












NARLAPREVIR
An antiviral agent that inhibits hepatitis C virus NS3 protease.
M.Wt: 707.96
Formula: C36H61N5O7S
CAS No.: 865466-24-6
SCH 900518;SCH900518;SCH-900518
3-Azabicyclo[3.1.0]hexane-2-carboxamide, N-[(1S)-1-[2-(cyclopropylamino)-2-
oxoacetyl]pentyl]-3-[(2S)-2-[[[[1-[[(1,1-dimethylethyl)sulfonyl]methyl]cyclohexyl]
amino]carbonyl]amino]-3,3-dimethyl-1-oxobutyl]-6,6-dimethyl-, (1R,2S,5S)-
2. (1R,2S,5S)-N-{(1S)-1-[2-(cyclopropylamino)-2-oxoacetyl]pentyl}-3-[(2S)-2-{[(1-{[(1,1-
dimethylethyl)sulfonyl]methyl}cyclohexyl)carbamoyl]amino}-3,3-dimethylbutanoyl]-6,6-
dimethyl-3-azabicyclo[3.1.0]hexane-2-carboxamide
3. (1R,2S,5S)-3-{N-[({1-[(tert-butylsulfonyl)methyl]cyclohexyl}amino)carbonyl]-3-methyl-L-
valyl}-N-{(1S)-1-[(cyclopropylamino)(oxo)acetyl]pentyl}-6,6-dimethyl-3-
azabicyco[3.1.0]hexane-2-carboxamide
Narlaprevir is a potent, Second Generation HCV NS3 Serine Protease Inhibitor.Narlaprevir is useful for Antiviral
Merck & Co. (Originator)
SCH-900518 had been in phase II clinical trials by Merck & Co. for the treatment of genotype 1 chronic hepatitis C; however, no recent development has been reported for this indication.
A potent oral inhibitor of HCV NS3 protease, SCH-900518 disrupts hepatitis C virus (HCV) polyprotein processing. When added to the current standard of care (SOC), peginterferon-alfa plus ribavirin, SCH-900518 is likely to increase the proportion of patients achieving undetectable HCV-RNA levels and sustained virologic response (SVR).
In 2012, the product was licensed by Merck & Co. to R-Pharm in Russia and the Commonwealth of Independent States (CIS) for the development and commercialization as treatment of hepatitis C (HCV)
PATENTS
WO 2011014494
WO 2010068714
(1 R,5S)-N-[1 (S)-[2-(cyclopropylamino)-1 ,2-dioxoethyl]pentyl]-3-[2(S)- [[[[1-[[1.1-dimethylethyl)sulfonyl]methyl]cyclohexyl]amino]carbonyl]amino]-3,3- dimethyl-1-oxobutyl]-6,6-dimethyl-3-azabicyclo[3.1.0]hexane-2(S)-carboxamide.

Identification of any publication in this section or any section of this application is not an admission that such publication is prior art to the present invention.
The compound of Formula I is generically and specifically disclosed in
Published U.S. Patent No.2007/0042968, published February 22, 2007 (the ‘968 publication), incorporated herein by reference.
Processes suitable for making the compound of Formula I are generally described in the ‘968 publication. In particular, the ‘968 publication discusses preparing a sulfone carbamate compound, for example, the compound of Formula 837 comprising a cyclic sulfone substituent (paragraphs [0395] through [0403]). The following reaction scheme describes the procedure:

The process disclosed in the ‘968 publication produces the intermediate alcohol in step S7 as a mixture of diastereomers at the hydroxyl group; while this chiral center is lost in the final step of the disclosed process, the alcohol intermediate as a mixture of isomers cannot be crystallized and required a volumetrically inefficient precipitative isolation that did not remove any impurities
,………………………………………………………………………………………………………………………

