 5% Pd / C to the reaction solution 17.7g (0.2w / w) was added, After three nitrogen substitution reduced pressure Atmosphere, Was Performed Three Times A Hydrogen Substituent. The Reaction Solution Was 18 Hours With Vigorous Stirring At Room Temperature To Remove The Pd / C And After The Completion Of The Reaction Vacuum Filtration. NaHCO The Resulting Filtrate 3 Aq. 441ML And (5.0V / W) Were Added For Liquid Separation, And The Organic Layer Was Extracted By The Addition Of EtOAc 200ML (2.3V / W) In The Aqueous Layer. NaHCO The Combined Organic Layer 3 Aq. 441ML And (5.0V / W) Were Added for liquid separation, and the organic layer was extracted addition of EtOAc 200mL (2.3v / w) in the aqueous layer. NaClaq the combined organic layers. 441mL and (5.0v / w) is added to liquid separation, was extracted by the addition EtOAc 200ML Of (2.3V / W) In The Aqueous Layer. The Combined Organic Layer On The Na 2 SO 4 Dried Addition Of 17.7 g of (0.2W / W), Then The Filtrate Was Concentrated Under Reduced Pressure Obtained Subjected To Vacuum Filtration By an evaporator, and pump up in the vacuum pump, D-Lys (Boc) -α-Boc-Pic- OMe (4) to give 62.7g (90.5% yield, HPLC purity 93.6%).
(3) Cbz-D-Leu -D-Lys (Boc) -α-Boc-Pic-OMe synthesis of (5)
in the four-necked flask of 2L, D-Lys (Boc) -α-Boc-Pic-OMe (4) was charged 57.7 g (120 mmol), was suspended in EtOAc 576mL (10v / w). HOBt 19.3g (126mmol) to this suspension, was added EDC · HCl 24.2g (126mmol) while cooling in an ice bath added Cbz-D-Leu-OH 33.4g (126mmol). After 20 minutes, after stirring the temperature was raised 5 hours at room temperature, further the EDC · HCl and stirred 1.15 g (6.00 mmol) was added 16 h. After completion of the reaction, it was added liquid separation 1N HCl 576mL (10v / w) . NaHCO to the resulting organic layer 3 Aq. 576ML (10V / W), Et 3 N 24.3 g of (240Mmol) was stirred for 30 minutes, and the mixture was separated. The organic layer HCl 576ML 1N (10V / W), NaHCO 3 Aq. 576mL (10v / w), NaClaq . Was washed successively with 576ML (10V / W), Na 2 SO 4 dried addition of 11.5g (0.2w / w). After the filtrate was concentrated under reduced pressure obtained subjected to vacuum filtration by an evaporator, and pump up in the vacuum pump, the Cbz-D-Leu-D- Lys (Boc) -α-Boc-Pic-OMe (5) 85.8g It was obtained as a white solid (98.7% yield, HPLC purity 96.9%).
(4) D-Leu-D -Lys (Boc) -α-Boc-Pic-OMe synthesis of (6)
in an eggplant-shaped flask of 1L, Cbz-D-Leu- D-Lys (Boc) -α-Boc-Pic -OMe the (5) 91.9g (125mmol) were charged, was added and dissolved 459mL (5.0v / w) the EtOAc. The 5% Pd / C to the reaction solution 18.4g (0.2w / w) was added, After three nitrogen substitution reduced pressure atmosphere, was performed three times a hydrogen substituent. The reaction solution was subjected to 8 hours with vigorous stirring at room temperature to remove the Pd / C and after the completion of the reaction vacuum filtration. NaHCO the resulting filtrate 3 Aq. 200mL (2.2v / w) were added to separate liquid, NaHCO to the organic layer 3 Aq. 200mL (2.2v / w), NaClaq . It was sequentially added washed 200mL (2.2v / w). To the resulting organic layer Na 2 SO 4 dried added 18.4g (0.2w / w), to the filtrate concentrated under reduced pressure obtained subjected to vacuum filtration by an evaporator, and a pump-up with a vacuum pump. The resulting amorphous solid was dissolved adding EtOAc 200mL (2.2v / w), was crystallized by the addition of heptane 50mL (1.8v / w). Was filtered off precipitated crystals by vacuum filtration, the crystals were washed with a mixed solvent of EtOAc 120mL (1.3v / w), heptane 50mL (0.3v / w). The resulting crystal 46.1g to added to and dissolved EtOAc 480mL (5.2v / w), was crystallized added to the cyclohexane 660mL (7.2v / w). Was filtered off under reduced pressure filtered to precipitate crystals, cyclohexane 120mL (1.3v / w), and washed with a mixed solvent of EtOAc 20mL (0.2v / w), and 30 ° C. vacuum dried, D-Leu- as a white solid D-Lys (Boc) -α- Boc-Pic-OMe (6) to give 36.6 g (48.7% yield, HPLC purity 99.9%).
(5) Synthesis of Cbz-D-Phe-D- Leu-D-Lys (Boc) -α-Boc-Pic-OMe (7)
to the four-necked flask of 1L, D-Leu-D- Lys (Boc) -α-Boc-Pic-OMe with (6) 35.8g (59.6mmol) was charged, it was suspended in EtOAc 358mL (10v / w). To this suspension HOBt 9.59g (62.6mmol), Cbz- D-Phe-OH 18.7g was cooled in an ice bath is added (62.6mmol) while EDC · HCl 12.0g (62.6mmol) It was added. After 20 minutes, a further EDC · HCl After stirring the temperature was raised 16 hours was added 3.09 g (16.1 mmol) to room temperature. After completion of the reaction, it was added and the organic layer was 1N HCl 358mL of (10v / w). NaHCO to the resulting organic layer 3 Aq. 358ML (10V / W), Et 3 N 12.1 g of (119Mmol) was stirred for 30 minutes, and the mixture was separated. The organic layer HCl 358ML 1N (10V / W), NaHCO 3 Aq. 358mL (10v / w), NaClaq . Was washed successively with 358ML (10V / W), Na 2 SO 4 dried addition of 7.16g (0.2w / w). After the filtrate was concentrated under reduced pressure obtained subjected to vacuum filtration by an evaporator, and pump up in the vacuum pump, Cbz-D-Phe-D -Leu-D-Lys (Boc) -α-Boc-Pic-OMe (7) was obtained 52.5g as a white solid (yield quant, HPLC purity 97.6%).
