











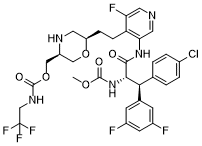

MK 8718
Cas 1582729-24-5 (free base); 1582732-29-3 (HCl).
MF: C30H30ClF6N5O4
MW: 673.1891
| INNOVATOR | Merck Sharp & Dohme Corp., Merck Canada Inc. |
((3S,6R)-6-(2-(3-((2S,3S)-2-amino-3-(4-chlorophenyl)-3-(3,5-difluorophenyl)propanamido)-5-fluoropyridin-4-yl)ethyl)morpholin-3-yl)methyl (2,2,2-trifluoroethyl)carbamate
MK-8718 is a potent, selective and orally bioavailable HIV protease inhibitor with a favorable pharmacokinetic profile with potential for further development.
A retrovirus designated human immunodeficiency virus (HIV), particularly the strains known as HIV type-1 (HIV-1) virus and type-2 (HIV-2) virus, is the etiological agent of acquired immunodeficiency syndrome (AIDS), a disease characterized by the destruction of the immune system, particularly of CD4 T-cells, with attendant susceptibility to opportunistic infections, and its precursor AIDS-related complex (“ARC”), a syndrome characterized by symptoms such as persistent generalized lymphadenopathy, fever and weight loss. This virus was previously known as LAV, HTLV-III, or ARV. A common feature of retrovirus replication is the extensive post-translational processing of precursor polyproteins by a virally encoded protease to generate mature viral proteins required for virus assembly and function. Inhibition of this processing prevents the production of normally infectious virus. For example, Kohl et al., Proc. Nat’l Acad. Sci. 1988, 85: 4686, demonstrated that genetic inactivation of the HIV encoded protease resulted in the production of immature, non-infectious virus particles. These results indicated that inhibition of the HIV protease represents a viable method for the treatment of AIDS and the prevention or treatment of infection by HIV.
Nucleotide sequencing of HIV shows the presence of a pol gene in one open reading frame [Ratner et al, Nature 1985, 313: 277]. Amino acid sequence homology provides evidence that the pol sequence encodes reverse transcriptase, an endonuclease, HIV protease and gag, which encodes the core proteins of the virion (Toh et al, EMBO J. 1985, 4: 1267; Power et al, Science 1986, 231 : 1567; Pearl et al, Nature 1987, 329: 351].
Several HIV protease inhibitors are presently approved for clinical use in the treatment of AIDS and HIV infection, including indinavir (see US 5413999), amprenavir (US5585397), saquinavir (US 5196438), ritonavir (US 5484801) and nelfmavir (US 5484926). Each of these protease inhibitors is a peptide-derived peptidomimetic, competitive inhibitor of the viral protease which prevents cleavage of the HIV gag-pol polyprotein precursor. Tipranavir (US 5852195) is a non-peptide peptidomimetic protease inhibitors also approved for use in treating HIV infection. The protease inhibitors are administered in combination with at least one and typically at least two other HIV antiviral agents, particularly nucleoside reverse transcriptase inhibitors such as zidovudine (AZT) and lamivudine (3TC) and/or non-nucleoside reverse transcriptase inhibitors such as efavirenz and nevirapine. Indinavir, for example, has been found to be highly effective in reducing HIV viral loads and increasing CD4 cell counts in HIV-infected patients, when used in combination with nucleoside reverse transcriptase inhibitors. See, for example, Hammer et al, New England J. Med. 1997, 337: 725-733 and Gulick et al, New England J. Med. 1997, 337: 734-739.
The established therapies employing a protease inhibitor are not suitable for use in all HIV-infected subjects. Some subjects, for example, cannot tolerate these therapies due to adverse effects. Many HIV-infected subjects often develop resistance to particular protease inhibitors. Furthermore, the currently available protease inhibitors are rapidly metabolized and cleared from the bloodstream, requiring frequent dosing and use of a boosting agent.
Accordingly, there is a continuing need for new compounds which are capable of inhibiting HIV protease and suitable for use in the treatment or prophylaxis of infection by HIV and/or for the treatment or prophylaxis or delay in the onset or progression of AIDS.

PATENT
https://www.google.co.in/patents/WO2014043019A1?cl=en
INTERMEDIATE 1
Synthesis of morpholine intermediate (tert-butyl ( ^S^-S-d tert- butyl(dimethyl)silylloxy|methyl)-2-(hydroxymethyl)morpholine-4-carboxylate)



Scheme 1

EXAMPLE 97
( S)- -(4-Chlorophenyl)-3,5-difiuoro-N-(5-fiuoro-4-{2-[(2R,5S)-5-({[(2,2,2- trifluoroethyl)carbamoyl]oxy}methyl)morpholin-2-yl]ethyl}pyridin-3-yl)-L-phenylalaninamide

Step 1. (2S,3S)-2-Azido-3-(4-chlorophenyl)-3-(3,5-difluorophenyl)propanoic acid

The title compound was prepared from 4-chlorocinnamic acid and 3,5- difluorophenylmagnesium bromide using the procedures given in steps 1-4 of Example 92.
Step 2. (2R,5S)-tert-butyl 2-(2-(3-((2S,3S)-2-azido-3-(4-chlorophenyl)-3-(3,5- difluorophenyl)propanamido)-5-fluoropyridin-4-yl)ethyl)-5-((((2,2,2- trifluoroethyl)carbamoyl)oxy)methyl)morpholine-4-carboxylate

The product from step 1 (105 mg, 0.31 mmol) and the product from step 4 of Example 89 (150 mg, 0.31 mmol) were dissolved in pyridine (1 mL) and the stirred solution was cooled to -10 °C in an ice/acetone bath. To the cold solution was added POCI3 dropwise (0.035 mL, 0.38 mmol). The mixture was stirred at -10 °C for 30 min. The reaction was quenched by the addition of saturated aqueous NaHC03 solution (1 mL) and the mixture was allowed to warm to ambient temperature. The mixture was diluted with water (10 mL) and extracted with dichloromethane (3 x 10 mL). The combined dichloromethane phases were dried (Na2S04), filtered, and the filtrate solvents were removed in vacuo. The residue was purified on a 12 g silica gel column using a gradient elution of 0-70% EtOAc:hexanes. Fractions containing product were combined and the solvents were removed in vacuo to give the title compound as a gum. (M+H)+ = 800.6.
Step 3. (2R,5S)-tert-butyl 2-(2-(3-((2S,3S)-2-amino-3-(4-chlorophenyl)-3-(3,5- difluorophenyl)propanamido)-5-fluoropyridin-4-yl)ethyl)-5-((((2,2,2- trifluoroethyl)carbamoyl)oxy)methyl)morpholine-4-carboxylate
The product from step 2 (150 mg, 0.19 mmol) and triphenylphosphine (74 mg, 0.28 mmol) were dissolved in THF (4 mL) and to the solution was added water (1 mL). The mixture was heated to reflux under a nitrogen atmosphere for 12 h. The mixture was cooled to ambient temperature and the solvents were removed in vacuo. The residue was purified on a 12 g silica gel column eluting with a gradient of 0-10% methanol: chloroform. Fractions containing product were combined and the solvents were removed in vacuo to give the title compound as a gum. (M+H)+ = 774.7. Step 4. ( S)- -(4-Chlorophenyl)-3,5-difluoro-N-(5-fluoro-4-{2-[(2R,5S)-5-({[(2,2,2- trifluoroethyl)carbamoyl]oxy}methyl)morpholin-2-yl]ethyl}pyridin-3-yl)-L-phenylala
The product from step 3 (60 mg, 0.078 mmol) was dissolved in a solution of 4M HCl in dioxane (1 mL, 4 mmol) and the solution was stirred at ambient temperature for 1 h. The solvent was removed under reduced pressure and the residue was dried in vacuo for 12 h to give an HCl salt of the title compound as a solid. LCMS: RT = 0.95 min (2 min gradient), MS (ES) m/z = 674.6 (M+H)+.
PAPER

A novel HIV protease inhibitor was designed using a morpholine core as the aspartate binding group. Analysis of the crystal structure of the initial lead bound to HIV protease enabled optimization of enzyme potency and antiviral activity. This afforded a series of potent orally bioavailable inhibitors of which MK-8718 was identified as a compound with a favorable overall profile.
Discovery of MK-8718, an HIV Protease Inhibitor Containing a Novel Morpholine Aspartate Binding Group
References
Discovery of MK-8718, an HIV Protease Inhibitor Containing a Novel Morpholine Aspartate Binding Group
Christopher J. Bungard*†, Peter D. Williams†, Jeanine E. Ballard†, David J. Bennett†, Christian Beaulieu‡, Carolyn Bahnck-Teets†, Steve S. Carroll†, Ronald K. Chang†, David C. Dubost†, John F. Fay†, Tracy L. Diamond†, Thomas J. Greshock†, Li Hao§, M. Katharine Holloway†, Peter J. Felock, Jennifer J. Gesell†, Hua-Poo Su†, Jesse J. Manikowski†, Daniel J. McKay‡, Mike Miller†, Xu Min†, Carmela Molinaro†, Oscar M. Moradei‡, Philippe G. Nantermet†, Christian Nadeau‡, Rosa I. Sanchez†, Tummanapalli Satyanarayana§, William D. Shipe†, Sanjay K. Singh§, Vouy Linh Truong‡, Sivalenka Vijayasaradhi§, Catherine M. Wiscount†, Joseph P. Vacca‡, Sheldon N. Crane‡, and John A. McCauley†
† Merck Research Laboratories, 770 Sumneytown Pike, PO Box 4, West Point, Pennsylvania 19486, United States
‡ Merck Frosst Centre for Therapeutic Research, 16711 TransCanada Highway, Kirkland, Quebec H9H 3L1, Canada
§ Albany Molecular Research Singapore Research Center, 61 Science Park Road #05-01, The Galen Singapore Science Park II, Singapore 117525
ACS Med. Chem. Lett., Article ASAP
DOI: 10.1021/acsmedchemlett.6b00135
Publication Date (Web): May 09, 2016
////MK-8718, HIV, protease, inhibitor
O=C(OC[C@H]1NC[C@@H](CCC(C(F)=CN=C2)=C2NC([C@@H](N)[C@@H](C3=CC=C(Cl)C=C3)C4=CC(F)=CC(F)=C4)=O)OC1)NCC(F)(F)F



 LinkedIn
LinkedIn Facebook
Facebook Twitter
Twitter GooglePlus
GooglePlus