http://www.google.com/patents/WO2008022462A1?cl=en
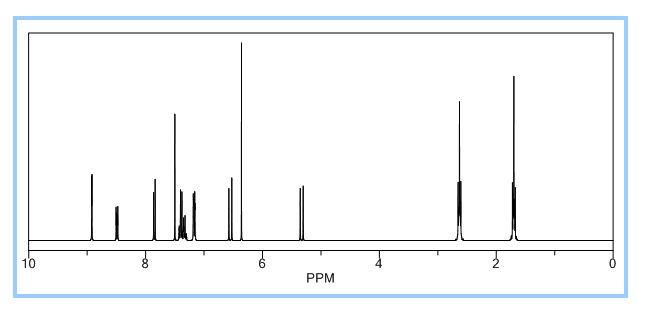
1 H-NMR Spectrum of Compound 35…………3-[3-(benzoylamino)-4-hydroxylphenyl] propanoic acid
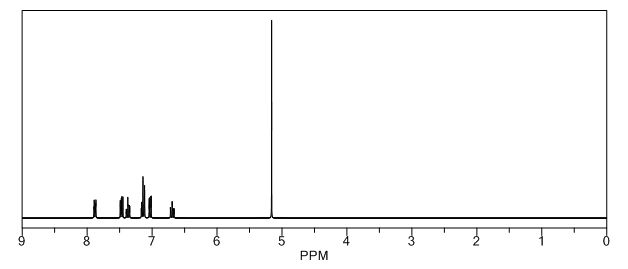
1H-NMR (Acetone-D6) δ: 2.60 (t, 2H, J = 7.4, H- 3), 2.84 (t, 2H, J = 7.9, H-2), 6.89 (d, IH, J = 8.2, H-8), 7.00 (dd, IH, J = 2.1 , 8.25, H- 9), 7.57 (m, 4H, H-5, H-4′, H-5′, H-61), 8.05 (d, 2H, J = 8.2, H-3′, H-7′), 9.07 (broad s, IH, NH), 9.54 (broad s, IH, OH), 10.58 (broad s, IH, CO2H).
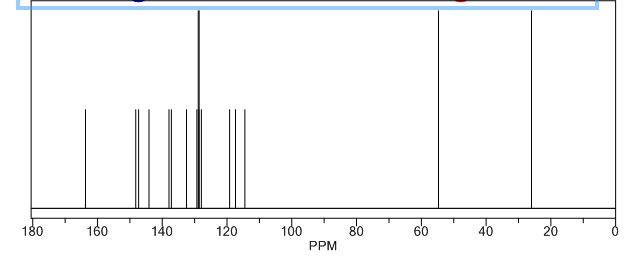
13C-NMR Spectrum of Compound 35
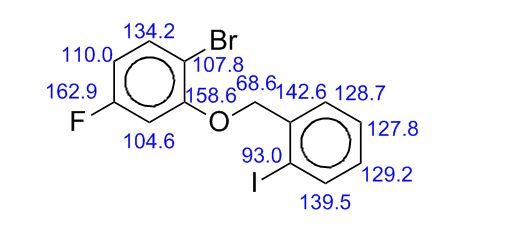
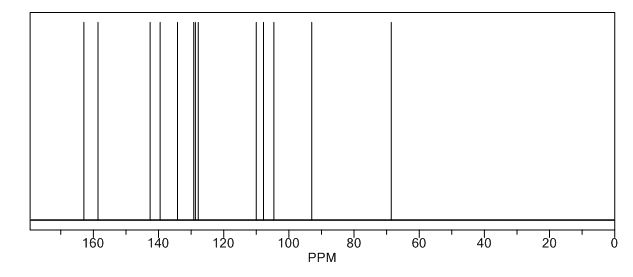
13C-NMR (Acetone- D6) δ: 30.87 (C- 3), 36.21 (C- 2), 118.69 (C- 8), 123.31 (C-5), 123.41 (C- 6), 126.88 (C- 9), 127.37 (C- 4), 128.54 (C-41, C-61), 129.61 (C-31, C-7′), 132.99 (C-51), 134.99 (C-21), 148.03 (C-7), 167.34 (C-I1), 173.94 (C-I ).