(6) D-Phe-D -Leu-D-Lys (Boc) synthesis of -α-Boc-Pic-OMe ( 8)
in an eggplant-shaped flask of 2L, Cbz-D-Phe- D-Leu-D-Lys ( Boc) -α-Boc-Pic- OMe (7) the 46.9g (53.3mmol) were charged, the 840ML EtOAc (18V / W), H 2 added to and dissolved O 93.8mL (2.0v / w) It was. The 5% Pd / C to the reaction mixture 9.38g (0.2w / w) was added, After three nitrogen substitution reduced pressure atmosphere, was performed three times a hydrogen substituent. The reaction solution was subjected to 10 hours with vigorous stirring at room temperature to remove the Pd / C and after the completion of the reaction vacuum filtration. NaHCO the resulting filtrate 3 Aq. 235mL (5.0v / w) were added to separate liquid, NaHCO to the organic layer 3 Aq. 235mL (5.0v / w), NaClaq . It was added sequentially cleaning 235mL (5.0v / w). To the resulting organic layer Na 2 SO 4 dried addition of 9.38g (0.2w / w), then the filtrate was concentrated under reduced pressure obtained subjected to vacuum filtration by an evaporator, pump up with a vacuum pump to D-Phe -D-Leu-D-Lys ( Boc) -α-Boc-Pic-OMe (7) was obtained 39.7g (yield quant, HPLC purity 97.3%).
351mL was suspended in (10v / w). To this suspension HOBt 7.92g (51.7mmol), Boc-D-Phe-OH HCl HCl
(8) D-Phe-D -Phe-D-Leu-D-Lys-Pic-OMe Synthesis Of Hydrochloric Acid Salt (1)
In An Eggplant-Shaped Flask Of 20ML Boc-D-Phe-D -Phe-D- Leu-D- lys (Boc) -α -Boc- Pic-OMe (9) and 2.00gg, IPA 3.3mL (1.65v / w), was suspended by addition of PhMe 10mL (5v / w). It was stirred at room temperature for 19 hours by addition of 6N HCl / IPA 6.7mL (3.35v / w). The precipitated solid was filtered off by vacuum filtration and dried under reduced pressure to a white solid of D-Phe-D-Phe- D- Leu-D-Lys-Pic- OMe 1.59ghydrochloride (1) (yield: 99 .0%, HPLC purity 98.2%) was obtained.
(9) D-Phe-D -Phe-D-Leu-D-Lys-Pic-OMe Purification Of The Hydrochloric Acid Salt (1)
In An Eggplant-Shaped Flask Of 20ML-D-Phe-D- Phe D-Leu -D-Lys- pic-OMe hydrochloride crude crystals (1) were charged 200mg, EtOH: MeCN = 1: after stirring for 1 hour then heated in a mixed solvent 4.0 mL (20v / w) was added 40 ° C. of 5 , further at room temperature for 2 was time stirring slurry. Was filtered off by vacuum filtration, the resulting solid was dried under reduced pressure a white solid ((1) Purification crystals) was obtained 161 mg (80% yield, HPLC purity 99.2% ).
(10) D-Phe-D -Phe-D-Leu-D-Lys-Pic Synthesis (Using Purified
(1)) Of (A) To A Round-Bottomed Flask Of 10ML D-Phe-D-Phe-D- -D-Lys Leu-Pic-OMe Hydrochloride Salt (1) Was Charged With Purified Crystal 38.5Mg (0.0488Mmol), H 2 Was Added And Dissolved O 0.2ML (5.2V / W). 1.5H Was Stirred Dropwise 1N NaOH 197MyuL (0.197mmol) at room temperature. After completion of the reaction, concentrated under reduced pressure by an evaporator added 1N HCl 48.8μL (0.0488mmol), to obtain a D-Phe-D-Phe- D-Leu-D-Lys- Pic (A) (yield: quant , HPLC purity 99.7%).
Example 2
(Trifluoroacetic Acid (TFA)
Use) (1) D-Phe-D-Phe-D-Leu-D-Lys-Pic-OMe TFA Synthesis Of Salt (1)
TFA 18ML Eggplant Flask Of 50ML (18V / W) , 1- Dodecanethiol 1.6ML (1.6V / W), Triisopropylsilane 0.2ML (0.2V / W), H 2 Sequentially Added Stirring The O 0.2ML (0.2V / W) Did. The Solution To The Boc-D-Phe- D- Phe-D-Leu-D -Lys (Boc) -α-Boc-Pic-OMe the (9) 1.00g (1.01mmol) was added in small portions with a spatula. After completion of the reaction, concentrated under reduced pressure by an evaporator, it was added dropwise the resulting residue in IPE 20mL (20v / w). The precipitated solid was filtered off, the resulting solid was obtained and dried under reduced pressure to D-Phe-D-Phe- D-Leu -D-Lys-Pic-OMe · TFA salt as a white solid (1) (Osamu rate 93.0%, HPLC purity 95.2%).
(2) D-Phe-D -Phe-D-Leu-D-Lys-Pic synthesis of (A)
to a round-bottomed flask of 10mL D-Phe-D-Phe -D-Leu-D-Lys-Pic-OMe TFA were charged salt (1) 83mg (0.0843mmol), was added and dissolved H2O 431μL (5.2v / w). Was 12h stirring dropwise 1N NaOH 345μL (0.345mmol) at room temperature. After completion of the reaction, concentrated under reduced pressure by an evaporator added 1N HCl 84.3μL (0.0843mmol), to obtain a D-Phe-D-Phe- D-Leu-D-Lys-Pic (A) ( yield: quant, HPLC purity 95.4%).
Example
3 (HCl / EtOAc
Use) (1) In An Eggplant-Shaped Flask Of 30ML Boc-D-Phe-D -Phe-D-Leu-D-Lys (Boc) -Arufa-Boc-Pic-OMe (9) 1. It was charged with 00g (1.01mmol ), was added and dissolved EtOAc7.0mL (7.0v / w). 4N HCl / EtOAc 5.0mL (5.0v / w) was added after 24h stirring at room temperature, the precipitated solid was filtered off by vacuum filtration, washed with EtOAc 2mL (2.0v / w). The resulting solid D-Phe-D-Phe- D-Leu-D-Lys-Pic-OMe hydrochloride (1) was obtained 781mg of a white solid was dried under reduced pressure (the 96.7% yield, HPLC purity 95.4%).
(2) D-Phe-D -Phe-D-Leu-D-Lys-Pic (A) Synthesis of
eggplant flask of 10mL D-Phe-D-Phe -D-Leu-D-Lys-Pic-OMe hydrochloride were charged salt (1) 90 mg (0.112 mmol), H 2 was added and dissolved O 0.47mL (5.2v / w). Was 12h stirring dropwise 1N NaOH 459μL (0.459mmol) at room temperature. After completion of the reaction, concentrated under reduced pressure by an evaporator added 1N HCl 0.112μL (0.112mmol), was obtained D-Phe-D-Phe- D-Leu-D-Lys-Pic (A) ( yield: quant, HPLC purity 93.1%).
4 Example
Compound (1) Of The Compound By Hydrolysis Synthesis Of (The A) (Compound (1) Without
Purification) Eggplant Flask 10ML D-Phe-D-Phe -D-Leu-D-Lys-Pic-OMe (1) Charged Hydrochloride Were (Without Pre-Step Purification) 114.5Mg (0.142Mmol), H 2 Was Added And Dissolved O 595MyuL (5.2V / W). Was 14H Stirring Dropwise 1N NaOH 586MyuL (0.586Mmol) At Room Temperature. After Completion Of the reaction, concentrated under reduced pressure by an evaporator added 1N HCl 0.15μL (0.150mmol), was obtained D-Phe-D-Phe- D-Leu-D-Lys-Pic (A) (yield: quant, HPLC purity 95.2 %).
