












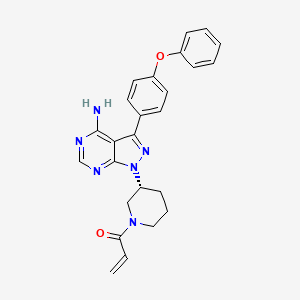
IBRUTINIB 依鲁替尼
A Btk protein inhibitor.
1-[(3R)-3-[4-Amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl]piperidin-1-yl]prop-2-en-1-one
1-((R)-3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)prop-2-en-1-one
| CAS number | 936563-96-1 |
|---|
- CRA-032765
- Ibrutinib
- Imbruvica
- Pc-32765
- PCI 32765
- PCI32765
- UNII-1X70OSD4VX
Company: Pharmacyclics
Approval Status: Approved February 2014US FDA:link
Treatment Area: chronic lymphocytic leukemia
Bruton’s tyrosine kinase (Btk) inhibitor
U.S. Patent No: 7,514,444 , 7,718,662
patent validity: December 2026
An orally bioavailable small-molecule inhibitor of Bruton’s tyrosine kinase (BTK) with potential antineoplastic activity. Ibrutinib binds to and inhibits BTK activity, preventing B-cell activation and B-cell-mediated signaling and inhibiting the growth of malignant B cells that overexpress BTK. BTK, a member of the src-related BTK/Tec family of cytoplasmic tyrosine kinases, is required for B cell receptor (BCR) signaling, plays a key role in B-cell maturation, and is overexpressed in a number of B-cell malignancies.
Imbruvica (ibrutinib) is an orally available, selective inhibitor of Bruton’s tyrosine kinase (Btk), a gene that is disrupted in the human disease X-linked agammaglobulenemia (XLA). BTK is a signaling molecule of the B-cell antigen receptor (BCR) and cytokine receptor pathways.
Imbruvica is specifically approved for chronic lymphocytic leukemia in patients who have received at least one prior therapy.
Imbruvica (Ibrutinib, previously known as PCI-32765) was approved as a “breakthrough therapy” on November 13, 2013 by the US Food and Drug Administration (FDA) for the treatment of mantle cell lymphoma (MCL), a rare and deadly form of blood cancer.
Ibrutinib, a first in class oral Bruton’s tyrosine kinase (Btk) inhibitor, was launched in the U.S. for the treatment of patients with mantle cell lymphoma in 2013, and for the treatment of chronic lymphocytic leukemia in 2014. In the E.U., the product candidate is awaiting registration for both indications. Additional phase III clinical trials are ongoing for the treatment of these indications in combination with bendamustine and rituximab and for the treatment of relapsed or refractory marginal zone lymphoma (MZL). Janssen and Pharmacyclics are conducting phase II clinical trials for the treatment of refractory follicular lymphoma. Early clinical development is also under way at Pharmacyclics for the treatment of recurrent B-cell lymphoma, relapsed/refractory MCL, and relapsed or relapsed and refractory multiple myeloma. The company filed an IND seeking approval to commence clinical evaluation of ibrutinib for the treatment of autoimmune disease. Preclinical studies had been under way for rheumatoid arthritis; however, no recent development has been reported. Ibrutinib is also active against Lyn and LCK tyrosine kinases.
In 2011, a codevelopment agreement was signed between the National Cancer Institute (NCI) and Pharmacyclics for the treatment of hematologic/blood cancer. Also in 2011, a worldwide codevelopment and comarketing agreement was signed by Janssen and Pharmacyclics for the treatment of cancer. In 2012, orphan drug designation was assigned in the U.S. and the E.U. for the treatment of CLL. This designation was also assigned by the FDA in 2012 for the treatment of mantle cell lymphoma. In 2013, several orphan drug designations were assigned in the U.S.; for the treatment of small lymphocytic lymphoma, for the treatment of Waldenstrom’s macroglobulinemia and for the treatment of diffuse large B-cell lymphoma. For this indication, orphan drug designation was assigned also in the E.U. the same year. In 2012, fast track designation was assigned by the FDA for the treatment of CLL. In 2013, breakthrough therapy designations were assigned to the compound in the U.S.: for the treatment (as monotherapy) of patients with chronic lymphocytic leukemia or small lymphocytic lymphoma, for the treatment of relapsed or refractory mantle cell lymphoma who have received prior therapy and for the treatment of Waldenstrom’s macroglobulinemia.
Imbruvica is supplied as a capsule for oral administration. The recommended dose is 420 mg taken orally once daily (three 140 mg capsules once daily). Capsules should be taken orally with a glass of water. Do not open, break, or chew the capsules.
The FDA approval of Imbruvica for chronic lymphocytic leukemia was based on an open-label, multi-center trial of 48 previously treated patients. Imbruvica was administered orally at 420 mg once daily until disease progression or unacceptable toxicity. The overall response rate (ORR) and duration of response (DOR) were assessed using a modified version of the International Workshop on CLL Criteria by an Independent Review Committee. The ORR was 58.3%, all partial responses. None of the patients achieved a complete response. The DOR ranged from 5.6 to 24.2+ months. The median DOR was not reached.
Imbruvica (ibrutinib) is an orally available, selective inhibitor of Bruton’s tyrosine kinase (Btk). Ibrutinib forms a covalent bond with a cysteine residue in the BTK active site, leading to inhibition of BTK enzymatic activity. BTK is a signaling molecule of the B-cell antigen receptor (BCR) and cytokine receptor pathways. BTK’s crole in signaling through the B-cell surface receptors results in activation of pathways necessary for B-cell trafficking, chemotaxis, and adhesion.
Ibrutinib (USAN,[1] also known as PCI-32765 and marketed in the U.S. under the name Imbruvica) is an anticancer drug targeting B-cell malignancies. It was approved by the US FDA in November 2013 for the treatment of mantle cell lymphoma[2] and in February 2014 for the treatment ofchronic lymphocytic leukemia.[3] It is an orally-administered, selective and covalent inhibitor of the enzyme Bruton’s tyrosine kinase (BTK).[4][5][6]Ibrutinib is currently under development by Pharmacyclics, Inc and Johnson & Johnson‘s Janssen Pharmaceutical division for additional B-cell malignancies including diffuse large B-cell lymphoma and multiple myeloma.[7][8][9]
Mechanism
In preclinical studies on chronic lymphocytic leukemia (CLL) cells, ibrutinib has been reported to promote apoptosis, inhibit proliferation, and also prevent CLL cells from responding to survival stimuli provided by the microenvironment.[12] In this study, treatment of activated CLL cells with ibrutinib resulted in inhibition of Btk tyrosine phosphorylation and also effectively abrogated downstream survival pathways activated by this kinase including ERK1/2, PI3K, and NF-κB. Additionally, ibrutinib inhibited proliferation of CLL cells in vitro, effectively blocking survival signals provided externally to CLL cells from the microenvironment including soluble factors (CD40L, BAFF, IL-6, IL-4, and TNF-α), fibronectin engagement and stromal cell contact.
In early clinical studies, the activity of ibrutinib has been described to include a rapid reduction in lymphadenopathy accompanied by a transient lymphocytosis, suggesting that the drug might have direct effects on cell homing or migration to factors in tissue microenvironments.[13]
Ibrutinib has been reported to reduce CLL cell chemotaxis towards the chemokines CXCL12 and CXCL13, and inhibit cellular adhesion following stimulation at the B cell receptor.[14][15] Together, these data are consistent with a mechanistic model whereby ibrutinib blocks BCR signaling, which drives cells into apoptosis and/or disrupts cell migration and adherence to protective tumour microenvironments.
History
Ibrutinib was first designed and synthesized at Celera Genomics which reported in 2007 a structure-based approach for creating a series of small molecules that inactivate BTK through covalent binding to cysteine-481 near the ATP binding domain of BTK.[4] These small molecules irreversibly inhibited BTK by using a Michael acceptor for binding to the target cysteine. In April 2006, Pharmacyclics acquired Celera’s small molecule BTK inhibitor discovery program, which included a compound, PCI-32765 that was subsequently chosen for further preclinical development based on the discovery of anti-lymphoma properties in vivo.[16] Since 2006, Pharmacyclics’ scientists have advanced the molecule into clinical trials and identified specific clinical indications for the drug. It also has potential effects against autoimmune arthritis.[17] It was approved by the US FDA on November 13, 2013 for the treatment of mantle cell lymphoma.[2] On Feb. 12, 2014, the U.S. Food and Drug Administration expanded the approved use of the drug ibrutinib to chronic lymphocytic leukemia (CLL). [18]
Ibrutinib is an inhibitor of Bruton’s tyrosine kinase (BTK). It is a white to off-white solid with the empirical formula C25H24N6O2 and a molecular weight 440.50. Ibrutinib is freely soluble in dimethyl sulfoxide, soluble in methanol and practically insoluble in water.
The chemical name for ibrutinib is 1-[(3R)-3-[4-amino-3-(4-phenoxyphenyl)-1Hpyrazolo[ 3,4-d]pyrimidin-1-yl]-1-piperidinyl]-2-propen-1-one and has the following structure:
 |
IMBRUVICA (ibrutinib) capsules for oral administration are supplied as white opaque capsules that contain 140 mg ibrutinib as the active ingredient. Each capsule also contains the following inactive ingredients: croscarmellose sodium, magnesium stearate, microcrystalline cellulose, sodium lauryl sulfate. The capsule shell contains gelatin, titanium dioxide and black ink. Each white opaque capsule is marked with “ibr 140 mg” in black ink.
PCI-32765 (ibrutinib) is disclose d in U.S. Patent No. 7,514,444, issued on April 7, 2009, and has the following structur

Ibrutinib is an orally available drug that targets Bruton’s tyrosine kinase (BTK).
Ibrutinib is an irreversible small molecule BTK inhibitor that is in Ph Ib/II of clinical trials in a variety of B-cell malignancies including chronic lymphocytic leukemia (CLL), small lymphocytic lymphoma (SLL), mantle cell lymphoma (MCL), diffuse large B-cell lymphoma (DLBCL) and multiple myeloma (cancer of plasma cells, a type of white blood cell present in bone marrow). At present ibrutinib is administered orally in clinical trials, via the gastrointestinal tract, at high clinical doses (420 mg/day or 840 mg/day) to patients with CLL and SLL to obtain the desired thereapeutic effect. The need for such high doses of ibrutinib may be due to low bioavailability (the oral bioavailability of ibrutinib is reported to be 22.8% in rats) and may be responsible for the adverse side effects associated with the use of ibrutinib such as nausea or emesis, dizziness and diarrhea. Moreover, low bioavailability results in more variable absorption and potential variability of the desired therapeutic response.
As stated above, at present ibrutinib is administered orally, via the gastrointestinal tract, at high clinical doses (420 mg/day or 840 mg/day) to patients to obtain the desired clinical benefit. It is presently disclosed that when ibrutinib is administered intraduodenally versus via the gastrointestinal tract in rats, the oral bioavailability of ibrutinib unexpectedly increased from 21 % to 100% as determined by AUC.
This unexpected increase in oral bioavailability of ibrutinib can translate into a number of desirable practical benefits. The increase in oral bioavailability should enable administration of ibrutinib at a significantly lower therapeutically effective dose than is currently being used. The lower variability associated with this greater bioavailability should lead to a more reliable therapeutic response as well as more predictable drug absorption.
And avoidance of exposure of Ibtrutinib to the stomach and/or use of lower therapeutically effective dose of ibrutinib can reduce or altogether eliminate potential adverse side effects of this drug such as diahrrea, nausea or emesis, and dizziness. U.S. Patent No. 7,514,444, mentioned above, discloses administration of 0.02-5000 mg/kg andl-1500 mg of ibrutinib/per day and in clinical trials 420 or 840 mg/day of ibrutinib is being administered to the patients with CLL and SLL.
There is no reasonable expectation in the art that ibrutinib can be adminstered orally at lower efficacious doses to the patients with CLL and SLL, particularly as evidenced by the 420 or 840 mg/day of ibrutinib being administered in clinical trials to those patients. Moreover, other than for active agents that are unstable in the stomach or at acidic pH delivery of any active agent with low bioavailability further along in the gastrointestinal tract reduces the path length for drug absorption and would be expected to reduce bioavailability. Therefore, it was unexpected to achieve delivery of ibruntinib directly to the small intestine with greater bioavailability.
PC1-32765 (Ibrutinib), chemical name: 1_ [(3R) _3-[4_-3 – (4 – phenoxy-phenyl)-1H-pyrazolo [3,4-d] pyrimidine – 1 – yl] – 1-piperidinyl]-2 – propen-1 – one, and its structural formula is as follows:

PC1-32765 is an oral medication that inhibits B cell as the main receptor tyrosine kinase signaling and promote cell death process, preventing cell migration and adhesion in malignant B cells.
US20080108636 basic patent has been disclosed a synthetic route:
This synthetic route with 4 – phenoxy-benzoic acid as raw material, after eight-step reaction the final product, the following reaction steps:

The above method has the following disadvantages:
1, eight single-step reaction, long route, the economy is bad; i1, to use synthetic intermediates 4:00 trimethylsilyl diazomethane (TMSCHN2), this material easy to blow up, the risk coefficient is large, so large-scale production greatly reduces the possibility;
ii1, synthetic intermediates 7:00, set out to use polymer-supported triphenylphosphine, non-industrial raw materials used, the price is expensive, the cost of smell;
iv, the final step of acylation, the selectivity is poor, a large amount of negative product, purification is difficult, amplification reaction is difficult.
In summary, the route material is not common, expensive step, high costs, the reaction dangerous side reactions, purification difficult, limiting the possibility of industrial production of the route.
………………………
Polymorphs
EXAMPLES
[00438] The following ingredients, formulations, processes and procedures for practicing the methods disclosed herein correspond to that described above. Example 1; Preparation of Crystalline Forms of l-((R)-3-(4-amino-3-(4-phenoxyphenyl)- lH-pyrazolo[3,4-dlpyrimidin-l-yl)piperidin-l-yl)prop-2-en-l-one (Compound 1)
Form A – Route 1:
[00439] Amorphous Compound 1 (ca. 15 mg) was measured into a vial. Ten volumes (150 μΐ) of solvent [methyl tert-butyl ether (MTBE), diisopropyl ether (DIPE), ethyl acetate, isopropyl acetate, isopropyl alcohol, methyl isobutyl ketone (MIBK), methyl ethyl ketone (MEK), acetone, methanol, nitromethane, 10% aqueous acetone, or 10% aqueous isopropyl alcohol] were added to the vial. The vial was sealed and placed in a shaker at 50 °C for one hour. If a slurry was obtained, an additional thirty volumes (total of 600 μΐ) of solvent was added, then the slurry was returned to 50 °C for another hour. If the sample remained as a slurry at this point, no further solvent was added. The solution/slurry was stirred at 50 °C for one hour, then cooled to 0 °C at 0.1 °C/min, then held at 0 °C overnight. If a slurry was obtained, the solids were filtered under vacuum to provide Compound 1 , Form A; the solution was returned to ambient temperature for slow evaporation through a pin-hole to furnish Compound 1, Form A.
“Compound 1” or “l-((R)-3-(4-amino-3-(4-phenoxyphenyl)-lH-pyrazolo[3,4- d]pyrimidin- 1 -yl)piperidin- 1 -yl)prop-2-en- 1 -one” or “1 – {(3i?)-3-[4-amino-3-(4-phenoxyphenyl)- lH-pyrazolo[3,4-JJpyrimidin-l-yl]piperidin-l-yl}prop-2-en-l-one” or “2-Propen- 1 -one, 1- [(3R)-3-[4-amino-3-(4-phenoxyphenyl)- lH-pyrazolo[3,4-<f]pyrimidin- 1 -yl] – 1 -piperidinyl-” or ibrutinib or any other suitable name refers to the compound with the following structure:


………….
Synthesis
Synthesis of Compound 3—Btk Activity Probe
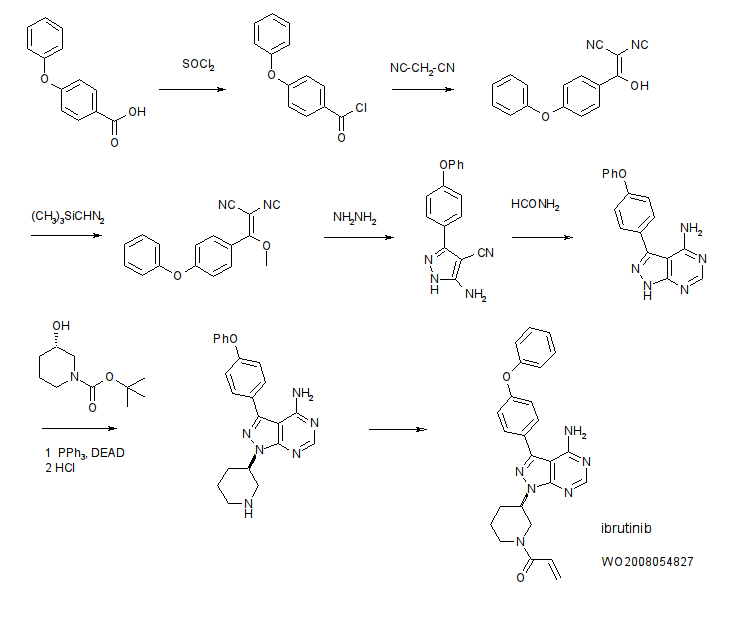
4-Amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidine (Intermediate 2) is prepared. Briefly, 4-phenoxybenzoic acid (48 g) is added to thionyl chloride (100 mL) and heated under gentle reflux for 1 hour. Thionyl chloride was removed by distillation, the residual oil was dissolved in toluene and volatile material removed at 80° C./20 mbar. The resulting acid chloride was dissolved in toluene (200 mL) and tetrahydrofuran (35 mL). Malononitrile (14.8 g) was added and the solution and stirred at −10° C. while adding diisopropylethylethylamine (57.9 g) in toluene (150 mL), while maintaining the temperature below 0° C. After 1 hour at 0° C., the mixture was stirred at 20° C. overnight. Amine hydrochloride is removed by filtration and the filtrate evaporated in vacuo. The residue was taken up in ethyl acetate and washed with 1.25 M sulphuric acid, then with brine and dried over sodium sulfate. Evaporation of the solvents gave a semisolid residue which was treated with a portion of ethyl acetate to give 4.1 g of 1,1-dicyano-2-hydroxy-2-(4-phenoxyphenyl)ethene as a white solid (m.p. 160-162° C.). The filtrate on evaporation gave 56.58 (96%) of 1,1-dicyano-2-hydroxy-2-(4-phenoxyphenyl)ethene as a grey-brown solid, which was sufficiently pure for further use.
1,1-Dicyano-2-hydroxy-2-(4-phenoxyphenyl)ethene (56.5 g) in acetonitrile (780 mL) and methanol (85 mL) is stirred under nitrogen at 0° C. while adding diisopropylethylamine (52.5 mL) followed by 2M trimethylsilyldiazomethane (150 mL) in THF. The reaction is stirred for 2 days at 20° C., and then 2 g of silica is added (for chromatography). The brown-red solution is evaporated in vacuo, the residue dissolved in ethyl acetate and washed well with water then brine, dried and evaporated. The residue is extracted with diethyl ether (3×250 mL), decanting from insoluble oil. Evaporation of the ether extracts gives 22.5 g of 1,1-dicyano-2-methoxy-2-(4-phenoxyphenyl)ethene as a pale orange solid. The insoluble oil is purified by flash chromatography to give 15.0 g of a red-orange oil.
1,1-Dicyano-2-methoxy-2-(4-phenoxyphenyl)ethene (22.5 g) and 1,1-dicyano-2-methoxy-2-(4-phenoxyphenyl)ethene oil (15 g) are treated with a solution of hydrazine hydrate (18 mL) in ethanol (25 mL) and heated on the steambath for 1 hour. Ethanol (15 mL) is added followed by water (10 mL). The precipitated solid is collected and washed with ethanol:water (4:1) and then dried in air to give 3-amino-4-cyano-5-(4-phenoxyphenyl)pyrazole as a pale orange solid.
3-Amino-4-cyano-5-(4-phenoxyphenyl)pyrazole (29.5 g) is suspended in formamide (300 mL) and heated under nitrogen at 180° C. for 4 hours. The reaction mixture is cooled to 30° C. and water (300 mL) is added. The solid is collected, washed well with water, then with methanol and dried in air to give of 4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidine (Intermediate 2).
Synthesis of 1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl (Intermediate 4); a) triphenylphosphine (TPP), diisopropyl diazodicarboxylate (DIAD), tetrahydrofuran (THF); b) TFA/CH2Cl2.

To a solution of 1-boc-3-(S)-hydroxypiperidine (3.98 g, 19.8 mmol) and triphenylphosphine (5.19 g, 19.8 mmol) in THF (150 ml) was added DIAD (3.9 ml, 19.8 mmol). The yellow solution was stirred 1 minute then Intermediate 2 (4.0 g, 13.2 mmol) was added and the reaction was heated with a heat gun (3-5 minutes) until the solid had dissolved. After stirring for 1 hour at room temperature, the solvent was removed and the resulting brown oil was subjected to flash chromatography (30% then 50% THF/hexanes) to provide 4.45 g (69%) of Intermediate 3 (trace of triphenylphosphine oxide is present) as a light brown foam.
To a solution of Intermediate 3 (4.4 g, 9.0 mmol) in CH2Cl2 (20 ml) was added TFA (2.8 ml, 36.2 mmol). After stirring 2 hrs at room temperature, the solvent was removed and the residue was partitioned between ethyl acetate (250 ml) and dilute aq. K2CO3. The organic layer was dried (MgSO4), filtered and concentrated to 70 ml. The resulting solution was stirred and 4.0M HCl in dioxane (4 ml) was added to provide a thick light orange precipitate. The precipitate was collected by filtration and washed with ethyl acetate (50 ml). The material was then partitioned between ethyl acetate (300 ml) and dilute aq. K2CO3. The organic layer was dried (MgSO4), filtered and concentrated to provide 2.78 g (80%) of Intermediate 4 as a light yellow foam.
……………………
SYNTHESIS
Compounds described herein may be prepared using the synthetic methods described herein as a single isomer or a mixture of isomers.
A non-limiting example of a synthetic approach towards the preparation of compounds of any of Formula (A), (B), (C) or (D) is shown in Scheme I.

Halogenation of commercially available 1H-pyrazolo[3,4-d]pyrimidin-4-amine provides an entry into the synthesis of compounds of Formula (A), (B), (C) and/or (D). In one embodiment, 1H-pyrazolo[3,4-d]pyrimidin-4-amine is treated with N-iodosuccinamide to give 3-iodo-1H-pyrazolo[3,4-d]pyrimidin-4-amine. Metal catalyzed cross coupling reactions are then carried out on 3-iodo-1H-pyrazolo[3,4-d]pyrimidin-4-amine. In one embodiment, palladium mediated cross-coupling of a suitably substituted phenyl boronic acid under basic conditions constructs intermediate 2. Intermediate 2 is coupled with N-Boc-3-hydroxypiperidine (as non-limiting example) via Mitsunobu reaction to give the Boc (tert-butyloxycarbonyl) protected intermediate 3. After deprotection with acid, coupling with, but not limited to, an acid chloride, such as, but not limited to, acryloyl chloride, completes the synthesis to give compound 4.
Example 1 Synthesis of Compounds Preparation of 4-Amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidine (Intermediate 2)
4-Amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidine (Intermediate 2) is prepared as disclosed in International Patent Publication No. WO 01/019829. Briefly, 4-phenoxybenzoic acid (48 g) is added to thionyl chloride (100 mL) and heated under gentle reflux for 1 hour. Thionyl chloride is removed by distillation, the residual oil dissolved in toluene and volatile material removed at 80° C./20 mbar. The resulting acid chloride is dissolved in toluene (200 mL) and tetrahydrofuran (35 mL). Malononitrile (14.8 g) is added and the solution and stirred at −10° C. while adding diisopropylethylethylamine (57.9 g) in toluene (150 mL), while maintaining the temperature below 0° C. After 1 hour at 0° C., the mixture is stirred at 20° C. overnight. Amine hydrochloride is removed by filtration and the filtrate evaporated in vacuo. The residue is taken up in ethyl acetate and washed with 1.25 M sulphuric acid, then with brine and dried over sodium sulfate. Evaporation of the solvents gives a semisolid residue which is treated with a little ethyl acetate to give 4.1 g of 1,1-dicyano-2-hydroxy-2-(4-phenoxyphenyl)ethene as a white solid (m.p. 160-162° C.). The filtrate on evaporation gives 56.58 (96%) of 1,1-dicyano-2-hydroxy-2-(4-phenoxyphenyl)ethene as a grey-brown solid, which is sufficiently pure for further use.
1,1-Dicyano-2-hydroxy-2-(4-phenoxyphenyl)ethene (56.5 g) in acetonitrile (780 mL) and methanol (85 mL) is stirred under nitrogen at 0° C. while adding diisopropylethylamine (52.5 mL) followed by 2M trimethylsilyldiazomethane (150 mL) in THF. The reaction is stirred for 2 days at 20° C., and then 2 g of silica is added (for chromatography). The brown-red solution is evaporated in vacuo, the residue dissolved in ethyl acetate and washed well with water then brine, dried and evaporated. The residue is extracted with diethyl ether (3×250 mL), decanting from insoluble oil. Evaporation of the ether extracts gives 22.5 g of 1,1-dicyano-2-methoxy-2-(4-phenoxyphenyl)ethene as a pale orange solid. The insoluble oil is purified by flash chromatography to give 15.0 g of a red-orange oil.
1,1-Dicyano-2-methoxy-2-(4-phenoxyphenyl)ethene (22.5 g) and 1,1-dicyano-2-methoxy-2-(4-phenoxyphenyl)ethene oil (15 g) are treated with a solution of hydrazine hydrate (18 mL) in ethanol (25 mL) and heated on the steambath for 1 hour. Ethanol (15 mL) is added followed by water (10 mL). The precipitated solid is collected and washed with ethanol:water (4:1) and then dried in air to give 3-amino-4-cyano-5-(4-phenoxyphenyl)pyrazole as a pale orange solid.
3-Amino-4-cyano-5-(4-phenoxyphenyl)pyrazole (29.5 g) is suspended in formamide (300 mL) and heated under nitrogen at 180° C. for 4 hours. The reaction mixture is cooled to 30° C. and water (300 mL) is added. The solid is collected, washed well with water, then with methanol and dried in air to give of 4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidine.
Example 1a Synthesis of 1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)prop-2-en-1-one (Compound 4)

-
- Synthesis of compound 4; a) polymer-bound triphenylphosphine (TPP), diisopropyl diazodicarboxylate (DIAD), tetrahydrofuran (THF); b) HCl/dioxane; then acryloyl chloride, triethylamine (TEA).
Compounds described herein were synthesized by following the steps outlined in Scheme 1. A detailed illustrative example of the reaction conditions shown in Scheme 1 is described for the synthesis of 1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)prop-2-en-1-one (Compound 4).
101 mg of 4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidine and 330 mg of polymer-bound triphenylphosphine(TPP) (polymerlab) were mixed together with 5 mL of tetrahydrofuran (THF). tert-Butyl 3-hydroxypiperidine-1-carboxylate (200 mg; 2.0 equivalents) was added to the mixture followed by the addition of diisopropyl diazodicarboxylate (0.099 mL). The reaction mixture was stirred at room temperature overnight. The reaction mixture was filtered to remove the resins and the reaction mixture was concentrated and purified by flash chromatography (pentane/ethyl acetate=1/1) to give intermediate 3 (55 mg).
Intermediate 3 (48.3 mg) was treated with 1 mL of 4N HCl in dioxane for 1 hour and then concentrated to dryness. The residue was dissolved in dichloromethane and triethylamine (0.042 mL) was added followed by acryl chloride (0.010 mL). The reaction was stopped after 2 hours. The reaction mixture washed with 5% by weight aqueous citric acid and then with brine. The organic layer was dried with MgSO4, and concentrated. Flash chromatography (with CH2Cl2/MeOH=25/1) gave 22 mg of compound 4 as a white solid. MS (M+1): 441.2; 1H-NMR (400 MHz): 8.26, s, 1H, 7.65, m, 2H, 7.42, m, 2H, 7.1-7.2, m, 5H, 6.7-6.9, m, 1H, 6.1, m, 1H, 5.5-5.7, m, 1H, 4.7, m, 1H, 4.54, m, 0.5H, 4.2, m, 1H, 4.1, m, 0.5H, 3.7, m, 0.5H, 3.2, 1,1H, 3.0, m, 0.5H, 2.3, m, 1H, 2.1, m, 1H, 1.9, m, 1H, 1.6, m, 1H.
Example 1b Synthesis of 1-((R)-3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)prop-2-en-1-one (Compound 13)

The synthesis of compound 13 was accomplished using a procedure analogous to that described in Example 1a. EM (calc.): 440.2; MS (ESI) m/e (M+1H)+: 441.1, (M−1H)−: 439.2.
Example 1c Synthesis of 1-((S)-3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)prop-2-en-1-one (Compound 14)

The synthesis of compound 14 was accomplished using a procedure analogous to that described for Example 1a. EM (calc.): 440.2; MS (ESI) m/e (M+1H)+: 441.5, (M−1H)−: 439.2.
……………….
Synthesis of Compounds Example 1 Preparation of 4-Amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidine (2a)
4-Amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidine (Intermediate 2) is prepared as disclosed in International Patent Publication No. WO 01/019829. Briefly, 4-phenoxybenzoic acid (48 g) is added to thionyl chloride (100 mL) and heated under gentle reflux for 1 hour. Thionyl chloride is removed by distillation, the residual oil dissolved in toluene and volatile material removed at 80° C./20 mbar. The resulting acid chloride is dissolved in toluene (200 mL) and tetrahydrofuran (35 mL). Malononitrile (14.8 g) is added and the solution and stirred at −10° C. while adding diisopropylethylethylamine (57.9 g) in toluene (150 mL), while maintaining the temperature below 0° C. After 1 hour at 0° C., the mixture is stirred at 20° C. overnight. Amine hydrochloride is removed by filtration and the filtrate evaporated in vacuo. The residue is taken up in ethyl acetate and washed with 1.25 M sulphuric acid, then with brine and dried over sodium sulfate. Evaporation of the solvents gives a semisolid residue which is treated with a little ethyl acetate to give 4.1 g of 1,1-dicyano-2-hydroxy-2-(4-phenoxyphenyl)ethene as a white solid (m.p. 160-162° C.). The filtrate on evaporation gives 56.58 (96%) of 1,1-dicyano-2-hydroxy-2-(4-phenoxyphenyl)ethene as a grey-brown solid, which is sufficiently pure for further use.
1,1-Dicyano-2-hydroxy-2-(4-phenoxyphenyl)ethene (56.5 g) in acetonitrile (780 mL) and methanol (85 mL) is stirred under nitrogen at 0° C. while adding diisopropylethylamine (52.5 mL) followed by 2M trimethylsilyldiazomethane (150 mL) in THF. The reaction is stirred for 2 days at 20° C., and then 2 g of silica is added (for chromatography). The brown-red solution is evaporated in vacuo, the residue dissolved in ethyl acetate and washed well with water then brine, dried and evaporated. The residue is extracted with diethyl ether (3×250 mL), decanting from insoluble oil. Evaporation of the ether extracts gives 22.5 g of 1,1-dicyano-2-methoxy-2-(4-phenoxyphenyl)ethene as a pale orange solid. The insoluble oil is purified by flash chromatography to give 15.0 g of a red-orange oil.
1,1-Dicyano-2-methoxy-2-(4-phenoxyphenyl)ethene (22.5 g) and 1,1-dicyano-2-methoxy-2-(4-phenoxyphenyl)ethene oil (15 g) are treated with a solution of hydrazine hydrate (18 mL) in ethanol (25 mL) and heated on the steambath for 1 hour. Ethanol (15 mL) is added followed by water (10 mL). The precipitated solid is collected and washed with ethanol:water (4:1) and then dried in air to give 3-amino-4-cyano-5-(4-phenoxyphenyl)pyrazole as a pale orange solid.
3-Amino-4-cyano-5-(4-phenoxyphenyl)pyrazole (29.5 g) is suspended in formamide (300 mL) and heated under nitrogen at 180° C. for 4 hours. The reaction mixture is cooled to 30° C. and water (300 mL) is added. The solid is collected, washed well with water, then with methanol and dried in air to give of 4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidine.
Example 1a Synthesis of 1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)prop-2-en-1-one (4)

Synthesis of compound 4; a) polymer-bound triphenylphosphine (TPP), diisopropyl diazodicarboxylate (DIAD), tetrahydrofuran (THF); b) HCl/dioxane; then acryloyl chloride, triethylamine (TEA)
Compounds described herein were synthesized by following the steps outlined in Scheme III. A detailed illustrative example of the reaction conditions shown in Scheme II is described for the synthesis of 1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)prop-2-en-1-one (Compound 4).
101 mg of 4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidine and 330 mg of polymer-bound triphenylphosphine (TPP) (polymerlab) were mixed together with 5 mL of tetrahydrofuran (THF). tert-Butyl 3-hydroxypiperidine-1-carboxylate (200 mg; 2.0 equivalents) was added to the mixture followed by the addition of diisopropyl diazodicarboxylate (0.099 mL). The reaction mixture was stirred at room temperature overnight. The reaction mixture was filtered to remove the resins and the reaction mixture was concentrated and purified by flash chromatography (pentane/ethyl acetate=1/1) to give intermediate 3a (55 mg).
Intermediate 3a (48.3 mg) was treated with 1 mL of 4N HCl in dioxane for 1 hour and then concentrated to dryness. The residue was dissolved in dichloromethane and triethylamine (0.042 mL) was added followed by acryl chloride (0.010 mL). The reaction was stopped after 2 hours. The reaction mixture was washed with 5% by weight aqueous citric acid and then with brine. The organic layer was dried with MgSO4, and concentrated. Flash chromatography (with CH2Cl2/MeOH=25/1) gave 22 mg of compound 4 as a white solid. MS (M+1): 441.2; 1H-NMR (400 MHz): 8.26, s, 1H, 7.65, m, 2H, 7.42, m, 2H, 7.1-7.2, m, 5H, 6.7-6.9, m, 1H, 6.1, m, 1H, 5.5-5.7, m, 1H, 4.7, m, 1H, 4.54, m, 0.5H, 4.2, m, 1H, 4.1, m, 0.5H, 3.7, m, 0.5H, 3.2, m, 1H, 3.0, m, 0.5H, 2.3, m, 1H, 2.1, m, 1H, 1.9, m, 1H, 1.6, m, 1H.
…………………….
SYNTHESIS
To solve the above problems, the present invention adopts a technical solution is: to provide a tyrosine kinase inhibitor PC1-32765 synthesis method, the reaction steps are as follows:

The beneficial effect of the present invention: The invention relates to a tyrosine kinase inhibitor synthesis of PC1-32765, as the B cell to inhibit the tyrosine kinase receptor signaling key, not only can inhibit the formation of blood cells and less side effects and mild reaction conditions, simple operation, easy purification, low cost, environmentally friendly, suitable for large-scale production.
A tyrosine kinase inhibitor PC1-32765 synthesis method comprising the steps of:
1, the compound 10 and the coupling reaction of compound 15 to give compound 6;
2, the compound 6 obtained by reacting compound 16 with compound 11 in the process, we have chosen a more perfect catalyst;
3, compound 11 to give compound 12 by protecting;
4, selective deprotection of Compound 12 Compound 13; 5, Compound 13 for Compound 17 only attack only remaining position to obtain a very pure compound 14;
6, take off the protecting group to obtain PC1-32765

Wherein the compound can 10,15,16,17 agent or industrial grade reagent compound or the use of methods and techniques related to synthesis.
Example 1 Preparation of Compound 6
Under nitrogen and the 0.1moL 1.5 equivalents of compound 10 Compound 15 and 800mL of dioxane was added to 2L reaction flask, and then 1.5 equivalents of sodium acetate was added and the catalyst PdC12 (PPh3) 2 0.2 equivalents, 50_60 ° C for 5 hours , filtered hot and the filter residue was washed three times with ethanol, the combined filtrate was concentrated to give a solid, rinsed with ethanol to give the pure product 16.2 g, yield 60%
Example 2 Preparation of Compound 6
Under nitrogen and the 0.1moL 1.5 equivalents of compound 10 Compound 15 and 2L 800mL DMF was added to the reaction flask, and then 1.5 equivalents of sodium acetate was added and the catalyst PdCl2 (PhCN) 2 0.2 equivalents, 50_60 ° C for 5 hours, hot filtered, the filter residue was washed three times with ethanol, the combined filtrate was concentrated to give a solid, which was rinsed with ethanol to give pure product 21.5 g, yield 71%.
Example 3 Preparation of Compound 11
The compound 0.1moL 1.2 equivalent of compound 6 and 16, and 2L IOOOmL THF was added to the reaction flask, 1.5 equivalents of cesium carbonate was added, refluxed for 24 hours, after the reaction, most of the solvent was concentrated and the remaining water was poured into a large, precipitated solid was filtered, washed with water to afford compound 36.9 g compound 11, yield 76%, used without further purification.
Example 4 Preparation of Compound 12
The compound will be to 0.1moL 11 and 1.2 equivalent of compound IOOOmL THF trifluoroacetyl chloride and the reaction was added to 2L flask, then triethylamine was added 2.5 ,30-40 0C for 24 hours, after the reaction, the solvent was concentrated, diluted with water, extracted with ethyl acetate, washed with water, saturated sodium chloride each time, and concentrated to obtain the product 50.1 g of ethyl acrylate, 86% yield, used directly in the next reaction.
Example 5 Preparation of Compound 13
The compound 0.1moL 12 and 500mL of methanol and 50mL 6N hydrochloric acid was added to IL reaction flask, stirred at room temperature for 3 hours to complete the reaction quickly, and a solid precipitates, filtered and the solid was washed several times with ethyl acetate, obtain 38.5 g of pure compound 13 in 80% yield.
Example 6 Preparation of Compound 14 ‘
The 0.1moL compound 13 and 1.2 equivalents of acrylic acid chloride was added to 2L of methylene chloride IL reaction flask ,20-40 ° C was added dropwise 1.2 equivalents of triethylamine was added dropwise, at room temperature for 3 hours after the reaction with two chloride extraction and concentrated to give the product 47.7 g, yield 89%. Without further purification.
Example 7 PC1-32765 Preparation
Compound 14 with the 0.1moL 160mL 800mL of methanol and a saturated solution of sodium carbonate small, 50_60 ° C for 5 hours,
After completion of the reaction was diluted with water, concentrated and then extracted with methylene chloride, concentrated to obtain crude product was recrystallized from toluene to give the final product 28.6 g, yield 65%. HPLC purity 98.6%, ee%> 98%.
The present invention relates to a tyrosine kinase inhibitor of the synthesis of PC1-32765, as the B cell to inhibit the tyrosine kinase receptor signaling key, not only can inhibit the formation of blood cells and less side effects, and the reaction conditions gentle, simple operation, easy purification, low cost, environmentally friendly, suitable for large-scale production.
…
Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase
ChemMedChem
Volume 2, Issue 1, pages 58–61, January 15, 2007
http://onlinelibrary.wiley.com/doi/10.1002/cmdc.200600221/full
http://www.wiley-vch.de/contents/jc_2452/2007/z600221_s.pdf
SYN OF COMPD 4
To 101 mg of a known intermediate 2 [WO 2001019829] and 330 mg polymer-bound Triphenylphosphine (polymerlab) in 5 ml THF, 200 mg (2.0 eq.) of 3-OH N-Boc piperidine was added followed by 0.099 ml diisopropyl diazodicarboxylate. The reaction mixture stirred at room temperature overnight. After filtered off resins, the reaction mixture was concentrated and purified with flash chromatography (pentane/ethyl acetate = 1/1) to give 55 mg of intermediate 3. This compound (48.3 mg) was treated with 1 ml of 4N HCl in dioxane for 1 hour and concentrated to dryness, which was dissolved in dichloromethane and 0.042 ml of triethylamine, followed by 0.010 ml of acryl chloride. The reaction was stopped after 2 hours. The reaction mixture was washed with 5wt% citric acid (aq.) and brine, dried with MgSO4, and concentrated. Flash chromatography with (CH2Cl2/MeOH = 25/1) gave 22 mg of compound 4 as white solids. MS (M+1): 441.2; 1H-NMR (400MHz): 8.26, s, 1H; 7.65, m, 2H; 7.42, m, 2H; 7.1-7.2, m, 5H; 6.7-6.9, m, 1H; 6.1, m, 1H; 5.5-5.7, m, 1H; 4.7, m, 1H; 4.54, m, 0.5H; 4.2, m, 1H; 4.1, m, 0.5H; 3.7, m, 0.5H; 3.2, m, 1H; 3.0, m, 0.5H; 2.3, m, 1H; 2.1, m, 1H; 1.9, m, 1H; 1.6, m, 1H
……………..
References
- Statement on a Nonproprietary Name Adopted by the USAN Council
- FDA Press Release
- Azvolinsky, PhD, Anna. “FDA Approves Ibrutinib for Chronic Lymphocytic Leukemia”. Cancer Network. Retrieved 14 February 2014.
- Pan, Z; Scheerens, H; Li, SJ; Schultz, BE; Sprengeler, PA; Burrill, LC; Mendonca, RV; Sweeney, MD; Scott, KC; Grothaus, Paul G.; Jeffery, Douglas A.; Spoerke, Jill M.; Honigberg, Lee A.; Young, Peter R.; Dalrymple, Stacie A.; Palmer, James T. (2007). “Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase”. ChemMedChem 2 (1): 58–61.doi:10.1002/cmdc.200600221. PMID 17154430.
- Celera Genomics Announces Sale of Therapeutic Programs to Pharmacyclics
- United States patent 7514444
- Janssen Biotech, Inc. Announces Collaborative Development and Worldwide License Agreement for Investigational Anti-Cancer Drug, PCI-32765
- Clinical trials involve PCI-32765
- Clinical trials involve ibrutinib
- “Imbruvica (Ibrutinib)”. Medscape Reference. WebMD. Retrieved 13 January 2014.
- “IMBRUVICA (ibrutinib) capsule [Pharmacyclics, Inc]”. DailyMed. Pharmacyclics, Inc. November 2013. Retrieved 13 January 2013.
- Herman SE, Gordon AL, Hertlein E, Ramanunni A, Zhang X, Jaglowski S, Flynn J, Jones J, Blum KA, Buggy J.J., Hamdy A, Johnson AJ, Byrd JC, SE (2011). “Bruton’s tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765”. Blood 117 (23): 6287–6296. doi:10.1182/blood-2011-01-328484. PMC 3122947. PMID 21422473.
- The Bruton’s tyrosine kinase (BTK) inhibitor PCI-32765 (P) in treatment-naive (TN) chronic lymphocytic leukemia (CLL) patients (pts): Interim results of a phase Ib/II study” J Clin Oncol 30, 2012 (suppl; abstr 6507)
- Ponader S, Chen SS, Buggy JJ, Balakrishnan K, Gandhi V, Wierda WG, Keating MJ, O’Brien S, Chiorazzi N, Burger JA (2012). The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo 119. Blood. pp. 1182–1189.
- de Rooij MF, Kuil A, Geest CR, Eldering E, Chang BY, Buggy JJ, Pals ST, Spaargaren M (2012). “The clinically active BTK inhibitor PCI-32765 targets B-cell receptor (BCR)- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia”. Blood 2012: 2590–2594.
- Honigberg, LA; Smith, AM; Sirisawad, M; Verner, E; Loury, D; Chang, B; Li, S; Pan, Z; Thamm, DH; Miller, RA; Buggy, JJ (2010). “The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy”.Proceedings of the National Academy of Sciences of the United States of America 107 (29): 13075–80. doi:10.1073/pnas.1004594107. PMC 2919935. PMID 20615965.
- Chang, BY; Huang, MM; Francesco, M; Chen, J; Sokolove, J; Magadala, P; Robinson, WH; Buggy, JJ (2011). “The Bruton tyrosine kinase inhibitor PCI-32765 ameliorates autoimmune arthritis by inhibition of multiple effector cells”. Arthritis Research & Therapy 13 (4): R115.doi:10.1186/ar3400. PMC 3239353. PMID 21752263.
- http://cancer.osu.edu/mediaroom/releases/Pages/Ohio-State-Cancer-Research-Played-a-Significant-Role-in-FDA-Approval-of-Important-New-CLL-Drug.aspx#sthash.3o9uyt78.dpuf
- BTK inhibitor PCI-32765, National Cancer Institute Drug Dictionary
- Official Imbruvica patient website
MORE
1) E · Werner, L · Honeywell Berg, Z · Pan; Bruton tyrosine kinase inhibitor; drugs circulating Company; filing date: 2006.12.28, open) No. (Notice: CN101610676A CN101610676B, CN101805341A, CN101805341B, CN102746305A, CN102887900A
2) Pan, Z ; Scheerens, H; Li, SJ; Schultz, BE; Sprengeler, PA; Burrill, LC; Mendonca, RV; Sweeney, MD; Scott, KC; Grothaus, Paul G.; Jeffery, Douglas A.; Spoerke, Jill M. ..; Honigberg, Lee A.; Young, Peter R.; Dalrymple, Stacie A.; Palmer, James T. (2007) “Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase” ChemMedChem 2 (1): 58-61 ( article link )
3) Celera Genomics Announces Sale of Therapeutic Programs to Pharmacyclics , April 10, 2006
4) Honigberg; Lee, Verner; Erik, Pan; Zhengying; Inhibitors of Bruton’s tyrosine kinase, U.S. Patent 7,514,444 ; US 20080108636 A1; CA2663116A1, CN101610676B, CN101805341A, CN101805341B, CN102746305A, CN102887900A, EP2081435A2, EP2081435A4, EP2201840A1, EP2201840B1, EP2443929A1, EP2526771A1, EP2526933A2, EP2526933A3, EP2526934A2, EP2526934A3, EP2529621A1, EP2529622A1, EP2530083A1, EP2532234A1, EP2532235A1, US7514444, US7732454, US7825118, US7960396, US8008309, US8088781, US8158786, US8232280, US8236812, US8399470, US8476284, US8497277, US8501751, US8552010, US20080076921, US20080108636, US20080139582, US20090181987, US20100004270, US20100022561, US20100041677, US20100324050, US20100331350, US20110008257, US20110039868, US20110184001, US20110257203, US20110281322, US20120088912, US20120095026, US20120108612, US20120115889, US20120122894, US20120129821, US20120129873, US20120135944, US20120214826, US20120252821, US20120252822, US20120277254, US20120283276, US20120283277, US20130005745, WO2008039218A2, WO2008039218A3
5) Buggy, Joseph J. Chang, Betty Y.; Methods and Compositions . for inhibition of Bone resorption, PCT Int Appl, WO2013003629, 03 Jan 2013.
6) Wei Chen, David J. Loury, Tarak D. Mody; Preparation of pyrazolo-pyrimidine Compounds as Inhibitors of Bruton’s tyrosine kinase; U.S. Patent Number 7,718,662 , 18 May 2010; Also published as CA2776543A1, CN102656173A, EP2393816A2, EP2393816A4, EP2650294A1, US7741330, US20110086866, WO2011046964A2, WO2011046964A3
7) Wei Chen, David J. Loury, Tarak D. Mody; Inhibitors of Bruton’s tyrosine kinase; WO2013010136 A3
8) John C. Byrd, et al;. Targeting BTK with Relapsed Ibrutinib in Chronic Lymphocytic Leukemia; N Engl J Med 2013; 369:32-42
9) Michael L. Wang, MD, et al, Targeting with BTK. Ibrutinib in Refractory or Relapsed Mantle-Cell Lymphoma; N Engl J Med 2013; 369:507-516
| US8236812 | 8-8-2012 | INHIBITORS OF BRUTON’S TYROSINE KINASE |
| US8232280 | 7-32-2012 | INHIBITORS OF BRUTON’S TYROSINE KINASE |
| US8158786 | 4-18-2012 | INHIBITORS OF BRUTON’S TYROSINE KINASE |
| US8088781 | 1-4-2012 | INHIBITORS OF BRUTONS TYROSINE KINASE |
| US2011281322 | 11-18-2011 | INHIBITORS OF BRUTON’S TYROSINE KINASE |
| US2011224235 | 9-16-2011 | INHIBITORS OF BRUTON’S TYROSINE KINASE FOR THE TREATMENT OF SOLID TUMORS |
| US8008309 | 8-31-2011 | INHIBITORS OF BRUTON’S TYROSINE KINASE |
| US7960396 | 6-15-2011 | INHIBITORS OF BRUTON’S TYROSINE KINASE |
| US2011086866 | 4-15-2011 | INHIBITORS OF BRUTON’S TYROSINE KINASE |
| US2011039868 | 2-18-2011 | INHIBITORS OF BRUTON’S TYROSINE KINASE |
| US2010324050 | 12-24-2010 | INHIBITORS OF BRUTON’S TYROSINE KINASE |
| US7825118 | 11-3-2010 | INHIBITORS OF BRUTON’S TYROSINE KINASE |
| US2010254905 | 10-8-2010 | INHIBITORS OF BRUTON’S TYROSINE KINASE |
| US7732454 | 6-9-2010 | INHIBITORS OF BRUTON’S TYROSINE KINASE |
| US2010041677 | 2-19-2010 | INHIBITORS OF BRUTON’S TYROSINE KINASE |
| US2010022561 | 1-29-2010 | INHIBITORS OF BRUTON’S TYROSINE KINASE |
| US7514444 | 4-8-2009 | INHIBITORS OF BRUTON’S TYROSINE KINASE |
| US2008214501 | 9-5-2008 | BRUTON’S TYROSINE KINASE ACTIVITY PROBE AND METHOD OF USING |
| US7718662 | 16 Oct 2009 | 18 May 2010 | Pharmacyclics, Inc. | Pyrazolo-pyrimidine inhibitors of bruton’s tyrosine kinase |
| US7741330 | 16 Oct 2009 | 22 Jun 2010 | Pharmacyclics, Inc. | Pyrazolo-pyrimidine inhibitors of Bruton’s tyrosine kinase |
| US7982036 | 17 Oct 2008 | 19 Jul 2011 | Avila Therapeutics, Inc. | 4,6-disubstitued pyrimidines useful as kinase inhibitors |
| US7989465 | 20 Apr 2009 | 2 Aug 2011 | Avila Therapeutics, Inc. | 4,6-disubstituted pyrimidines useful as kinase inhibitors |
| US8008309 * | 7 Jul 2009 | 30 Aug 2011 | Pharmacyclics, Inc. | Inhibitors of bruton’s tyrosine kinase |
| US8088781 | 20 Jan 2009 | 3 Jan 2012 | Pharmacyclics, Inc. | Inhibitors of brutons tyrosine kinase |
| US8158786 | 15 Jun 2011 | 17 Apr 2012 | Pharmacyclics, Inc. | Inhibitors of Bruton’s tyrosine kinase |
| US8232280 * | 21 Jan 2011 | 31 Jul 2012 | Pharmacyclics, Inc. | Inhibitors of bruton’S tyrosine kinase |
| US8236812 | 21 Sep 2010 | 7 Aug 2012 | Pharmacyclics, Inc. | Inhibitors of bruton’s tyrosine kinase |
| US8329901 | 1 Feb 2011 | 11 Dec 2012 | Celgene Avilomics Research, Inc. | 4,6-disubstitued pyrimidines useful as kinase inhibitors |
| US8338439 | 29 Dec 2009 | 25 Dec 2012 | Celgene Avilomics Research, Inc. | 2,4-disubstituted pyrimidines useful as kinase inhibitors |
| US8377946 | 21 Jun 2012 | 19 Feb 2013 | Pharmacyclics, Inc. | Pyrazolo[3,4-d]pyrimidine and pyrrolo[2,3-d]pyrimidine compounds as kinase inhibitors |
| US8399470 | 30 Jan 2012 | 19 Mar 2013 | Pharmacyclics, Inc. | Inhibitors of bruton’s tyrosine kinase |
| US8445498 | 1 Feb 2011 | 21 May 2013 | Celgene Avilomics Research, Inc. | 4,6-disubstituted pyrimidines useful as kinase inhibitors |
| US8450335 | 26 Jun 2009 | 28 May 2013 | Celgene Avilomics Research, Inc. | 2,4-disubstituted pyrimidines useful as kinase inhibitors |
| US8476284 | 16 Dec 2011 | 2 Jul 2013 | Pharmacyclics, Inc. | Inhibitors of Bruton’s tyrosine kinase |
| US8481733 * | 19 May 2009 | 9 Jul 2013 | OSI Pharmaceuticals, LLC | Substituted imidazopyr- and imidazotri-azines |
| US8497277 | 6 Dec 2011 | 30 Jul 2013 | Pharmacyclics, Inc. | Inhibitors of Bruton’s tyrosine kinase |
| US8501724 | 30 Apr 2012 | 6 Aug 2013 | Pharmacyclics, Inc. | Purinone compounds as kinase inhibitors |
| US8501751 | 7 Jul 2009 | 6 Aug 2013 | Pharmacyclics, Inc. | Inhibitors of Bruton’s tyrosine kinase |
| US8552010 | 18 Jun 2012 | 8 Oct 2013 | Pharmacyclics, Inc. | Inhibitors of Bruton’S tyrosine kinase |
| US8563563 | 30 Jan 2012 | 22 Oct 2013 | Pharmacyclics, Inc. | Inhibitors of bruton’s tyrosine kinase |
| US8563568 | 8 Aug 2011 | 22 Oct 2013 | Celgene Avilomics Research, Inc. | Besylate salt of a BTK inhibitor |
| US8609679 | 7 Nov 2012 | 17 Dec 2013 | Celgene Avilomics Research, Inc. | 2,4-diaminopyrimidines useful as kinase inhibitors |
| US20090286768 * | 19 May 2009 | 19 Nov 2009 | Osi Pharmaceuticals, Inc. | Substituted imidazopyr- and imidazotri-azines |
| US20110184001 * | 21 Jan 2011 | 28 Jul 2011 | Pharmacyclics, Inc. | Inhibitors of bruton’s tyrosine kinase |
| US20120088912 * | 29 Sep 2011 | 12 Apr 2012 | Pharmacyclics, Inc. | Inhibitors of bruton’s tyrosine kinase |
| US20120252822 * | 23 May 2012 | 4 Oct 2012 | Pharmacyclics, Inc. | Inhibitors of bruton’s tyrosine kinase |
| US20120277254 * | 5 Jul 2012 | 1 Nov 2012 | Pharmacyclics, Inc. | Inhibitors of bruton’s tyrosine kinase |
| WO2010123870A1 * | 20 Apr 2010 | 28 Oct 2010 | Avila Therapeutics, Inc. | Heteroaryl compounds and uses thereof |
| WO2011031979A1 * | 10 Sep 2010 | 17 Mar 2011 | Cylene Pharmaceuticals Inc. | Pharmaceutically useful heterocycle-substituted lactams |
| WO2013155347A1 | 11 Apr 2013 | 17 Oct 2013 | Izumi Raquel | Bruton’s tyrosine kinase inhibitors for hematopoietic mobilization |
| WO2014004707A1 | 26 Jun 2013 | 3 Jan 2014 | Principia Biopharma Inc. | Formulations comprising ibrutinib |