……………………………………………………………………………………………………………………
Preparation of Compound VIJ

LDA was made by slowly charging n-butyl lithium (2.5 M, 159 kg) to diisopropyl amine (60 kg) dissolved in THF (252 kg), keeping the temperature at about −20° C., followed by agitation at this temperature for about 30 min. To this solution was charged cyclohexane carboxylic acid, methyl ester (70 kg), keeping the temperature below −10° C. The mixture was agitated at this temperature for about 2 h. To the resulting enolate was charged TMSCI (64.4 kg). The mixture was agitated at −10 to −20° C. for about 30 min, and then heated to about 25° C. and held at this temperature to allow for conversion to the silylenol ether Compound VIH. The reaction mixture was solvent exchanged to n-heptane under vacuum, keeping the temperature below 50° C., resulting in the precipitation of solids. The solids were filtered and washed with n-heptane, and the wash was combined with the n-heptane reaction mixture. The n-heptane mixture of Compound VIH was concentrated under vacuum and diluted with CH2Cl2.
In a separate reactor was charged CH2Cl2 (461 kg) and anhydrous ZnBr2 (14.5 kg). The temperature of the zinc slurry was adjusted to about 20° C. To the zinc slurry was simultaneously charged the solution of Compound VIH and 2-chloromethylsulfanyl-2-methyl-propane (63.1 kg, ref: Bioorg. Med. Chem. Lett, 1996, 6, 2053-2058), keeping the temperature below 45° C. After complete addition, the mixture was agitated for about 1.5 h at 35 to 45° C., after which the reaction mixture was cooled to 10 to 15° C. A solution of dilute aqueous HCl was then charged, keeping the temperature between 0 and 15° C., followed by a separation of the aqueous and organic layers (desired compound in organic layer). The organic layer was washed with aqueous NaHCO3 and water. The organic layer was solvent exchanged to methanol by vacuum distillation, keeping the temperature below 35° C., and kept as a solution in methanol for further processing to Compound VIK. Active Yield of Compound VIJ=69.7 kg (molar yield=57.9%).
Preparation of Compound VIK

To a fresh reactor was charged Compound VIJ (99.8 kg active in a methanol solution), water (270 kg), NaOH (70 kg), and methanol (603 kg). The mixture was heated to −70° C. and agitated at this temperature for about 16 h. Upon conversion to the sodium salt of Compound VIK, the reaction mixture was concentrated under vacuum, keeping the temperature below 55° C., and then cooled to about 25° C. Water and MTBE were then charged, agitiated, and the layers were separated (product in the aqueous layer). The product-containing aqueous layer was further washed with MTBE.
CH2Cl2 was charged to the aqueous layer and the temperature was adjusted to ˜10° C. The resultant mixture was acidified to a pH of about 1.5 with HCl, agitated, settled, and separated (the compound was in the organic layer). The aqueous layer was extracted with CH2Cl2, and the combined organic layers were stored as a CH2Cl2 solution for further processing to Compound VID. Active yield of Compound VIK=92.7 kg (molar yield=98.5 kg). MS Calculated: 230.13; MS Found (ES−, M−H): 229.11.
Preparation of Compound VID

To a reactor was charged water (952 kg), Oxone® (92.7 kg), and Compound VIK (92.7 kg active as a solution in CH2Cl2). The reaction mixture was agitated for about 24 h at a temperature of about 15° C., during which time Compound VIK oxidized to sulfone Compound VID. The excess Oxone® was quenched with aqueous Na2S2O5, the reaction mixture was settled and the layers separated; the aqueous layer was back-extracted with CH2Cl2, and the combined product-containing organic layers were washed with water.
The resultant solution was then concentrated under vacuum. To precipitate Compound VID, n-heptane was charged, and the resulting slurry was agitated for about 60 min at a temperature of about 30° C. The reaction mixture was filtered, and the wet cake was washed with n-heptane. The wet cake was redissolved in CH2Cl2, followed by the addition of n-heptane. The resultant solution was then concentrated under vacuum, keeping the temperature below 35° C., to allow for product precipitation. The resultant solution was cooled to about 0° C. and agitated at this temperature for about 1 h. The solution was filtered, the wet cake was washed with n-heptane, and dried under vacuum at about 45° C. to yield 68.7 kg Compound VID (molar yield=65.7%). MS Calculated: 262.37; MS Found (ES−, M−H): 261.09
Preparation of Compound VI