Example 1 Comparative
Path Not Via The Compound (1) (Using Whole Guard Boc-D-Phe-D-Phe-D-Leu-D-Lys (Boc) -Alpha-Boc-Pic-OMe
(A)) (1) D–Boc Phe- D-Phe-D-Leu-D-Lys (Boc) -Arufa-Boc-Pic-OH Synthesis Of
Eggplant Flask Of 30ML Boc-D-Phe-D -Phe-D-Leu-D- Lys (Boc) -α- Boc-Pic -OMe (9) were charged 1.00g (1.00mmol), was added and dissolved MeOH 5.0mL (5.0v / w). After stirring for four days by the addition of 1N NaOH 1.1 mL (1.10mmol) at room temperature, further MeOH 5.0mL (5.0v / w), 1N NaOH 2.0mL the (2.0mmol) at 35 ℃ in addition 3h and the mixture was stirred. After completion of the reaction, 1 N HCl 6.1 mL was added, After distilling off the solvent was concentrated under reduced pressure was separated and the organic layer was added EtOAc 5.0mL (5.0mL) .NaClaq. 5.0mL (5.0v / w) Wash the organic layer was added, the organic layer as a white solid was concentrated under reduced pressure to Boc-D-Phe-D- Phe-D-Leu-D-Lys (Boc) – α-Boc-Pic-OH 975.1mg (99.3% yield, HPLC purity 80.8% )
(2) D-Phe-D -Phe-D-Leu-D-Lys-Pic synthesis of (A)
to a round-bottomed flask of 20mL Boc-D-Phe-D -Phe-D-Leu-D-Lys (Boc) It was charged -α-Boc-Pic-OH ( 10) 959mg (0.978mmol), was added and dissolved EtOAc 4.9mL (5.0v / w). And 4h stirring at room temperature was added dropwise 4N HCl / EtOAc 4.9mL (5.0mL) at room temperature. After completion of the reaction, it was filtered under reduced pressure, a white solid as to give D-Phe-D-Phe- D-Leu-D-Lys-Pic the (A) (96.4% yield, HPLC purity 79.2%) .
If not via the compound of the present invention (1), the purity of the compound obtained (A) was less than 80%.
Programmed Cell Death 1 or PD-1 (also referred to as PDCD1) is a 50 to 55 kD type I membrane glycoprotein (Shinohara T et al, Genomics, 1994, Vol. 23, No. 3, pp. 704-706). PD-1 is a receptor of the CD28 superfamily that negatively regulates T cell antigen receptor signalling by interacting with the specific ligands and is suggested to play a role in the maintenance of self tolerance.
PD-1 peptide relates to almost every aspect of immune responses including autoimmunity, tumour immunity, infectious immunity, transplantation immunity, allergy and immunological privilege.
The PD-1 protein’s structure comprise of—
an extracellular IgV domain followed by
a transmembrane region and
an intracellular tail
The intracellular tail contains two phosphorylation sites located in an immunoreceptor tyrosine-based inhibitory motif and an immunoreceptor tyrosine-based switch motif, which suggests that PD-1 negatively regulates TCR signals. Also, PD-1 is expressed on the surface of activated T cells, B cells, and macrophages, (Y. Agata et al., Int Immunol 8, 765, May 1996) suggesting that compared to CTLA-4 ((Cytotoxic T-Lymphocyte Antigen 4, also known as CD152 (Cluster of differentiation 152) is a protein that also plays an important regulatory role in the immune system), PD-1 more broadly negatively regulates immune responses.
PD-1 has two ligands, PD-L1 (Programmed Death Ligand for PDCD1L1 or B7-H1) (Freeman G J et al, Journal of Experimental Medicine, 2000, Vol. 19, No. 7, pp. 1027-1034) and PD-L2 (Programmed Death Ligand 2 or PDCD1L2 or B7-DC) (Latchman Y et al, Nature Immunology, 2001, Vol. 2, No. 3, pp. 261-267), which are members of the B7 family. PD-L1 is known to be expressed not only in immune cells, but also in certain kinds of tumour cell lines (such as monocytic leukaemia-derived cell lines, mast cell tumour-derived cell lines, hematoma-derived cell lines, neuroblastoma-derived cell lines, and various mammary tumour-derived cell lines) and in cancer cells derived from diverse human cancer tissues (Latchman Y et al, Nature Immunology, 2001, Vol. 2, No. 3, pp. 261-267) and on almost all murine tumour cell lines, including PA1 myeloma, P815 mastocytoma, and B16 melanoma upon treatment with IFN-γ (Y. Iwai et al., Proc Natl Acad Sci USA 99, 12293, Sep. 17, 2002 and C. Blank et al., Cancer Res 64, 1140, February, 2004). Similarly PD-L2 expression is more restricted and is expressed mainly by dendritic cells and a few tumour cell lines. PD-L2 expression has been verified in Hodgkin’s lymphoma cell lines and others. There is a hypothesis that some of the cancer or tumour cells take advantage from interaction between PD-1 and PD-L1 or PD-L2, for suppressing or intercepting T-cell immune responses to their own (Iwai Y et al, Proceedings of the National Academy of Science of the United States of America, 2002, Vol. 99, No. 19, pp. 12293-12297).
Tumour cells and virus (including HCV and HIV) infected cells are known to express the ligand for PD-1 (to create Immunosuppression) in order to escape immune surveillance by host T cells. It has been reported that the PD-1 gene is one of genes responsible for autoimmune diseases like systemic lupus erythematosis (Prokunina et al, Nature Genetics, 2002, Vol. 32, No. 4, 666-669). It has also been indicated that PD-1 serves as a regulatory factor for the onset of autoimmune diseases, particularly for peripheral self-tolerance, on the ground that PD-1-deficient mice develop lupus autoimmune diseases, such as glomerulonephritis and arthritis (Nishimura H et al, International Immunology, 1998, Vol. 10, No. 10, pp. 1563-1572; Nishimura H et al, Immunity, 1999, Vol. 11, No. 2, pp. 141-151), and dilated cardiomyopathy-like disease (Nishimura H et al, Science, 2001, Vol. 291, No. 5502, pp. 319-332).
Hence, in one approach, blocking the interaction of PD-1 with its ligand (PD-L1, PD-L2 or both) may provide an effective way for specific tumour and viral immunotherapy.
Wood et al in U.S. Pat. No. 6,808,710 discloses method for down modulating an immune response comprising contacting an immune cell expressing PD-1 with an antibody that binds to PD-1, in multivalent form, such that a negative signal is transduced via PD-1 to thereby down modulate the immune response. Such an antibody may be a cross-linked antibody to PD-1 or an immobilized antibody to PD-1.