13C-NMR Spectrum of Compound 35
COSY-NMR Spectrum of Compound 35
COSY-NMR Spectrum of Compound 35
HETCOR-NMR Spectrum of Compound 35
3-[3-(benzoylamino)-4-hydroxylphenyl] propanoic acid 35:
To a solution of 32 (222 mg, 1.06 mmol, leq.) dissolved in THF (20 mL) was added the catalyst 10 % palladium-on-charcoal (15 % by mass, 33 mg). The resulting mixture was then placed on a hydrogenator, flushed (5 times) with hydrogen and left to agitate under pressure (36 psi.) overnight (12 hrs) while recharging hydrogen pressure twice (36 psi.) until hydrogen up-take by reaction mixture stopped (pressure did not decrease for 1-2 hrs.). The reaction mixture was vacuum filtered through Celite ‘ rinsing with THF. To the filtered solution containing 33 was directly added BzCl (154 mg, 1.1 mmol, 1 eq.) and left to stir at room temperature for 30 min. Then 10 % HCl (25 mL) was added and stirring continued an additional 5 min. followed by extraction with CIT2Cl2 (2 x 35 mL). The organic fractions were combined, dried (MgSO4), and evaporated off solvent. The resulting mixture was re-crystallized with Hexane/ Acetone to afford an off white solid (250 mg) with an 83 % yield from compound 32. Molecular Formula – C16Hi5NO4. Formula Weight – 285.295 g mole“1.
FT-IR (KBR disk) cm” 1 : 3201 (NH, OH), 1692 (CO2H), 1636 (NHAc).
1H-NMR (Acetone-D6) δ: 2.60 (t, 2H, J = 7.4, H- 3), 2.84 (t, 2H, J = 7.9, H-2), 6.89 (d, IH, J = 8.2, H-8), 7.00 (dd, IH, J = 2.1 , 8.25, H- 9), 7.57 (m, 4H, H-5, H-4′, H-5′, H-61), 8.05 (d, 2H, J = 8.2, H-3′, H-7′), 9.07 (broad s, IH, NH), 9.54 (broad s, IH, OH), 10.58 (broad s, IH, CO2H).
13C-NMR (Acetone- D6) δ: 30.87 (C- 3), 36.21 (C- 2), 118.69 (C- 8), 123.31 (C-5), 123.41 (C- 6), 126.88 (C- 9), 127.37 (C- 4), 128.54 (C-41, C-61), 129.61 (C-31, C-7′), 132.99 (C-51), 134.99 (C-21), 148.03 (C-7), 167.34 (C-I1), 173.94 (C-I ).
/////////////////////////////////////////
1H-NMR Spectrum of Compound (+/-V36
13 C-NMR Spectrum of Compound (+/-V36


N-(l-oxaspiro[4.5]deca-6,9-dien-2,8-dion-7-yl)acetamide (+/-)-36: To a solution of 34 (122 mg, .547 mmol, 1 eq.) dissolved in acetone (10 mL, 0 0C) was added PIFA (306 mg, .71 1 mmol, 1.3 eq.) in one portion and stirred for 20-25 minutes (confirmed by tic: [1 : 1] EtOAc/Hexane). The reaction mixture was diluted with ethyl acetate (15 mL), washed with cold water (10 mL), dried organic fraction (MgSO4) and evaporated off solvent to afford a Tan solid. The crude product was purified by re-dissolving with CHCI3, filtering of the solution through Celite ®, evaporating off the solvent and placing it under vacuum overnight to afford an off white solid (120 mg, 98 % yield). Molecular Formula – C1 1Hi iNO4. Formula Weight – 221.209 g mole“1. FT-IR (KBR disk) cm“1: 3333 (NH), 1777 (lactone), 1668 (amide), 1650 (ketone), 1620 (α, β-conjugation to ketone). 1H-NMR (CDCl3) δ: 2.17 (s, 3H, H-2′), 2.44 (m, 2H, H-4), 2.81 (m, 2H, H-3), 6.35 (d, IH, J = 10.0, H-9), 6.94 (dd, IH, J = 3.1, 10.0, H- 10), 7.75 (d, IFI, J = 3.1, H-6), 7.99 (broad s, IH, NH). 13C-NMR (CDCl3) δ: 24.86 (C- 2′), 28.36 (C- 4), 32.91 (C- 3), 79.76 (C-5), 124.30 (C- 6), 127.12 (C- 9), 131.55 (C- 7), 148.37 (C-10), 169.51 (C-I’), 175.46 (C-2), 179.40 (C- 8).