To a reactor was charged Compound VID (68.4 kg), toluene (531 kg), and Et3N (31 kg). The reaction mixture was atmospherically refluxed under Dean-Stark conditions to remove water (target KF <0.05%). The reaction temperature was adjusted to 80° C., DPPA (73.4 kg) was charged over 7 h, and the mixture was agitated for an additional 2 h. After conversion to isocyanate Compound VIE via the azide, the reaction mixture was cooled to about 0 to 5° C. and quenched with aqueous NaHCO3. The resultant mixture was agitated, settled and the layers were separated. The aqueous layer was extracted with toluene, and the combined isocyante Compound VIE organic layers were washed with water.
In a separate vessel was charged L-tert- Leucine (L-Tle, 30.8 kg), water (270 kg), and Et3N (60 kg). While keeping the temperature at about 5° C., the toluene solution of Compound VIE was transferred to the solution of L-Tle. The reaction mixture was stirred at 0 to 5° C. for about 5 h, at which time the mixture was heated to 15 to 20° C. and agitated at this temperature for 2 h to allow for conversion to urea Compound VI.
The reaction was quenched by the addition of aqueous NaOH, keeping the temperature between 0 and 25° C. The reaction mixture was separated, and the organic layer was extracted with water. The combined Compound VI-containing aqueous layers were washed with toluene, and acidified to pH 2 by the addition of HCl, at which time the product precipitated from solution. The reaction mixture was filtered, washed with water and dried under vacuum at 65 to 70° C. to yield 79.7 kg crude Compound VI (molar yield 52.7%). MS Calculated: 390.54; MS Found (ES−, M−H): 389.20.
Compound VI is further purified by slurrying in CH3CN at reflux (about 80° C.), followed by cooling to RT. Typical recovery is 94%, with an increase in purity from about 80% to 99%.
Preparation of Compound Va

To a reactor was charged Compound VI (87.6 kg), Compound VII-1 (48.2 kg), HOBt (6 kg) and CH3CN (615 kg). The reaction mixture was cooled to about 5° C., and NMM (35 kg) and EDCi (53.4 kg) were charged. The reaction was heated to 20 to 25° C. for about 1 h, and then to 35 to 40° C., at which time water was charged to crystallize Compound Va. The reaction mixture was cooled to 5° C. and held at this temperature for about 4 h. Compound Va was filtered and washed with water. XRD data for the hydrated polymorph of Va is as follows:
The Compound Va wet cake was charged to a fresh vessel and was dissolved in ethyl acetate at 25 to 30° C. The solution was washed with an aqueous HCl solution, aqueous K2CO3 solution, and brine. The solution was then concentrated under vacuum, keeping the temperature between 35 to 50° C. Additional ethyl acetate was charged, and the solution was heated to 65 to 70° C. While keeping the temperature at 65 to 70° C., n-heptane was charged, followed by cooling the resultant solution to 0 to 5° C. Compound Va was filtered and washed with an ethyl acetate/n-heptane mix.
The wet cake was dried under vacuum between 55 to 60° C. to yield 96.6 kg crystalline Compound Va (molar yield 79.2%). MS Calculated: 541.32; MS Found (ES+, M+H): 542.35.
Preparation of Compound IUB

Pyridine (92 L) was charged to the reactor and was cooled to 5° C. To the cooled pyridine was slowly charged malonic acid (48.5 kg) and valeraldehyde (59 L), keeping the temperature below 25° C. The reaction was stirred between 25 to 35° C. for at least 60 h. After this time, H2SO4 was charged to acidify, keeping the temperature below 30° C. The reaction mixture was then extracted into MTBE. The organic layer was washed with water. In a separate reactor was charged water and NaOH. The MTBE solution was charged to the NaOH solution, keeping the temperature below 25° C., and the desired material was extracted into the basic layer. The basic layer was separated and the organic layer was discarded. MTBE was charged, the mixture was agitated, settled, and separated, and the organic layer was discarded. To the resultant solution (aqueous layer) was charged water and H2SO4 to acidify, keeping the temperature between 10 to 15° C. To the acidified mixture was charged MTBE, keeping the temperature below 25° C. The resultant solution was agitated, settled, and separated, and the aqueous layer was discarded. The product-containing organic layer was washed with water and was concentrated under vacuum, keeping the temperature below 70° C., to yield 45.4 kg Compound IIIB (molar yield=76.2%) as an oil. Compound Reference: Concellon, J. M.; Concellon, C J. Org. Chem., 2006, 71, 1728-1731
Preparation of Compound IIIC