Freeman et al in U.S. Pat. No. 6,936,704 and its divisional patent U.S. Pat. No. 7,038,013 discloses isolated nucleic acids molecules, designated B7-4 nucleic acid molecules, which encode novel B7-4 polypeptides, isolated B7-4 proteins, fusion proteins, antigenic peptides and anti-B7-4 antibodies, which co-stimulates T cell proliferation in vitro when the polypeptide is present on a first surface and an antigen or a polyclonal activator that transmits an activating signal via the T-cell receptor is present on a second, different surface.
There are some reports regarding substances inhibiting immunosuppressive activity of PD-1, or interaction between PD-1 and PD-L1 or PD-L2, as well as the uses thereof. A PD-1 inhibitory antibody or the concept of a PD-1 inhibitory peptide is reported in WO 01/14557, WO 2004/004771, and WO 2004/056875. On the other hand, a PD-L1 inhibitory antibody or a PD-L1 inhibitory peptide is reported in WO 02/079499, WO 03/042402, WO 2002/086083, and WO 2001/039722. A PD-L2 inhibitory antibody or a PD-L2 inhibitory peptide is reported in WO 03/042402 and WO 02/00730.
WO2007005874 describes isolated human monoclonal antibodies that specifically bind to PD-L1 with high affinity. The disclosure provides methods for treating various diseases including cancer using anti-PD-L1 antibodies.
US2009/0305950 describes multimers, particularly tetramers of an extracellular domain of PD-1 or PD-L1. The application describes therapeutic peptides.
Further, the specification mentions that peptides can be used therapeutically to treat disease, e.g., by altering co-stimulation in a patient. An isolated B7-4 or PD-1 protein, or a portion or fragment thereof (or a nucleic acid molecule encoding such a polypeptide), can be used as an immunogen to generate antibodies that bind B7-4 or PD-1 using standard techniques for polyclonal and monoclonal antibody preparation. A full-length B7-4 or PD-1 protein can be used, or alternatively, the invention provides antigenic peptide fragments of B7-4 or PD-1 for use as immunogens. The antigenic peptide of B7-4 or PD-1 comprises at least 8 amino acid residues and encompasses an epitope of B7-4 or PD-1 such that an antibody raised against the peptide forms a specific immune complex with B7-4 or PD-1. Preferably, the antigenic peptide comprises at least 10 amino acid residues, more preferably at least 15 amino acid residues, even more preferably at least amino acid residues, and most preferably at least 30 amino acid residues.
Freeman et al in U.S. Pat. No. 7,432,059 appears to disclose and claim methods of identifying compounds that up modulate T cell activation in the presence of a PD-1-mediated signal. Diagnostic and treatment methods utilizing compositions of the invention are also provided in the patent.
Further, Freeman et al in U.S. Pat. No. 7,709,214 appears to cover methods for up regulating an immune response with agents that inhibit the interactions between PD-L2 and PD-1.
Despite existence of many disclosures as discussed above, however, a significant unmet medical need still exists due to the lack of effective peptides or modified peptides as therapeutic agents as alternatives in the therapeutic area. It is known that synthetic peptides offer certain advantages over antibodies such as ease of production with newer technologies, better purity and lack of contamination by cellular materials, low immunogenicity, improved potency and specificity. Peptides may be more stable and offer better storage properties than antibodies. Moreover, often peptides possess better tissue penetration in comparison with antibodies, which could result in better efficacy. Peptides can also offer definite advantages over small molecule therapeutics counterparts such as lesser degree of toxicity and lower probability of drug-drug interaction.
The present invention therefore may provide the solution for this unmet medical need by offering novel synthetic peptide and its derivatives which are based on the PD1 ectodomain.
Aurigene team: (from left) Brahma Reddy V, Thomas Antony, Murali Ramachandra, Venkateshwar Rao G, Wesley Roy Balasubramanian, Kishore Narayanan, Samiulla DS, Aravind AB, and Shekar Chelur
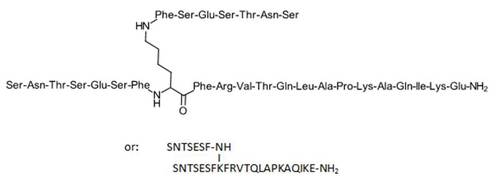
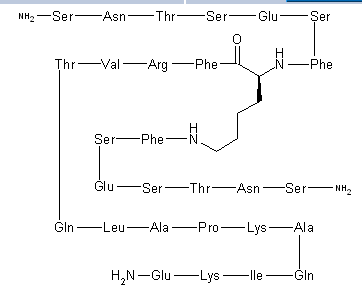
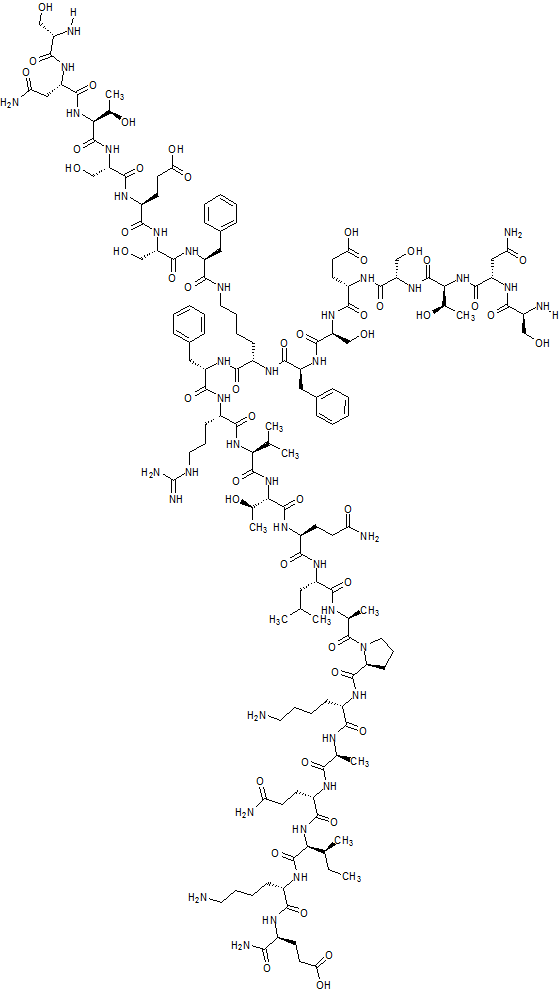
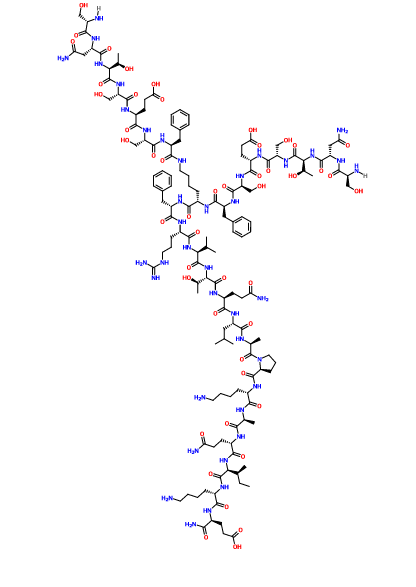
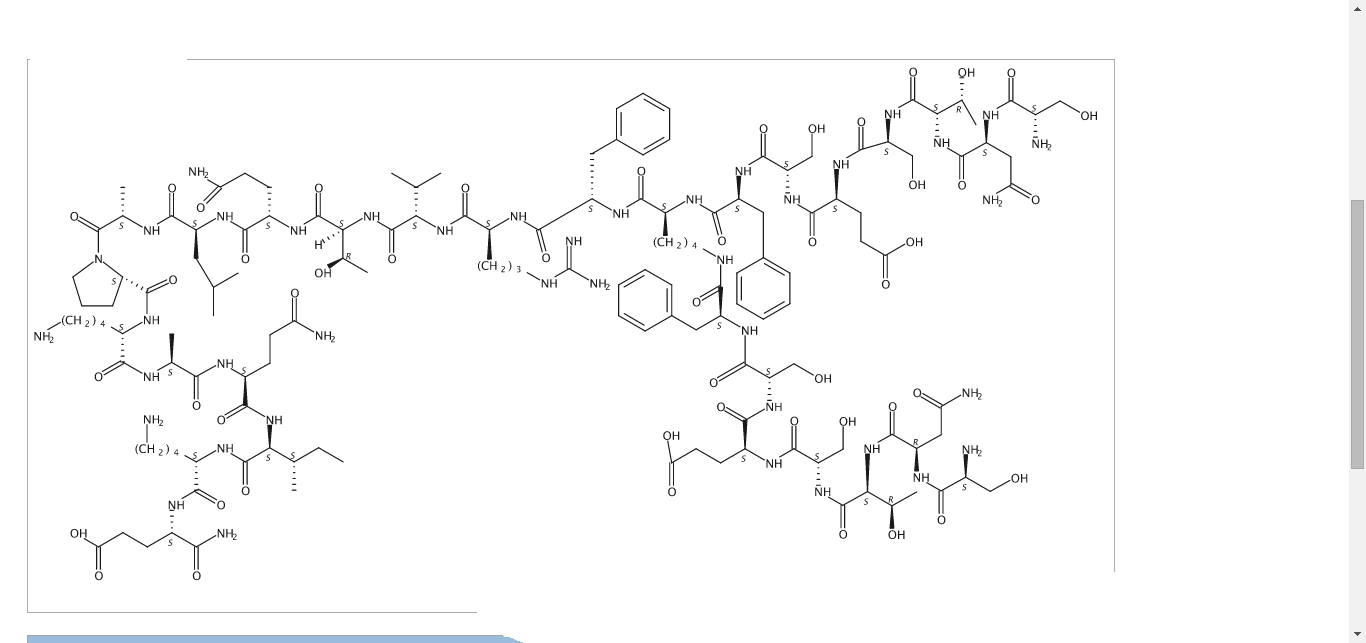
SNTSESFK(SNTSESF)FRVTQLAPKAQIKE-NH2 (SEQ ID NO: 49)
Example 2 Synthesis of
Synthesis of Linear Fragment—Fmoc-FRVTQLAPKAQIKE