////////////////////
1H-NMR Spectrum of Compound 32


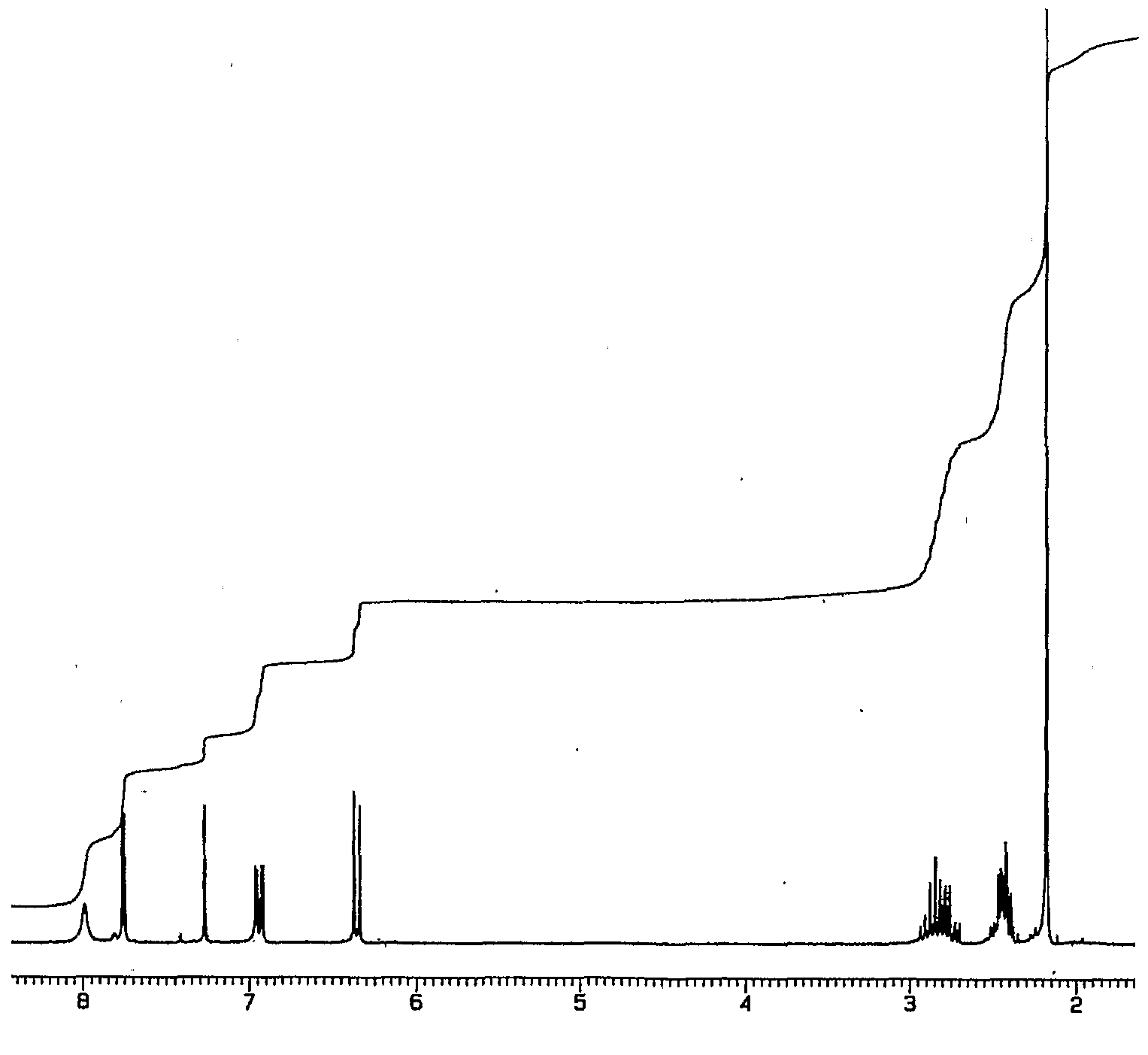
(2E)-3-(4-hydroxyl-3-nitrophenyl) acrylic acid 32: To a solution of 4- hydroxyl-3-nitrobenzaldehyde (1.073 g, 6.43 mmol, 1 eq.) dissolved in pyridine (25 mL) was added piperidine (25 drops) and the resulting mixture was stirred (4-5 min.). Malonic acid (1.671 g, 16.1 mmol, 2.5 eq.) was then added in one portion and the resulting mixture was warmed (60-63 0C) and stirred overnight (12-14 hrs, confirmed by tic: EtOAc, mini work up, 10 % HCl and EtOAc). The reaction was cooled and acidified (50 % HCl) until yellow precipitate formed (pH~2). This yellow precipitate was extracted with ethyl acetate (2 x 150 niL). The organic fractions were combined and washed with brine (150 mL), dried (MgSO4), and the solvent was evaporated to afford a yellow solid. Removed excess solvent by vacuum and used without further purification (1.250 g, 93 % yield). Molecular Formula – CgH7NO5. Formula Weight – 209.156 g mole“1. FT-IR (KBR disk) cm“1: 2942 (OH), 1684 (CO2H), 1626 (C=C), 1533,1270 (NO2). 1FI-NMR (Acetone-D6) δ: 2.87 (broad s, IH, OH), 6.58 (d, IH, J= 16.0, H-2), 7.27 (d, IH, J= 8.8, H-8), 7.70 (d, IH, J= 16.4, H-3), 8.08 (d, IH, J= 2.2, 8.5, H-9), 8.40 (d, IFI, J = 2.2, FI-5), 10.67 (broad s, I H, CO2FI). The13C-NMR of this compound agrees with the previously published data.52
Con Dao Island, Vietnam
Con Dao Island, Vietnam
This 16-island archipelago is a “pocket of paradise,” says Robert Reid, a travel editor at Lonely Planet.
Getting there: Take a 45-minute flight from Ho Chi Minh City.
What to do: The diving is among the best in Vietnam. Take scuba lessons as a couple or discover the nearby secluded beaches of Bai Dat Doc and Dam Trau.
Where to stay: Six Senses resort offers luxury villas on the East Vietnam Sea. The resort has an in-house spa offering traditional Vietnamese healing practices; it also boasts outdoor treatment rooms and a yoga and meditation pavilion. Inquire for rates.
wikitravel.org/en/Con_Dao
Con Dao is an island off the southern coast of Vietnam. … The Con Dao Islands separated from the mainland about 15,000 years ago. This has resulted in the …
![]() .
.
 airport
airport












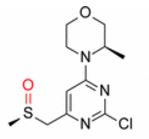

 , Bradley Adams, Helen Benson, Julie Demeritt†∥, Steven McKown, Keith Mulholland, Amy Robertson, Paul Siedlecki, Paula Tomlin, and Kevin Vare
, Bradley Adams, Helen Benson, Julie Demeritt†∥, Steven McKown, Keith Mulholland, Amy Robertson, Paul Siedlecki, Paula Tomlin, and Kevin Vare
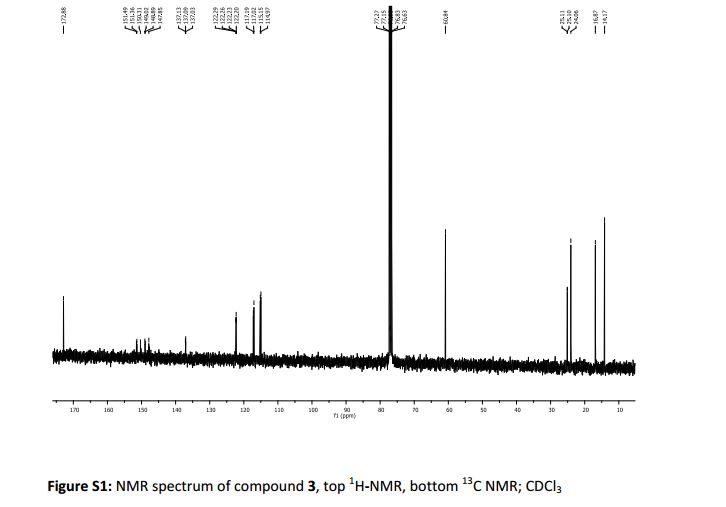

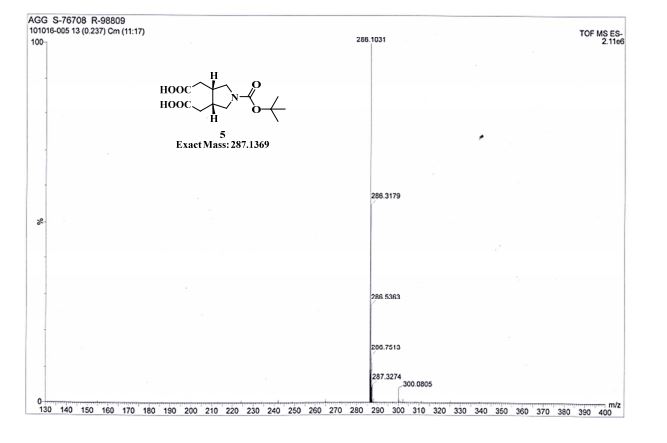
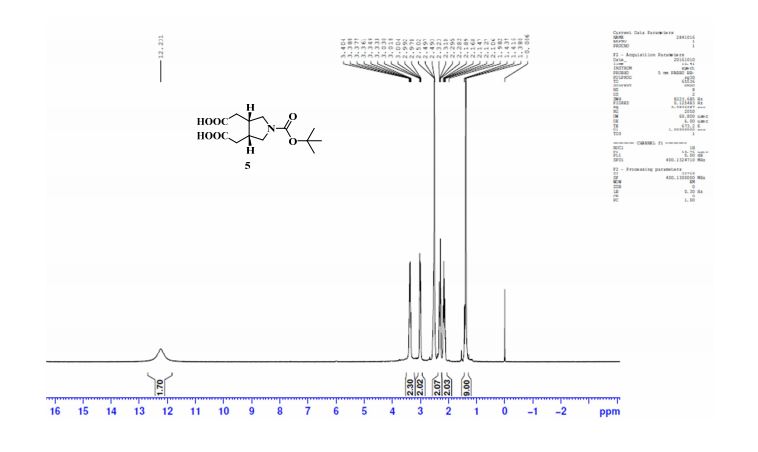
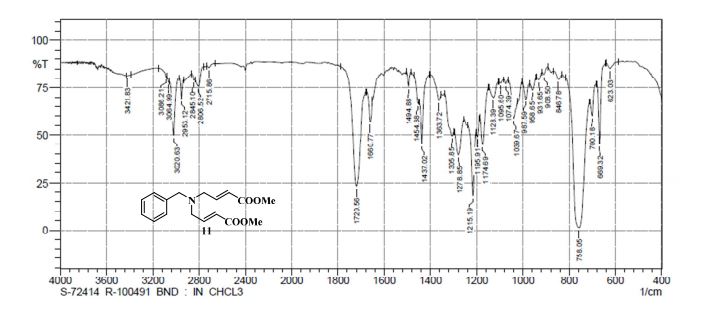
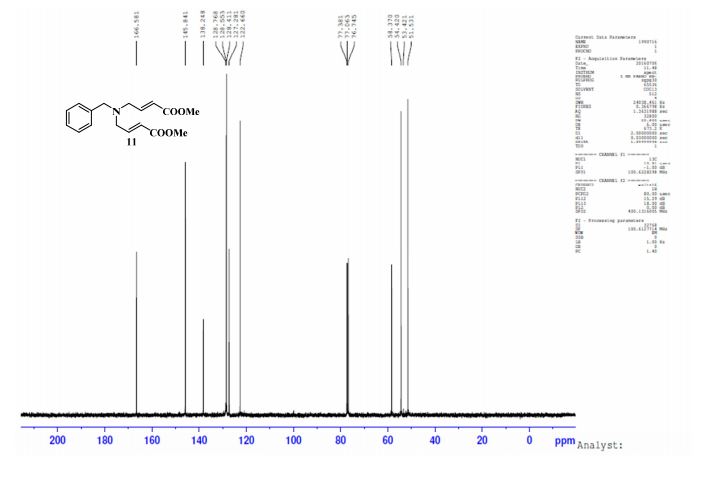
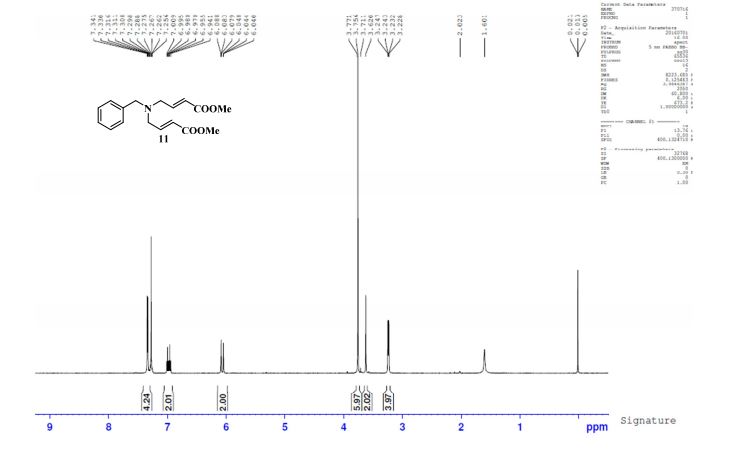
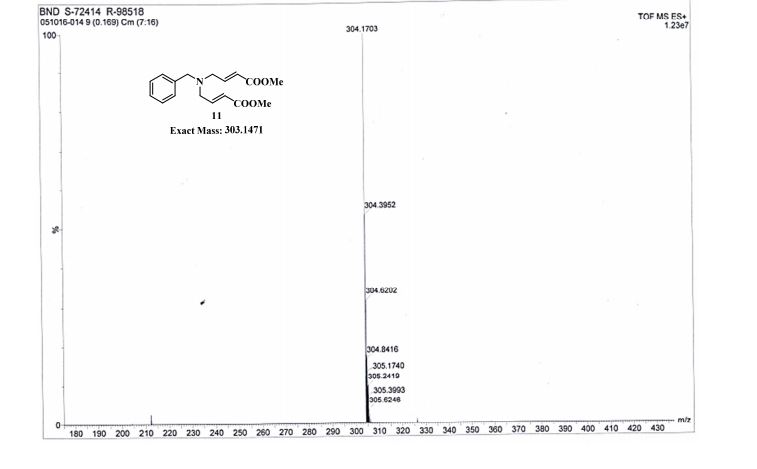
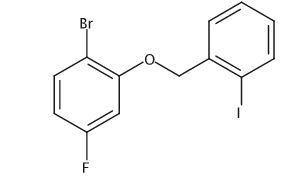
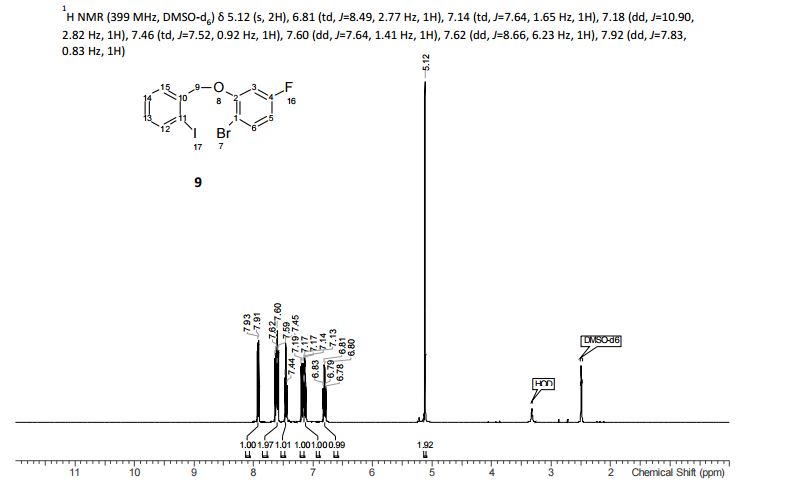
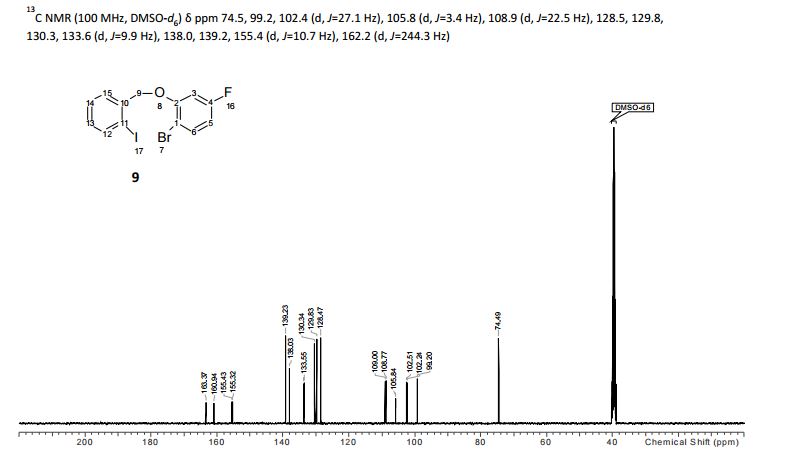
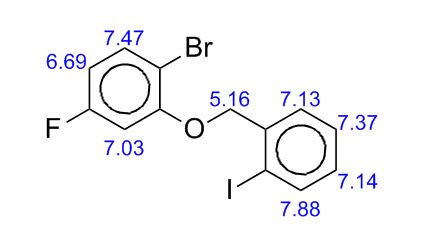
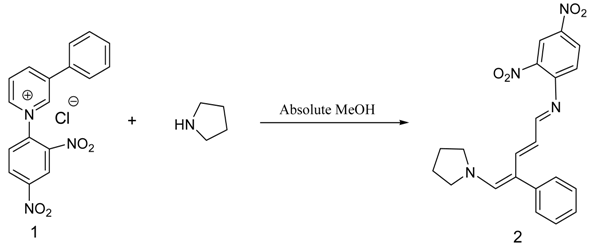
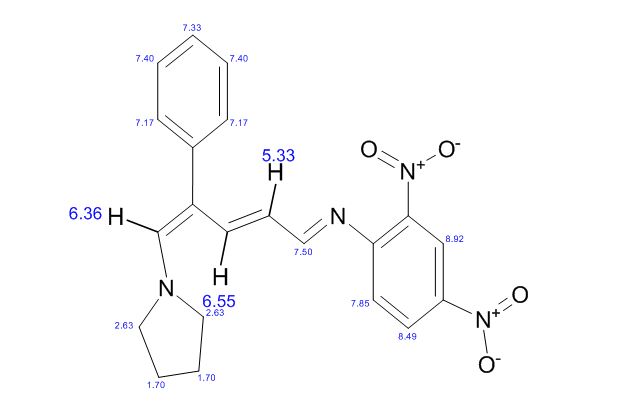




















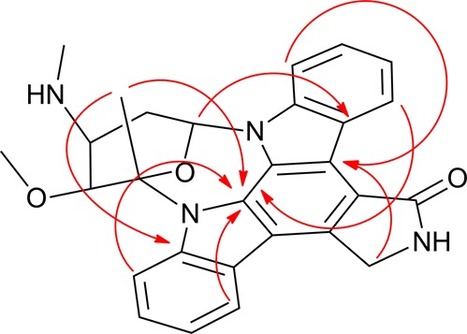























 .
.

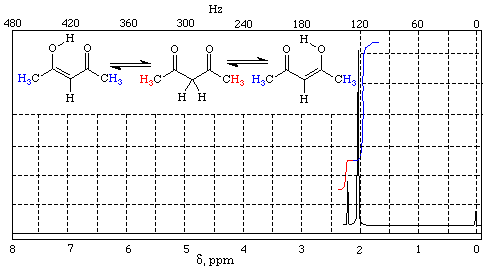












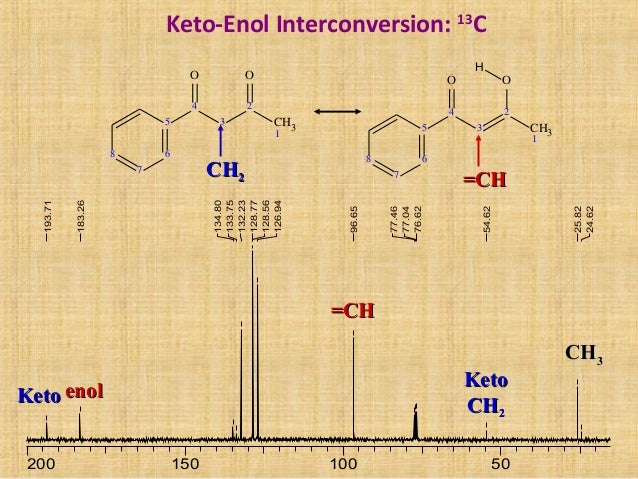 .
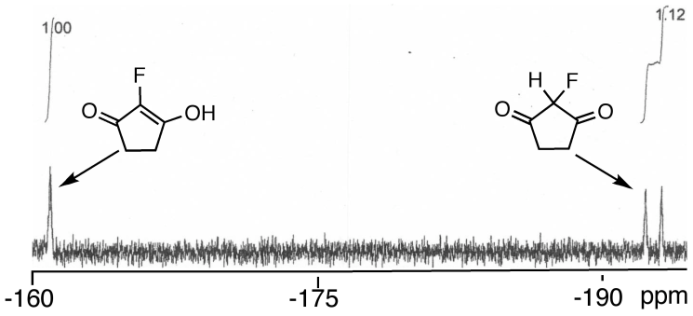
.

 .
.
 ISKCON
ISKCON



.jpg)