To a pressure vessel was charged Compound IIIB (9.1 kg), heptane (9 L), and H2SO4 (0.5 kg). The pressure vessel was sealed and isobutylene (13.7 kg) was charged, keeping the temperature between 19 to 25° C. The reaction mixture was agitated at this temperature for about 18 h. The pressure was released, and a solution of K2CO3 was charged to the reaction mixture, which was agitated and settled, and the bottom aqueous layer was then separated. The resultant organic solution was washed with water and distilled under vacuum (temp below 45° C.) to yield 13.5 kg Compound IIIC (molar yield=88.3%) as a yellow oil.
Preparation of Compound IIID
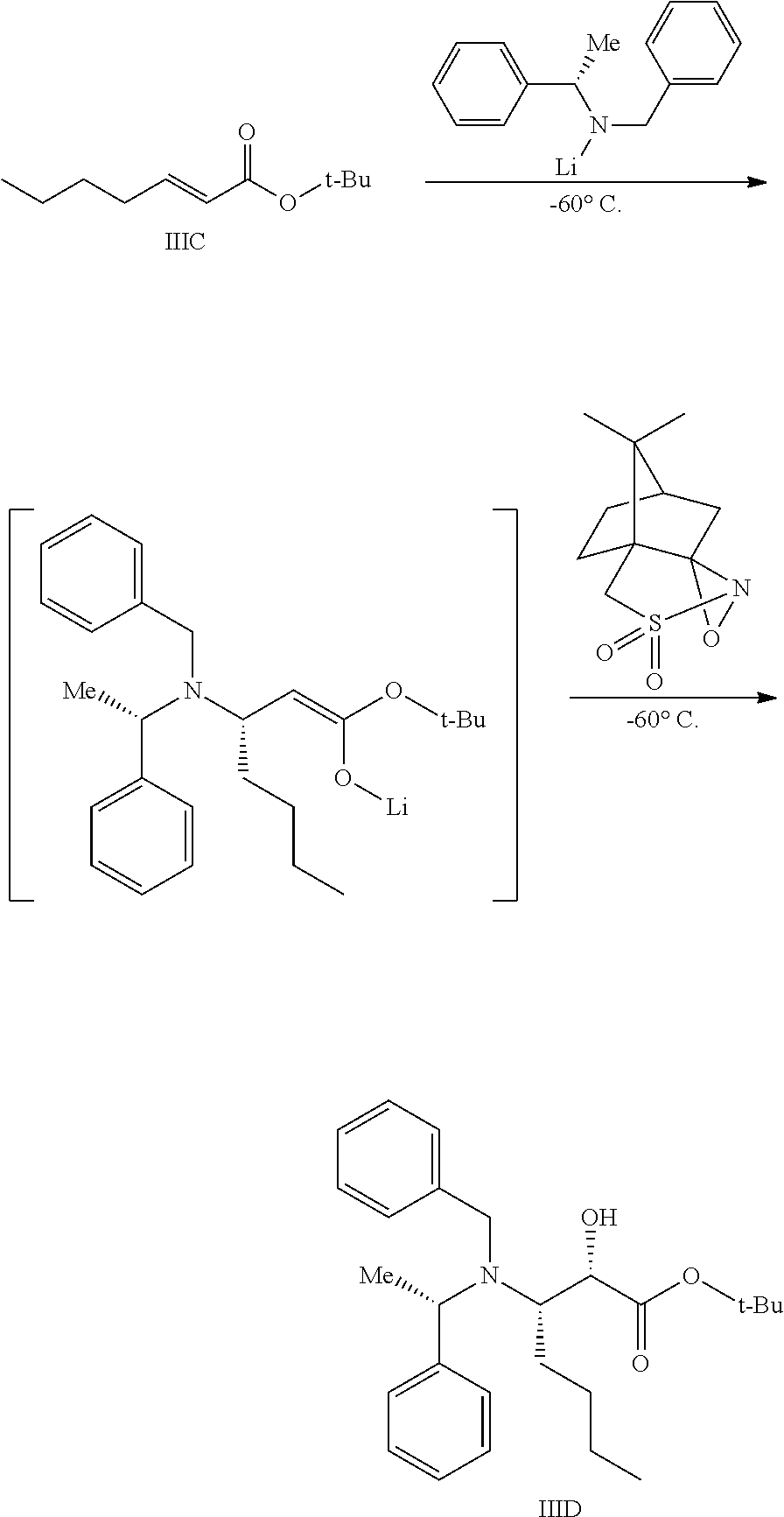
To a reactor capable of maintaining a temperature of −60° C. was charged (S)-benzyl-1-phenyl ethylamine (18 kg) and THF (75 L). The reaction mixture was cooled to −60° C. To the mixture was charged n-hexyl lithium (42 L of 2.3 M in heptane) while maintaining a temperature of −65 to −55° C., followed by a 30 min agitation within this temperature range. To the in situ-formed lithium amide was charged Compound IIIC over 1 h, keeping the temperature between −65 to −55° C. . The reaction mixture was agitated at this temperature for 30 min to allow for conversion to the enolate intermediate. To the resultant reaction mixture was charged (+)-camphorsulfonyl oxaziridine (24 kg) as a solid, over a period of 2 h, keeping the temperature between −65 to −55° C. . The mixture was agitated at this temperature for 4 h.
The resultant reaction mixture was quenched by the addition of acetic acid (8 kg), keeping the temperature between −60 to −40° C. The mixture was warmed to 20 to 25° C., then charged into a separate reactor containing heptane. The resultant mixture was concentrated under vacuum, keeping the temperature below 35° C. Heptane and water were charged to the reaction mixture, and the precipitated solids were removed by filtration (the desired compound is in the supernatant). The cake was washed with heptane and this wash was combined with the supernatant. The heptane/water solution was agitated, settled, and separated to remove the aqueous layer. An aqueous solution of H2SO4 was charged, and the mixture was agitated, settled, and separated. The heptane layer was washed with a solution of K2CO3.
The heptane layer was concentrated under reduced pressure, keeping the temperature below 45° C., and the resulting oil was diluted in toluene, yielding 27.1 kg (active) of Compound IIID (molar yield=81.0%). MS Calculated: 411.28; MS Found (ES+, M+H): 412.22.
A similar procedure for this step was reported in: Beevers, R, et al, Bioorg. Med. Chem. Lett. 2002, 12, 641-643.
Preparation of Compound IDE

Toluene (324 L) and a toluene solution of Compound IIID (54.2 kg active) was charged to the reactor. TFA (86.8 kg) was charged over about 1.5 h, keeping the temperature below 50° C. The reaction mixture was agitated for 24 h at 50° C. The reaction mixture was cooled to 15° C. and water was charged. NaOH was slowly charged, keeping the temperature below 20° C., to adjust the batch to a pH between 5.0 and 6.0. The reaction mixture was agitated, settled, and separated; the aqueous layer was discarded. The organic layer was concentrated under vacuum, keeping the temperature below 40° C., and the resulting acid intermediate (an oil), was dissolved in 2-MeTHF.
In a separate reactor, 2-MeTHF (250 L), HOBt (35.2 kg), and EDCi-HCl (38.0 kg) were charged and the mixture was adjusted to a temperature between 0 to 10° C. DIPEA (27.2 kg) was charged, keeping the mixture within this temperature range. The mixture was agitated for 5 min, followed by the addition of cyclopropyl amine (11.4 kg), keeping the temperature between 0 to 10° C.
To this solution was charged the 2-MeTHF/ acid intermediate solution, keeping the resultant solution between 0 to 10° C. The resultant mixture was heated to 25 to 35° C., and was agitated at this temperature for about 4 h. The reaction mixture was cooled to about 20° C., and was washed with aqueous citric acid, aqueous K2CO3, and water. The solvent was exchanged to n-heptane, and the desired compound was crystallized from a mix of n-heptane and toluene by cooling to 0° C. The crystalline product was filtered, washed with n-heptane, and dried to yield 37.1 kg Compound IIIE (molar yield=70.7%). MS Calculated: 394.26; MS Found (ES+, M+H): 395.22.
Preparation of Compound III