Desiccated CLEAR-Amide resin ((100-200 mesh) 0.4 mmol/g, 0.5 g) was distributed in 2 polyethylene vessels equipped with a polypropylene filter. The linear peptide synthesis on solid phase were carried out automatically, using Symphony parallel synthesizer (PTI) using the synthesis programs mentioned in the table below. Swelling, C-terminal amino acid [Fmoc-Glu(OtBu)-OH] attachment and capping of the peptidyl resin was carried out as per the protocol in Table I. Subsequent amino acid coupling was carried out as mentioned in Table II. The amino acids used in the synthesis were Fmoc Phe-OH, Fmoc-Arg(Pbf)-OH, Fmoc-Val-OH, Fmoc-Thr(OtBu)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Lys(Boc)-OH, Fmoc-Leu-OH, Fmoc-Ala-OH, Fmoc-Pro-OH, Fmoc-Ile-OH. After the completion of Fmoc-Phe-OH coupling the resin was taken out form peptide synthesiser and manual coupling was carried out as follows
Fmoc-Phe-OH peptidyl resin from automated synthesiser was pooled in to a glass vessel with frit. The Fmoc group of the peptidyl resin was deprotected by treating it twice with 20% (v/v) piperidine/DMF solution for 5 and 15 min (10 m L). The resin was washed with DMF (6×15 m L), DCM (6×15 m L) and DMF (6×15 m L). Kaiser test on peptide resin aliquot upon completion of Fmoc-deprotection was positive. Fmoc-Lys (Fmoc)-OH (0.48 g; 4 equiv. 0.8 m mol) in dry DMF was added to the deprotected resin and coupling was initiated with DIC (0.15 m L; 5 equiv, 1 m mol) and HOBT (0.08 g; 5 equiv, 0.6 m mol) in DMF. The concentration of each reactant in the reaction mixture was approximately 0.4 M. The mixture was rotated on a rotor at room temperature for 3 h. Resin was filtered and washed with DMF (6×15 mL), DCM (6×15 mL) and DMF (6×15 mL). Kaiser test on peptide resin aliquot upon completion of coupling was negative. The Fmoc group on the peptidyl resin is deprotected by treating it twice with 20% (v/v) piperidine/DMF solution for 5 and 15 min (15 mL). The resin was washed with DMF (6×15 mL), DCM (6×15 mL) and DMF (6×15 mL). Kaiser test on peptide resin aliquot upon completion of Fmoc-deprotection was positive. After the deprotection of Fmoc group on Fmoc-Lys(Fmoc)-attached peptidyl resin the peptide chain growth was carried out from both the free amino terminus suing 8 equivalent excess of amino acid (1.6 m mol, 8 equivalent excess of HOBt (0.22 g, 1.6 m mol) and 10 equivalent excess of DIC (0.32 m L, 2 m mol) relative to resin loading. The coupling was carried out at room temperature for 3 h. The amino acids coupled to the peptidyl resin were; Fmoc-Phe-OH (0.62 g; 8 equiv, 1.6 m mol), Fmoc-Ser (OtBu)-OH (0.62 g; 8 equiv, 1.6 m mol), Fmoc-Glu (OtBu)-OH (0.68 g; 8 equiv, 1.6 m mol), Fmoc-Ser (OtBu)-OH (0.62 g; 8 equiv, 1.6 m mol), Fmoc-Thr (OtBu)-OH (0.64 g; 8 equiv, 1.6 m mol), Fmoc-Asn (Trt)-OH (0.95 g; 8 equiv, 1.6 m mol) and N-terminus amino acids as Boc-Ser (OtBu)-OH (0.41 g; 8 equiv, 1.6 m mol) The peptidyl resin was cleaved as mentioned in procedure for cleavage using cleavage cocktail A to yield (565 mg), 70% yield. The crude material was purified by preparative HPLC on Zorbax Eclipse XDB-C18 column (9.4 mm×250 mm, 5 μm) with buffer A: 0.1% TFA/Water, buffer B: Acetonitrile. The peptide was eluted by gradient elution 0-5 min=5-10% buffer B, 10-20 min=29% buffer B with a flow rate of 7 mL/min. HPLC: (method 1): RT-12 min (96%); LCMS Calculated Mass: 3261.62, Observed Mass: 1631.6 [M/2+H]+; 1088 [M/3+H]+); 816.2[M/4+H]+;
Compound 8 (SEQ ID NO: 49) SNTSESFK(SNTSESF)FRVTQLAPKAQIKE-NH2
Example 2Synthesis of Sequence Shown in SEQ ID NO: 49
Synthesis of Linear Fragment—Fmoc-FRVTQLAPKAQIKE
Desiccated CLEAR-Amide resin ((100-200 mesh) 0.4 mmol/g, 0.5 g) was distributed in 2 polyethylene vessels equipped with a polypropylene filter. The linear peptide synthesis on solid phase were carried out automatically, using Symphony parallel synthesizer (PTI) using the synthesis programs mentioned in the table below. Swelling, C-terminal amino acid [Fmoc-Glu(OtBu)-OH] attachment and capping of the peptidyl resin was carried out as per the protocol in Table I. Subsequent amino acid coupling was carried out as mentioned in Table II. The amino acids used in the synthesis were Fmoc Phe-OH, Fmoc-Arg(Pbf)-OH, Fmoc-Val-OH, Fmoc-Thr(OtBu)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Lys(Boc)-OH, Fmoc-Leu-OH, Fmoc-Ala-OH, Fmoc-Pro-OH, Fmoc-Ile-OH. After the completion of Fmoc-Phe-OH coupling the resin was taken out form peptide synthesiser and manual coupling was carried out as follows.
Fmoc-Phe-OH peptidyl resin from automated synthesiser was pooled in to a glass vessel with frit. The Fmoc group of the peptidyl resin was deprotected by treating it twice with 20% (v/v) piperidine/DMF solution for 5 and 15 min (10 mL). The resin was washed with DMF (6×15 mL), DCM (6×15 mL) and DMF (6×15 mL). Kaiser test on peptide resin aliquot upon completion of Fmoc-deprotection was positive.
Fmoc-Lys (Fmoc)-OH (0.48 g; 4 equiv. 0.8 mmol) in dry DMF was added to the deprotected resin and coupling was initiated with DIC (0.15 mL; 5 equiv, 1 mmol) and HOBT (0.08 g; 5 equiv, 0.6 mmol) in DMF. The concentration of each reactant in the reaction mixture was approximately 0.4 M. The mixture was rotated on a rotor at room temperature for 3 h. Resin was filtered and washed with DMF (6×15 mL), DCM (6×15 mL) and DMF (6×15 mL). Kaiser test on peptide resin aliquot upon completion of coupling was negative. The Fmoc group on the peptidyl resin is deprotected by treating it twice with 20% (v/v) piperidine/DMF solution for 5 and 15 min (15 mL). The resin was washed with DMF (6×15 mL), DCM (6×15 mL) and DMF (6×15 mL). Kaiser test on peptide resin aliquot upon completion of Fmoc-deprotection was positive.
After the deprotection of Fmoc group on Fmoc-Lys(Fmoc)-attached peptidyl resin the peptide chain growth was carried out from both the free amino terminus suing 8 equivalent excess of amino acid (1.6 mmol, 8 equivalent excess of HOBt (0.22 g, 1.6 mmol) and 10 equivalent excess of DIC (0.32 mL, 2 mmol) relative to resin loading. The coupling was carried out at room temperature for 3 h. The amino acids coupled to the peptidyl resin were; Fmoc-Phe-OH (0.62 g; 8 equiv, 1.6 mmol), Fmoc-Ser (OtBu)-OH (0.62 g; 8 equiv, 1.6 mmol), Fmoc-Glu (OtBu)-OH (0.68 g; 8 equiv, 1.6 mmol), Fmoc-Ser (OtBu)-OH (0.62 g; 8 equiv, 1.6 mmol), Fmoc-Thr (OtBu)-OH (0.64 g; 8 equiv, 1.6 mmol), Fmoc-Asn (Trt)-OH (0.95 g; 8 equiv, 1.6 m mol) and N-terminus amino acids as Boc-Ser (OtBu)-OH (0.41 g; 8 equiv, 1.6 mmol) The peptidyl resin was cleaved as mentioned in procedure for cleavage using cleavage cocktail A to yield (565 mg), 70% yield. The crude material was purified by preparative HPLC on Zorbax Eclipse XDB-C18 column (9.4 mm×250 mm, 5 μm) with buffer A: 0.1% TFA/Water, buffer B:Acetonitrile. The peptide was eluted by gradient elution 0-5 min=5-10% buffer B, 10-20 min=29% buffer B with a flow rate of 7 mL/min. HPLC: (method 1): RT—12 min (96%); LCMS Calculated Mass: 3261.62, Observed Mass: 1631.6 [M/2+H]+; 1088 [M/3+H]+;); 816.2[M/4+H]+.
Aurigene and Pierre Fabre Pharmaceuticals Announce a Licensing Agreement for a New Cancer Therapeutic in Immuno-oncology: AUNP12, an Immune Checkpoint Modulator Targeting the PD-1 Pathway
Pierre Fabre are thus reinforcing their oncology portfolio which already enjoys a combination of chemotherapies, monoclonal antibodies and immuno-conjugates assets at various development phases
CASTRES, France and BANGALORE, India, February 13, 2014 /PRNewswire/ —
Pierre Fabre, the third largest French pharmaceutical company, and Aurigene, a leading biotech company based in India, today announced that the two companies have entered into a collaborative license, development and commercialization agreement granting Pierre Fabre global Worldwide rights (excluding India) to a new immune checkpoint modulator, AUNP-12.
AUNP-12 offers a breakthrough mechanism of action in the PD-1 pathway compared to other molecules currently in development in the highly promising immune therapy cancer space. AUNP-12 is the only peptide therapeutic in this pathway and could offer more effective and safer combination opportunities with emerging and established treatment regimens. AUNP-12 will be in development for numerous cancer indications.
Under the terms of this agreement, Aurigene will receive an upfront payment from Pierre Fabre. Aurigene will also receive additional milestone payments based upon the continued development, regulatory progresses and commercialization of AUNP-12.
“We are pleased that Pierre Fabre see the PD-1 program as a strategic asset in their portfolio. Overall, the deal structure, in line with the financial terms that have been seen in this space, demonstrate the importance that Pierre Fabre attach to the program,” said CSN Murthy, CEO, Aurigene.
“The plans that Pierre Fabre have detailed for the development of this differentiated asset highlight the long-term opportunities for this novel cancer therapeutic,” added Murali Ramachandra, Sr VP, Research, Aurigene.
“This agreement, in the field of oncology, is fully consistent with our vision to build Pierre Fabre’s future in prescription drugs, from a combination of cutting-edge internal R&D capabilities and license partnerships with innovative biotech companies like Aurigene,” stated Bertrand Parmentier, CEO, Pierre Fabre.