To a pressure reactor was charged acetic acid (1.1 kg), methanol (55 kg), and Compound IIIE (10.9 kg). In a separate vessel, Pd/C (50% water wet, 0.5 kg) was suspended in methanol (5 kg). The Pd/C suspension was transferred to the solution containing Compound IIIE. The resultant mixture was pressurized to 80 psi with hydrogen, and agitated at 60° C. for 7 h. The reaction mixture was then purged with nitrogen, and the Pd/C catalyst was filtered off. The resultant solution was concentrated under vacuum and adjusted to about 20° C. MTBE was charged, and the resultant solution was brought to reflux. Concentrated HCl (3 L) was charged and the product was crystallized by cooling the reaction mixture to about 3° C. The desired compound was filtered, washed with MTBE, and dried under vacuum, keeping the temperature below 40° C. to yield 5.5 kg Compound III (molar yield=83.0%). MS Calculated (free base): 200.15; MS Found (ES+, M+H): 201.12.
Preparation of Compound II

Compound Va (119.3 kg) was dissolved in 2-MeTHF (720 kg) and water (180 kg). To this solution was charged 50% NaOH (21.4 kg) while maintaining a temperature between 20 and 30° C. The reaction mixture was then agitated for about 7 h at a temperature between 50 and 60° C. The reaction mixture was cooled to a temperature between 20 and 30° C.
The pH of the reaction mixture was adjusted to 1.5-3.0 with dilute phosphoric acid, maintaining a temperature between 20 and 30° C. The resultant mixture was agitated for 10 min, settled for 30 min, and the bottom aqueous layer was separated and removed. The top organic layer was washed with water, followed by concentration by atmospheric distillation.
The concentrated solution was solvent exchanged to CH3CN by continuous atmospheric distillation, and crystallized by cooling to 0° C. The crystalline product was filtered, washed with CH3CN, and dried under vacuum at a temperature between 45 and 55° C. to yield 97.9 kg Compound II (molar yield=83.7%). MS Calculated: 527.30; MS Found (ES+, M+H): 528.29.
Preparation of Compound IV

Compound II (21.1 kg), Compound III (9.9 kg), HOBt (3.2 kg) and EDCi (11.2 kg) were charged to the vessel, followed by CH3CN (63 kg), ethyl acetate (20 kg) and water (1.5 kg). The reaction mixture was agitated and the heterogeneous mixture was cooled to −5 to +5° C. DIPEA (11.2 kg) was charged to the reaction mixture, maintaining a temperature between −5 to +5° C. and the mixture was agitated at a temperature of −5 to +5° C. for 1 h. The resultant reaction mixture was warmed to 20 to 30° C. and agitated for 2 to 3 h.
The resultant product was extracted with aqueous HCl, aqueous K2CO3, and water.
The desired product was crystallized from ethyl acetate by cooling from reflux (78° C.) to about 0° C. The crystalline product was filtered and dried at 30° C. under vacuum to yield 23.1 kg Compound IV (molar yield=81.3%). MS Calculated: 709.44; MS Found (ES+, M+H): 710.47.
Preparation of Compound I

Compound IV (22.5 kg), TEMPO (5 kg), NaOAc (45 kg), methyl acetate (68 L), MTBE (158 L), water (23 L) and acetic acid (22.5 L) were charged to the reactor. The reaction mixture was stirred at 20-30° C. to allow for dissolution of the solids, and was then cooled to 5-15° C. NaOCl solution (1.4 molar equivalents) was charged to the reaction mixture, keeping the temperature at about 10° C. After complete addition of NaOCl, the reaction mixture was agitated at 10° C. for 2 h.
The reaction was quenched by washing with a buffered sodium ascorbate/HCl aqueous solution, followed by a water wash.
The reaction mixture was solvent exchanged to acetone under vacuum, keeping the temperature below 20° C.; the desired product was crystallized by the addition of water, and dried under vacuum, keeping the temperature below 40° C. to yield 18.6 kg Compound I (molar yield=82.7%). MS Calculated: 707.43: MS Found (ES+, M+H): 708.44.