“With this deal, Pierre-Fabre Pharmaceuticals are reinforcing their portfolio of oncology assets and capitalizing on their proven capabilities in developing biological compounds such as monoclonal antibodies and immuno-conjugates. We have been impressed by the science at Aurigene and encouraged by the differentiated profile reported for AUNP-12,” added Frédéric Duchesne, President, Pierre Fabre Pharmaceuticals.
About immuno-oncology
Immuno-oncology is an emerging field in cancer therapy, where the body’s own immune system is harnessed to fight against cancer. This approach of targeting cancer through immune response has had a breakthrough when robust and sustained responses were obtained only upon blocking the immune checkpoint targets (such as PD-1 and CTLA4). Recent successes in clinical trials performed with such therapies suggest that immunotherapy should be considered alongside surgery, chemotherapy, radiotherapyand targeted therapyas the fifth cornerstone of cancer treatment.
PD-1 (Programmed cell Death 1) is a receptor that negatively regulates T-cell activation by interacting with specific ligands PD-L1 and PD-L2. Tumor cells express these ligands and thereby escape from the action of T-cells.
About AUNP-12
AUNP-12is a branched 29-amino acid peptide sequence engineered from the PD-L1/ L2 binding domain of PD-1It blocks the PD-1/PD-L1, PD-1/PD-L2 and PD-L1/CD80 pathways.AUNP-12 is highly effective in antagonizing PD-1 signaling, with desirable in vivo exposure upon subcutaneous dosing. It inhibits tumor growth and metastasis in preclinical models of cancer and is well tolerated with no overt toxicity at any of the tested doses.
About Aurigene
Aurigene is a biotech focused on development of innovative small molecule and peptide therapeutics for Oncology and Inflammation; key focus areas for Aurigene are Immuno-oncology, Epigenetics and the Th17 pathway.Aurigene’s PD-1 program is the first of several peptide-based immune checkpoint programs that are at different stages of Discovery.
Aurigene has partnered with several big pharma and mid-pharma companies in the US and Europe, and has delivered multiple clinical compounds through these partnerships. With over 500 scientists, Aurigene has collaborated with 6 of the top 10 pharma companies.
Aurigene’s pre-clinical pipeline includes (1) Selective and pan-BET Bromodomain inhibitors (2) RoR gamma reverse agonists (3) EZH2 inhibitors (4) NAMPT inhibitors and (5) Several immune check point peptide inhibitor programs.
Pierre Fabre is a privately-owned health care company created in 1961 by Mr Pierre Fabre. It is the second largest French independent pharmaceutical group with 2013 sales amounting to about €2 billion (yet to be audited) across 140 countries. The company is structured around two divisions: Pharmaceuticals (Prescription drugs, OTC, Oral care) and Dermo-cosmetics. Prescription drugs are organized around four main franchises: oncology, dermatology, women’s health and neuropsychiatry. Pierre Fabre employs some 10000 people worldwide, including1 300 inR&D. The company allocates about 20% of its pharmaceuticals sales to R&D and relies on more than 25 years of experience in the discovery, development and global commercialization of innovative drugs in oncology. Pierre Fabre has a long commitment to oncology and immunology with major R&D centers in France: the Pierre Fabre immunology Centre (CIPF) in Saint Julien en Genevois and the Pierre Fabre Research Institute (IRPF) located on the Toulouse-Oncopole campus which has been officially recognized as a National Center of Excellence for cancer research since 2012.
P. Sasikumar, R. Shrimali, S. Adurthi, R. Ramachandra, L. Satyam, A. Dhudashiya, D. Samiulla, K. B. Sunilkumar and M. Ramachandra, “A novel peptide therapeutic targeting PD1 immune checkpoint with equipotent antagonism of both ligands and a potential for better management of immune-related adverse events,” Journal for ImmunoTherapy of Cancer, vol. 1, no. Suppl 1, O24, 2013.
P. G. N. Sasikumar, M. Ramachandra, S. K. Vadlamani, K. R. Vemula, L. K. Satyam, K. Subbarao, K. R. Shrimali and S. Kandepudu (Aurigene Discovery Technologies Ltd, Bangalore, India), “Immunosuppression modulating compounds”, US Patent application US 2011/0318373, 29 Dec 2011.
P. G. Sasikumar, L. K. Satyam, R. K. Shrimali, K. Subbarao, R. Ramachandra, S. Vadlamani, A. Reddy, A. Kumar, A. Srinivas, S. Reddy, S. Gopinath, D. S. Samiulla and M. Ramachandra, “Demonstration of anti-tumor efficacy in multiple preclinical cancer models using a novel peptide inhibitor (Aurigene-012) of the PD1 signaling pathway,” Cancer Research, vol. 72, no. 8 Suppl. 1, Abstract 2850, 2012.
P. G. N. Sasikumar, M. Ramachandra, S. K. Vadlamani, K. R. Shrimali and K. Subbarao, “Therapeutic compounds for immunomodulation” (Aurigene Discovery Technologies Ltd, Bangalore, India), PCT Patent Application WO 2012/168944, 13 Dec 2012.
P. G. N. Sasikumar and M. Ramachandra, “Immunomodulating cyclic compounds from the BC loop of human PD1” (Aurigene Discovery Technologies Ltd, Bangalore, India), PCT Patent Application WO/2013/144704, 3 Oct 2013.
P. G. N. Sasikumar, M. Ramachandra and S. S. S. Naremaddepalli, “Peptidomimetic compounds as immunomodulators” (Aurigene Discovery Technologies Ltd, Bangalore, India), US Patent Application US 2013/0237580, 12 Sep 2013.
A. H. Sharpe, M. J. Butte and S. Oyama (Harvard College), “Modulators of immunoinhibitory receptor PD-1, and methods of use thereof”, PCT Patent Application WO/2011/082400, 7 Jul 2011.
M. Cordingley, “Battle of PD-1 blockade is on”, February 7, 2014 : http://discoveryview.ca/battle-of-pd-1-blockade-is-on/ [Accessed 25 February 2014].
Mr. CSN Murthy began his career with ICICI Ventures, India’s first Venture Capital fund. He was subsequently a management consultant to the Pharma and Chemical sectors. Later, he worked in the Business Development and General Management functions in Pharmaceutical companies, including as the Chief Operating Officer of Gland Pharma Ltd. CSN holds a Bachelors degree in Chemical Engineering from the Indian Institute of Technology (IIT), Madras and an MBA from the Indian Institute of Management (IIM), Bangalore.
Dr.Thomas Antony
Associate Research Director, Aurigene Discovery Technologies Ltd.
Dr.Thomas Antony did his Ph.D in Biophysical Chemistry from University of Delhi and had his postdoctoral training at Jawaharlal Nehru University- Delhi, The University of Medicine and Dentistry of New Jersey- USA, and Max Planck Institute for Biophysical Chemistry- Germany. He is the recipient of many research fellowships, including Max Planck Fellowship and Humboldt Research Fellowship. He has more than 20 years of research experience. Dr.Thomas has published 24 research papers and he is the co-author of three international patents. His core area of expertise is in assay development and screening. At Aurigene, Dr.Thomas leads the Biochemistry and Structural Biology Divisions. He was the coordinator of Aurigene-University of Malaya collaboration programs.
Dr. Kavitha Nellore
Associate Research Director, Aurigene Discovery Technologies Ltd.
Dr. Kavitha Nellore obtained her PhD in Bioengineering from Pennsylvania State University, USA. During this time, she was a fellow of the Huck’s Institute of Life Sciences specializing in Biomolecular Transport Dynamics. She has been at Aurigene for more than a decade, and is currently leading a group of cell biologists at both Bangalore and Kuala Lumpur. At Aurigene, she leads multiple drug discovery programs in the therapeutic areas of inflammation, oncology and immuno-oncology. She plays a key role in target selection as well as validation efforts to add to Aurigene’s pipeline. Kavitha also played a key role in coordinating the Aurigene-University of Malaya collaboration.
To print: Click hereor Select File and then Print from your browser's menu
“Strategic partnerships will boost drug discovery activities in India”
Wednesday, May 16, 2007 08:00 IST
The Bangalore-based Aurigene Discovery Technologies Limited, an independent subsidiary of Dr Reddy’s, is now competing in a mature contract research organization space in the country. Established in 2003, Aurigene Discovery Technologies is a partnership focused collaborative discovery organisation. CSN Murthy is chief executive officer of the company. In an interaction with Nandita Vijay, Murthy cut a clear picture of the contract research scene. Excerpts:
What are the factors led to the scores of partnerships in research and drug discovery space?
India is a cost-effective destination but there is lack of expertise, as it has so far not witnessed a complete cycle of discovery, development and marketing of a compound. Therefore, the possible solution is to enter into strategic business partnerships to combine low costs and excellent global skills. Such alliances make a lot of business sense, because no Indian company can afford to invest in resources to research on clinical development of compounds or identification of new drug targets, which is a highly risky area. The Indian CROs have so far focused largely on chemistry-based projects. But of late, there has been some attempt by CROs to offer biology services. The chemistry services have taken off well and there could be at least 2,000 chemists working in Indian CROs. But the biology service is still in the incipient stage.
Biology demands expertise in areas such as DMPK, cell and assay biology and vivo expertise, which is not as common as chemistry expertise in India. There is also a paradigm shift in assignments for CROs, as they are gearing up to gain higher value from existing projects, including Intellectual Property (IP) generation. Probably, this is where Aurigene has a head start over other CROs. Although we are in research services, our success is in IP generated work. We work with world-class companies like Novo Nordisk, Rheoscience, Debiopharm, Forest Labs and Merck Serono on integrated discovery projects.
What do you think is the uniqueness of Aurigene that sets it apart from other companies?
We are the oldest and know the ins and outs of the Indian pharmaceutical market. Our strengths include modern infrastructure and resourceful, talented scientific pool. Right now, we are working with Forrest Labs to meet the first milestone. We are adding more customers and there is value-addition to projects from existing customers. We are also able to add more customers in the area of drug discovery. This has led to expansion in infrastructure and manpower.
It is understood that a major chunk of the business for CROs is from global customers. What is the reason for this?
That is true. Even Aurigene is not favoring Indian customers. The situation is similar to the information technology sector in the country where local giants like Wipro and Infosys augmented businesses through global partnerships. However, after two decades, they are now serving Indian customers. The same thing will happen in India in the CROs space. Right now, I doubt, if any Indian pharma or biotech company will look for collaboration or business development with Indian counterparts. It makes more business sense to work for global customers.
The whole concept of drug discovery outsourcing business has caught on in the last 18-month. Could you give us an overview of the market?
The reason for the sudden interest in drug discovery is the cost arbitrage. There is downward pressure on prices and upward pressure on costs. Globally, the market size for drug discovery is around $8 billon, excluding the licensing costs. I feel the overall market expansion is not going to happen in terms of increased research spends. The opportunities will come from shift in spend to India from Europe and US. The amount of outsourced value will actually increase substantially in the next few years. In the western countries, pharma giants are busy partnering with biotechnology companies to license early/late-stage compounds. In other words, the biotech companies offer novel technology platforms. In a situation such as this, while the former gets the license rights, the latter earn milestone payments and royalties.
In India, CROs operate differently. The innovation driven companies are taking on ‘collaborative drug discovery’ projects, leveraging the mutual strengths each has to offer the other (cost and expertise respectively). Therefore, the complexion of business is different from that of the Western countries. We would see large outsourcing companies positioning themselves as offshore partners for pharma-biotech majors. They could also go in for a BOT (Build Operate Transfer) model. Under this model, companies will set-up the facility and manage it to make it a ‘centre of excellence’, focusing on certain disease segments. Another option is that some companies can take up their own programmes to develop compounds and enter into licensing partnerships. This is similar to the US and Europe biotech style of working. Both of these practices are likely to happen in India.
How much do you acknowledge science and strategy in Aurigene’s growth?
Science will translate into good value if there is an appropriate business direction to it. The same is true of Aurigene. We are adding value by generating IPs, because it does not need massive infusion of manpower. It definitely costs less to do a discovery in Bangalore than in Boston. Although time taken to generate an IP is higher, it can create potential revenues.
What according to you is the future of the drug discovery sector?
It is at an interesting stage. There is lot of scope for large and medium cap companies to enter India. Pfizer has a large presence in Mumbai for clinical and analytical work. Novartis is setting up a new facility in Hyderabad to focus more on clinical development and analytics. Even Wyeth is reportedly moving towards an expanded relationship with its Indian partners. Bristol Myers Squib has already teamed up with Biocon’s subsidiary Syngene. Companies like Aurigene are gaining visibility. There has never been such a huge interest towards drug discovery partnerships. New players like Advinus and Jubilant have set-up large facilities in Bangalore and Pune, respectively.
/////////AUNP-12, Aurigene, Pierre Fabre Pharmaceuticals, Licensing Agreement, New Cancer Therapeutic, Immuno-oncology, AUNP 12, Immune Checkpoint Modulator Targeting the PD-1 Pathway, PEPTIDES
FEW MORE ACADEMIC COMPDS FROM PATENT, REDER DISCRETION NEEDED























 The Bangalore-based Aurigene Discovery Technologies Limited, an independent subsidiary of Dr Reddy’s, is now competing in a mature contract research organization space in the country. Established in 2003, Aurigene Discovery Technologies is a partnership focused collaborative discovery organisation. CSN Murthy is chief executive officer of the company. In an interaction with Nandita Vijay, Murthy cut a clear picture of the contract research scene. Excerpts:
The Bangalore-based Aurigene Discovery Technologies Limited, an independent subsidiary of Dr Reddy’s, is now competing in a mature contract research organization space in the country. Established in 2003, Aurigene Discovery Technologies is a partnership focused collaborative discovery organisation. CSN Murthy is chief executive officer of the company. In an interaction with Nandita Vijay, Murthy cut a clear picture of the contract research scene. Excerpts:




