











Sofosbuvir
Sovaldi
M.Wt: 529.45
Formula: C22H29FN3O9P
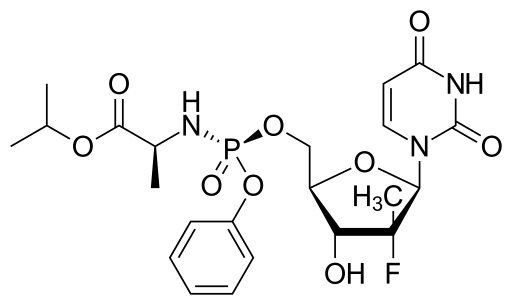
Isopropyl (2S)-2-[[[(2R,3R,4R,5R)-5-(2,4-dioxopyrimidin-1-yl)-4-fluoro-3-hydroxy-4-methyl-tetrahydrofuran-2-yl]methoxy-phenoxy-phosphoryl]amino]propanoate
A prodrug of 2′-deoxy-2′-alpha-F-2′-beta-C-methyluridine 5′-monophosphate.
GS-7977, PSI-7977
- GS 7977
- GS-7977
- PSI 7977
- PSI-7977
- Sofosbuvir
- Sovaldi
- UNII-WJ6CA3ZU8B
CAS Registry Number :1190307 -88-0
http://www.ama-assn.org/resources/doc/usan/sofosbuvir.pdf
Indications: Chronic hepatitis C (HCV GT1, GT2, GT3, GT4)
Mechanism: nucleoside NS5B polymerase inhibitor
approved Time: December 6, 2013
,U.S. Patent Number: 7964580,8415322,8334270,7429572;, patent validity: March 26, 2029 (U.S. Patent No.: 7,964,580 and 8,334,270), April 3, 2025 (U.S. Patent No.: 7,429,572 and 8,415,322)
US patent number 7964580, US patent number 8415322, US patent number 8334270,US patent number 7429572 Patent Expiration Date: March 26, 2029 for US patent number 7964580 and 8334270 (2028 in EU); April 3, 2025 for US patent number 7429572 and 8415322
Sales value (estimated): $ 1.9 billion (2014), 6600000000 USD (2016)
Drug Companies: Gilead Sciences, Inc. (Gilead Sciences)
WASHINGTON, Dec. 6, 2013 (AP) — Federal health officials have approved a highly anticipated hepatitis C drug from Gilead Sciences Inc. that is expected to offer a faster, more palatable cure to millions of people infected with the liver-destroying virus.
The Food and Drug Administration said Friday it approved the pill Sovaldi in combination with older drugs to treat the main forms of hepatitis C that affect U.S. patients.
Current treatments for hepatitis C can take up to a year of therapy and involve weekly injections of a drug that causes flu-like side effects. That approach only cures about three out of four patients. Sovaldi is a daily pill that in clinical trials cured roughly 90 percent of patients in just 12 weeks, when combined with the older drug cocktail.http://www.pharmalive.com/us-approves-breakthrough-hepatitis-c-drug
- The end of October 2013 saw a nod from the FDA given to Gilead’s New Drug Application for Sofosbuvir, a much needed treatment for hepatitis C.
- As a nucleotide analogue, Sofosbuvir is designed as a once daily treatment.
- There are roughly 170 million cases of hepatitis C around the world.
- A report in the Journal of the American Medical Association on August 28, 2013 revealed that the Sofosbuvir and Ribavirin combination treatment effectively cured many patients with the Hepatitis C Virus.
- The Sofosbuvir and Ribavirin drug combination was void of interferon-based treatments, which many patients are resistant too.
- More than 3 million Americans have chronic Hepatitis C Virus, and 22 percent of these patients are African American.
Sofosbuvir (brand names Sovaldi and Virunon) is a drug used for hepatitis C virus (HCV) infection, with a high cure rate.[1][2] It inhibits the RNA polymerase that the hepatitis C virus uses to replicate its RNA. It was discovered at Pharmasset and developed by Gilead Sciences.[3]
Sofosbuvir is a component of the first all-oral, interferon-free regimen approved for treating chronic Hepatitis C.[4]
In 2013, the FDA approved sofosbuvir in combination with ribavirin (RBV) for oral dual therapy of HCV genotypes 2 and 3, and for triple therapy with injected pegylated interferon (pegIFN) and RBV for treatment-naive patients with HCV genotypes 1 and 4.[4] Sofosbuvir treatment regimens last 12 weeks for genotypes 1, 2 and 4, compared to 24 weeks for treatment of genotype 3. The label furhter states that sofosbuvir in combination with ribavirin may be considered for patients infected with genotype 1 who are interferon-ineligible.[5] Sofosbuvir will cost $84,000 for 12 weeks of treatment and $168,000 for the 24 weeks, which some patient advocates have criticized as unaffordable.
Interferon-free therapy for treatment of hepatitis C eliminates the substantial side-effects associated with use of interferon. Up to half of hepatitis C patients cannot tolerate the use of interferon.[6]
Sofosbuvir is a prodrug that is metabolized to the active antiviral agent 2′-deoxy-2′-α-fluoro-β-C-methyluridine-5′-triphosphate.[7] Sofosbuvir is anucleotide analog inhibitor of the hepatitis C virus (HCV) polymerase.[8] The HCV polymerase or NS5B protein is a RNA-dependent RNA polymerase critical for the viral cycle.
The New Drug Application for Sofosbuvir was submitted on April 8, 2013 and received the FDA’s Breakthrough Therapy Designation, which grants priority review status to drug candidates that may offer major treatment advantages over existing options.[9]
On 6th December 2013, the U.S. Food and Drug Administration approved sofosbuvir for the treatment of chronic hepatitis C.[10]
Sofosbuvir is being studied in combination with pegylated interferon and ribavirin, with ribavirin alone, and with other direct-acting antiviral agents.[11][12] It has shown clinical efficacy when used either with pegylated interferon/ribavirin or in interferon-free combinations. In particular, combinations of sofosbuvir with NS5A inhibitors, such as daclatasvir or GS-5885, have shown sustained virological response rates of up to 100% in people infected with HCV.[13]
Data from the ELECTRON trial showed that a dual interferon-free regimen of sofosbuvir plus ribavirin produced a 24-week post-treatment sustained virological response (SVR24) rate of 100% for previously untreated patients with HCV genotypes 2 or 3.[14][15]
Data presented at the 20th Conference on Retroviruses and Opportunistic Infections in March 2013 showed that a triple regimen of sofosbuvir, ledipasvir, and ribavirin produced a 12-week post-treatment sustained virological response (SVR12) rate of 100% for both treatment-naive patients and prior non-responders with HCV genotype 1.[16] Gilead has developed a sofosbuvir + ledipasvir coformulation that is being tested with and without ribavirin.
Sofosbuvir will cost $84,000 for 12 weeks of treatment used for genotype 1 and 2, and $168,000 for the 24 weeks used for genotype 3.[17] This represents a substantial pricing increase from previous treatments consisting of interferon and ribavirin, which cost between $15,000 and $20,000.[18] The price is also significantly higher than that of Johnson & Johnson‘s recently approved drug simeprevir (Olysio), which costs $50,000 and also treats chronic hepatitis C.[18] The high cost of the drug has resulted in a push back from insurance companies and the like, includingExpress Scripts, which has threatened to substitute lower priced competitors, even if those therapies come with a more unfriendly dosing schedule.[18] Other treatments that have recently entered the market have not matched the efficacy of sofosbuvir, however, allowing Gilead to set a higher price until additional competition enters the market.[18] Patient advocates such as Doctors Without Borders have complained about the price, which is particularly difficult for underdeveloped countries to afford.[19]
sofosbuvir
- News: United States to approve potent oral drugs for hepatitis C, Sara Reardon, Nature, 30 October 2013
- Sofia MJ, Bao D, Chang W, Du J, Nagarathnam D, Rachakonda S, Reddy PG, Ross BS, Wang P, Zhang HR, Bansal S, Espiritu C, Keilman M, Lam AM, Steuer HM, Niu C, Otto MJ, Furman PA (October 2010). “Discovery of a β-d-2′-deoxy-2′-α-fluoro-2′-β-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus”. J. Med. Chem. 53 (19): 7202–18.doi:10.1021/jm100863x. PMID 20845908.
- “PSI-7977”. Gilead Sciences.
- Tucker M (December 6, 2013). “FDA Approves ‘Game Changer’ Hepatitis C Drug Sofosbuvir”. Medscape.
- “U.S. Food and Drug Administration Approves Gilead’s Sovaldi™ (Sofosbuvir) for the Treatment of Chronic Hepatitis C – See more at: http://www.gilead.com/news/press-releases/2013/12/us-food-and-drug-administration-approves-gileads-sovaldi-sofosbuvir-for-the-treatment-of-chronic-hepatitis-c#sthash.T9uTbSWK.dpuf”. Gilead. December 6, 2013.
- “Sofosbuvir is safer than interferon for hepatitis C patients, say scientists”. News Medical. April 25, 2013.
- Murakami E, Tolstykh T, Bao H, Niu C, Steuer HM, Bao D, Chang W, Espiritu C, Bansal S, Lam AM, Otto MJ, Sofia MJ, Furman PA (November 2010). “Mechanism of activation of PSI-7851 and its diastereoisomer PSI-7977”. J. Biol. Chem. 285 (45): 34337–47.doi:10.1074/jbc.M110.161802. PMC 2966047. PMID 20801890.
- Alejandro Soza (November 11, 2012). “Sofosbuvir”. Hepaton.
- “FDA Advisory Committee Supports Approval of Gilead’s Sofosbuvir for Chronic Hepatitis C Infection”. Drugs.com. October 25, 2013.
- “FDA approves Sovaldi for chronic hepatitis C”. FDA New Release. U.S. Food and Drug Administration. 2013-12-06.
- Murphy T (November 21, 2011). “Gilead Sciences to buy Pharmasset for $11 billion”.Bloomberg Businessweek.
- Asselah T (January 2014). “Sofosbuvir for the treatment of hepatitis C virus”. Expert Opin Pharmacother 15 (1): 121–30. doi:10.1517/14656566.2014.857656. PMID 24289735.
- “AASLD 2012: Sofosbuvir and daclatasvir dual regimen cures most people with HCV genotypes 1, 2, or 3”. News. European Liver Patients Association. 2012-11-21.
- AASLD: PSI-7977 plus Ribavirin Can Cure Hepatitis C in 12 Weeks without Interferon. Highleyman, L. HIVandHepatitis.com. 8 November 2011.
- Gane EJ, Stedman CA, Hyland RH, Ding X, Svarovskaia E, Symonds WT, Hindes RG, Berrey MM (January 2013). “Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C”.N. Engl. J. Med. 368 (1): 34–44. doi:10.1056/NEJMoa1208953. PMID 23281974.
- CROI 2013: Sofosbuvir + Ledipasvir + Ribavirin Combo for HCV Produces 100% Sustained Response. Highleyman, L. HIVandHepatitis.com. 4 March 2013.
- Campbell T (December 11, 2013). “Gilead’s Sofosbuvir Gets New Name, Price, Headaches”. The Motley Fool.
- Cohen, J. (2013). “Advocates Protest the Cost of a Hepatitis C Cure”. Science 342 (6164): 1302–1303. doi:10.1126/science.342.6164.1302. PMID 24337268. edit
The chemical structure
 approved by the FDA")
GS-7977, (S)-isopropyl 2-(((S)-(((2R,3R,4R,5R)-5-(2,4-dioxo-3,4- dihydropyrimidin^l(2H)-yl)-4-fluoro-3-hydroxy-4-methyltetrahydrofuran-2- yl)methoxy)(phenoxy)phosphoryl)amino)propanoate, available from Gilead Sciences, Inc., is described and claimed in U.S. Patent No. 7,964,580. (See also US 2010/0016251, US 2010/0298257, US 201 1/0251 152 and US 2012/0107278.) GS-7977 has the structure:

GS-7977 can be crystalline or amorphous. Examples of preparing crystalline and amorphous forms of GS-7977 are disclosed in US 2010/0298257 (US 12/783,680) and US 201 1/0251 152 (US 13/076,552),

Commerically available isopropylidine protected D-glyceraldehyde was reacted with (carbethoxyethylidene)triphenylmethylphosphorane gave the chiral pentenoate ester YP-1. Permanganate dihydroxylation of YP-1 in acetone gave the D-isomer diol YP-2. The cyclic sulfate YP-3 was obtained by first making the cyclic sulfite with thionyl chloride and then oxidizing to cyclic sulfate with sodium hypochlorite. Fluorination of YP-3 with triethylamine-trihydrofluoride(TEA-3HF) in the presence of triethylamine, followed by the hydrolysis of sulfate ester in the presence of concentrated HCl provided diol YP-4 which was benzoylated to give ribonolactone YP-5. Reduction of YP-5 with Red-Al followed by chlorination with sulfuryl chloride in the presence of catalytic amount of tetrabutylammonium bromide yielded YP-6. The conversion of YP-6 to benzoyl protected 2′-deoxyl-2′-alpha-F-2′-Beta-C-methylcytidine (YP-7) was achieved by using O-trimethyl silyl-N4-benzoylcytosine and stannic chloride. Preparation of the uridine nucleoside YP-8 was accomplished by first heating benzoyl cytidine YP-7 in acetic acid then treating with methoanolic ammonia to provide YP-8 in 78% yield.
The phosphoramidating reagent YP-9 was obtained by first reacting phenyldichlorophosphate with L-Alanine isopropyl ester hydrochloride and then with pentafluorophenol. Isolation of single Sp diastereomer YP-9 was achieved via crystallization-induced dynamic resolution in the presence of 20% MTBE/hexane at room temperature.
The uridine nucleoside YP-8 was treated with tert-butylmagnesium chloride in dry THF, followed by pentafluorophenyl Sp diastereomer YP-9 to furnish the Isopropyl (2S)-2-[[[(2R,3R,4R,5R)-5-(2,4-dioxopyrimidin-1-yl)-4-fluoro-3-hydroxy-4-methyl-tetrahydrofuran-2-yl]methoxy-phenoxy-phosphoryl]amino]propanoate (Sovaldi, sofosbuvir, GS-7977, PSI-7977)。
…………
US 7429572
US 8415322
US 7964580
US 8334270B
WO 2006012440
WO 2011123668
……………………………………………
In US 20050009737 published Jan. 13, 2005, J. Clark discloses fluoro-nucleoside derivatives that inhibit Hepatitis C Virus (HCV) NS5B polymerase. In particular, 4-amino-1-((2R,3R,4R,5R)-3-fluoro-4-hydroxy-5-hydroxymethyl-3-methyl-tetrahydro-faran-2-yl)-1H-pyrimidin-2-one (18) was a particularly potent inhibitor of HCV polymerase as well as the polymerase of other Flaviviridae.

In WO2006/012440 published Feb. 2, 2006, P. Wang et al disclose processes for the preparation of 18. Introduction of the cytosine is carried out utilizing the Vorbruggen protocol. In US 20060122146 published Jun. 8, 2006, B.-K. Chun et al. disclose and improved procedures for the preparation of the 2-methyl-2-fluoro-lactone 10. In the latter disclosure the nucleobase is glycosylated by reacting with ribofuranosyl acetate which is prepared by reduction of 10 with LiAlH(O-tert-Bu)3 followed by acetylaton of the intermediate lactol which was treated with an O-trimethylsilyl N4-benzoylcytosine in the presence of SnCl4 to afford the O,O,N-tribenzoylated nucleoside.
……………………………………………………………….
http://www.google.nl/patents/US20080139802
The present process as described in SCHEME A and the following examples contain numerous improvements which have resulted in higher yields of the desired nucleoside. The asymmetric hydroxylation of 22 was discovered to be best carried out with sodium permanganate in the presence of ethylene glycol, sodium bicarbonate in acetone which afforded the diol in 60-64% on pilot plant scale. The sodium permanganate procedure avoids introduction of osmium into the process stream. Further more the stereospecific hydroxylation can be accomplished without using an expensive chiral ligand. The requisite olefin is prepared from (1S,2S)-1,2-bis-((R)-2,2-dimethyl-[1,3]dioxolan-4-yl)-ethane-1,2-diol (20) (C. R. Schmid and J. D. Bryant, Org. Syn. 1995 72:6-13) by oxidative cleavage of the diol and treating the resulting aldehyde with 2-(triphenyl-λ5-phosphanylidene)-propionic acid ethyl ester to afford 22.

(i) NaIO4, NaHCO3, DCM; (ii) MeC(═PPh3)CO2Et; (iii) acetone-NaMnO4 (aq), ethylene glycol, NaHCO3, −10 to 0° C.; aq. NaHSO3 (quench); (iv) i-PrOAc, MeCN, TEA, SOCl2; (v) i-PrOAc, MeCN, NaOCl; (vi) TEA-3HF, TEA; (vii) HCl (aq)-BaCl2-aq; (viii) (PhCO)2O, DMAP, MeCN, (ix) RED-AL/TFE (1:1), DCM; (x) SO2Cl2-TBAB, DCM; (xi) 32, SnCl4-PhCl; (xii) MeOH-MeONa
EXAMPLE 3 (2S,3R)-3-[(4R)-2,2-dimethyl-[1,3]dioxolan-4-yl]-2,3-dihydroxy-2-methyl-propionic acid ethyl ester (24)

A suspension of 22 (10 kg, CAS Reg. No. 81997-76-4), ethylene glycol (11.6 kg), solid NaHCO3 (11.8 kg) and acetone (150 L) is cooled to ca.-15° C. A solution of 36% aqueous NaMnO4 (19.5 kg) is charged slowly (over 4 h) to the suspension maintaining reaction temperature at or below −10° C. After stirring for 0.5 h at −10° C., an aliquot of the reaction mixture (ca. 5 mL) is quenched with 25% aqueous sodium bisulfite (ca. 15 mL). A portion of resulting slurry is filtered and submitted for GC analysis to check the progress of the reaction. When the reaction is complete, the reaction mixture is quenched by slow addition (over 40 min) of cooled (ca. 0° C.) 25% aqueous NaHSO3 (60 L). The temperature of the reaction mixture is allowed to reach 4° C. during the quench. CELITE® (ca. 2.5 kg) is then slurried in acetone (8 kg) and added to the dark brown reaction mixture. The resulting slurry is aged at RT to obtain light tan slurry. The slurry is filtered, and the filter cake is washed with acetone (3×39 kg). The combined filtrate is concentrated by vacuum distillation (vacuum approximately 24 inches of Hg; max pot temperature is 32° C.) to remove the acetone. The aqueous concentrate is extracted with EtOAc (3×27 kg), and the combined organic extracts were washed with water (25 L). The organic phase is then concentrated by atmospheric distillation and EtOAc is replaced with toluene. The volume of the batch is adjusted to ca. 20 L. Heptane (62 kg) is added and the batch cooled to ca. 27° C. to initiate crystallization. The batch is then cooled to −10° C. After aging overnight at −10° C., the product is filtered, washed with 10% toluene in heptane and dried at 50° C. under vacuum to afford 6.91 kg (59.5%) of 24 (CARN 81997-76-4) as a white crystalline solid.
EXAMPLE 4 (3R,4R,5R)-3-Fluoro-4-hydroxy-5-hydroxymethyl-3-methyl-dihydro-furan-2-one (10)

steps 1 & 2—A dry, clean vessel was charged with 24 (6.0 kg), isopropyl acetate (28.0 kg), MeCN (3.8 kg) and TEA (5.4 kg). The mixture was cooled to 5-10° C., and thionyl chloride (3.2 kg) was added slowly while cooling the solution to maintain the temperature below 20° C. The mixture was stirred until no starting material was left (GC analysis). The reaction was typically complete within 30 min after addition is complete. To the mixture was added water (9 kg) and after stirring, the mixture was allowed to settle. The aqueous phase was discarded and the organic phase was washed with a mixture of water (8 kg) and saturated NaHCO3 (4 kg) solution. To the remaining organic phase containing 36 was added MeCN (2.5 kg) and solid NaHCO3 (3.1 kg). The resulting slurry was cooled to ca. 10° C. Bleach (NaOCl solution, 6.89 wt % aqueous solution, 52.4 kg, 2 eq.) was added slowly while cooling to maintain temperature below 25° C. The mixture was aged with stirring over 90-120 min at 20-25° C., until the reaction was complete (GC analysis). After completion of the reaction, the mixture was cooled to ca. 10° C. and then quenched with aqueous Na2SO3 solution (15.1% w/w, 21 kg) while cooling to maintain temperature below 20° C. The quenched reaction mixture was filtered through a cartridge filter to remove inorganic solids. The filtrate was allowed to settle, and phases are separated and the aqueous phase is discarded. The organic layer was washed first with a mixture of water (11 kg) and saturated NaHCO3 solution (4.7 kg), then with of saturated NaHCO3 solution (5.1 kg). DIPEA (220 mL) was added to the organic phase and the resulting solution was filtered through CELITE® (bag filter) into a clean drum. The reactor was rinsed with isopropyl acetate (7 kg) and the rinse is transferred to the drum. The organic phase was then concentrated under vacuum (25-28 inches of Hg) while maintaining reactor jacket temperature at 45-50° C. to afford 26 as an oil (˜10 L). Additional DIPEA (280 mL) was added and the vacuum distillation was continued (jacket temperature 50-55° C.) until no more distillate was collected. (batch volume ca. 7 L).
step 3—To the concentrated oil from step 2 containing 26 was added TEA (2.34 kg) and TEA-trihydrofluoride (1.63 kg). The mixture was heated to 85° C. for 2 h. The batch was sampled to monitor the progress of the reaction by GC. After the reaction was complete conc. HCl (2.35 kg) was added to the mixture and the resulting mixture heated to ca. 90° C. (small amount of distillate collected). The reaction mixture was stirred at ca. 90° C. for 30 min and then saturated aqueous BaCl2solution (18.8 kg) was added. The resulting suspension was stirred at about 90° C. for 4 h. The resulting mixture was then azeotropically dried under a vacuum (9-10 inches of Hg) by adding slowly n-propanol (119 kg) while distilling off the azeotropic mixture (internal batch temperature ca. 85-90° C.). To the residual suspension was added toluene (33 kg) and vacuum distillation was continued to distill off residual n-propanol (and traces of water) to a minimum volume to afford 28.
step 4—To the residue from step 3 containing 28 was added MeCN (35 kg) and ca. 15 L was distilled out under atmospheric pressure. The reaction mixture was cooled to ca. 10° C. and then benzoyl chloride (8.27 kg) and DMAP (0.14 kg) are added. TEA (5.84 kg) was added slowly to the reaction mixture while cooling to maintain temperature below 40° C. The batch was aged at ca. 20° C. and the progress of the benzoylation is monitored by HPLC. After completion of the reaction, EtOAc (30 kg) was added to the mixture and the resulting suspension is stirred for about 30 min. The reaction mixture was filtered through a CELITE® pad (using a nutsche filter) to remove inorganic salts. The solid cake was washed with EtOAc (38 kg). The combined filtrate and washes were washed successively with water (38 kg), saturated NaHCO3 solution (40 kg) and saturated brine (44 kg). The organic phase was polish-filtered (through a cartridge filter) and concentrated under modest vacuum to minimum volume. IPA (77 kg) was added to the concentrate and ca. 25 L of distillate was collected under modest vacuum allowing the internal batch temperature to reach ca. 75° C. at the end of the distillation. The remaining solution was then cooled to ca. 5° C. over 5 h and optionally aged overnight. The precipitate was filtered and washed with of cold (ca. 5° C.) IPA (24 kg). The product was dried under vacuum at 60-70° C. to afford 6.63 kg (70.7% theory of 10 which was 98.2% pure by HPLC.
EXAMPLE 1 Benzoic acid 3-benzoyloxy-5-(4-benzoylamino-2-oxo-2H-pyrimidin-1-yl)-4-fluoro-4-methyl-tetrahydro-furan-2-ylmethyl ester (14)

Trifluoroethanol (4.08 kg) is added slowly to a cold solution (−15° C.) of RED-AL® solution (12.53 kg) and toluene (21.3 kg) while maintaining the reaction temperature at or below −10° C. After warming up to RT (ca. 20° C.), the modified RED-AL reagent mixture (30.1 kg out of the 37.6 kg prepared) is added slowly to a pre-cooled solution (−15° C.) of fluorolactone dibenzoate 10 (10 kg) in DCM (94.7 kg) while maintaining reaction temperature at or below −10° C. After reduction of the lactone (monitored by in-process HPLC), a catalytic amount of tetrabutylammonium bromide (90 g) is added to the reaction mixture. Sulfiiryl chloride (11.86 kg) is then added while maintaining reaction temperature at or below 0° C. The reaction mixture is then heated to 40° C. until formation of the chloride is complete (ca. 4 h) or warmed to RT (20-25° C.) and stirred over night (ca. 16 h). The reaction mixture is cooled to about 0° C., and water (100 L) is added cautiously while maintaining reaction temperature at or below 15° C. The reaction mixture is then stirred at RT for ca. 1 h to ensure hydrolytic decomposition of excess sulfuryl chloride and the phases are separated. The organic layer is washed with a dilute solution of citric acid (prepared by dissolving 15.5 kg of citric acid in 85 L of water) and then with dilute KOH solution (prepared by dissolving 15 kg of 50% KOH in 100 L of water). The organic phase is then concentrated and solvents are replaced with chlorobenzene (2×150 kg) via atmospheric replacement distillation. The resulting solution containing 30 is dried azeotropically.
A suspension of N-benzoyl cytosine (8.85 kg), ammonium sulfate (0.07 kg) and hexamethyldisilazane (6.6 kg) in chlorobenzene (52.4 kg) is heated to reflux (ca. 135° C.) and stirred (ca. 1 h) until the mixture becomes a clear solution. The reaction mixture is then concentrated in vacuo to obtain 32 as a syrupy liquid. The anhydrous solution of 30 in chlorobenzene (as prepared) and stannic chloride (28.2 kg) is added to this concentrate. The reaction mixture is maintained at about 70° C. until the desired coupling reaction is complete (ca. 10 h) as determined by in-process HPLC. Upon completion, the reaction mixture is cooled to RT and diluted with DCM (121 kg). This solution is added to a suspension of solid NaHCO3 (47 kg) and CELITE® (9.4 kg) in DCM (100.6 kg). The resulting slurry is cooled to 10-15° C., and water (8.4 kg) is added slowly to quench the reaction mixture. The resulting suspension is very slowly (caution: gas evolution) heated to reflux (ca. 45° C.) and maintained for about 30 min. The slurry is then cooled to ca. 15° C. and filtered. The filter cake is repeatedly reslurried in DCM (4×100 L) and filtered. The combined filtrate is concentrated under atmospheric pressure (the distillate collected in the process is used for reslurrying the filter cake) until the batch temperature rises to about 90° C. and then allowed to cool slowly to about −5° C. The resulting slurry is aged for at least 2 h at −5° C. The precipitated product is filtered and washed with IPA (30 kg+20 kg), and oven-dried in vacuo at about 70° C. to afford 8.8 kg (57.3%) of 1-(2-deoxy-2-fluoro-2-methyl-3-5-O-dibenzoyl-β-D-ribofuranosyl)-N-4-benzoylcytosine (14, CAS Reg No. 817204-32-3) which was 99.3% pure.
EXAMPLE 2 4-Amino-1-(3-fluoro-4-hydroxy-5-hydroxymethyl-3-methyl-tetrahydro-furan-2-yl)-1H-pyrimidin-2-one (18)

A slurry of 14 (14.7 kg) in MeOH (92.6 kg) is treated with catalytic amounts of methanolic sodium methoxide (0.275 kg). The reaction mixture is heated to ca. 50° C. and aged (ca. 1 h) until the hydrolysis is complete. The reaction mixture is quenched by addition of isobutyric acid (0.115 kg). The resulting solution is concentrated under moderate vacuum and then residual solvents are replaced with IPA (80 kg). The batch is distilled to a volume of ca. 50 L. The resulting slurry is heated to ca. 80° C. and then cooled slowly to ca. 5° C. and aged (ca. 2 h). The precipitated product is isolated by filtration, washed with IPA (16.8 kg) and dried in an oven at 70° C. in vacuo to afford 6.26 kg (88.9%) of 18 which assayed at 99.43% pure.
………………………………………………………………………
https://www.google.com/patents/US8334270
EXAMPLE 4 Preparation of 2′-deoxy-2′-fluoro-2′-C-methyluridine

2′-Deoxy-2′-fluoro-2′-C-methylcytidine (1.0 g, 1 eq) (Clark, J., et al., J. Med. Chem., 2005, 48, 5504-5508) was dissolved in 10 ml of anhydrous pyridine and concentrated to dryness in vacuo. The resulting syrup was dissolved in 20 ml of anhydrous pyridine under nitrogen and cooled to 0° C. with stirring. The brown solution was treated with benzoyl chloride (1.63 g, 3 eq) dropwise over 10 min. The ice bath was removed and stirring continued for 1.5 h whereby thin-layer chromatography (TLC) showed no remaining starting material. The mixture was quenched by addition of water (0.5 ml) and concentrated to dryness. The residue was dissolved in 50 mL of dichloromethane (DCM) and washed with saturated NaHCO3 aqueous solution and H2O. The organic phase was dried over NaSO4 and filtered, concentrated to dryness to give N4,3′,5′-tribenzoyl-2′-Deoxy-2′-fluoro-2′-C-methylcytidine (2.0 g, Yield: 91%).
N4,3′,5′-tribenzoyl-2′-Deoxy-2′-fluoro-2′-C-methylcytidine (2.0 g, 1 eq) was refluxed in 80% aqueous AcOH overnight. After cooling and standing at room temperature (15° C.), most of the product precipitated and then was filtered through a sintered funnel. White precipitate was washed with water and co-evaporated with toluene to give a white solid. The filtrate was concentrated and co-evaporated with toluene to give additional product which was washed with water to give a white solid. Combining the two batches of white solid gave 1.50 g of 3′,5′-dibenzoyl-2′-Deoxy-2′-fluoro-2′-C-methyluridine (Yield: 91%).
To a solution of 3′,5′-dibenzoyl-2′-Deoxy-2′-fluoro-2′-C-methyluridine (1.5 g, 1 eq) in MeOH (10 mL) was added a solution of saturated ammonia in MeOH (20 mL). The reaction mixture was stirred at 0° C. for 30 min, and then warmed to room temperature slowly. After the reaction mixture was stirred for another 18 hours, the reaction mixture was evaporated under reduced pressure to give the residue, which was purified by column chromatography to afford pure compound 2′-deoxy-2′-fluoro-2′-C-methyluridine (500 mg, Yield: 60%).
Example numbers 13-54 and 56-66 are prepared using similar procedures described for examples 5-8. The example number, compound identification, and NMR/MS details are shown below:
| entry 25 |
 |
| entry 251H NMR (DMSO-d6) δ 1.13-1.28 (m, 12H), 3.74-3.81 (m, 2H), 3.95-4.08 (m, 1H), 4.20-4.45 (m, 2H), 4.83-4.87 (m, 1H), 5.52-5.58 (m, 1H),5.84-6.15 (m, 3H), 7.17-7.23 (m, 3H), 7.35-7.39 (m, 2H), 7.54-7.57(m, 1H), 11.50 (s. 1H); MS, m/e 530.2 (M + 1)+ | ||||||
…………………………………..
Synthesis of diastereomerically pure nucleotide phosphoramidates.
Ross BS, Reddy PG, Zhang HR, Rachakonda S, Sofia MJ.
J Org Chem. 2011 Oct 21;76(20):8311-9. doi: 10.1021/jo201492m. Epub 2011 Sep 26.
The HCV NS5B nucleoside and non-nucleoside inhibitors.
Membreno FE, Lawitz EJ.
Clin Liver Dis. 2011 Aug;15(3):611-26. doi: 10.1016/j.cld.2011.05.003. Review.
Sofia MJ, Bao D, Chang W, Du J, Nagarathnam D, Rachakonda S, Reddy PG, Ross BS, Wang P, Zhang HR, Bansal S, Espiritu C, Keilman M, Lam AM, Steuer HM, Niu C, Otto MJ, Furman PA.
J Med Chem. 2010 Oct 14;53(19):7202-18. doi: 10.1021/jm100863x.
Mechanism of activation of PSI-7851 and its diastereoisomer PSI-7977.
Murakami E, Tolstykh T, Bao H, Niu C, Steuer HM, Bao D, Chang W, Espiritu C, Bansal S, Lam AM, Otto MJ, Sofia MJ, Furman PA.
J Biol Chem. 2010 Nov 5;285(45):34337-47. doi: 10.1074/jbc.M110.161802. Epub 2010 Aug 26.
Michael J. Sofia,Donghui Bao, Wonsuk Chang, Jinfa Du, Dhanapalan Nagarathnam, Suguna Rachakonda, P. Ganapati Reddy, Bruce S. Ross, Peiyuan Wang, Hai-Ren Zhang, Shalini Bansal, Christine Espiritu, Meg Keilman, Angela M. Lam, Holly M. Micolochick Steuer, Congrong Niu, Michael J. Otto, and Phillip A. Furman; Discovery of a β-D-2’-Deoxy-2’-a-fluoro-2’-β-C-methyluridine Nucleotide Prodrug (PSI-7977) for the Treatment of Hepatitis C Virus; J. Med. Chem. 2010, 53, 7202–7218; Pharmasset, Inc.
Bruce S. Ross, P. Ganapati Reddy , Hai-Ren Zhang , Suguna Rachakonda , and Michael J. Sofia; Synthesis of Diastereomerically Pure Nucleotide Phosphoramidates; J. Org. Chem., 2011, 76 (20), pp 8311–8319; Pharmasset, Inc.
Peiyuan Wang, Byoung-Kwon Chun, Suguna Rachakonda, Jinfa Du, Noshena Khan, Junxing Shi, Wojciech Stec, Darryl Cleary, Bruce S. Ross and Michael J. Sofia; An Efficient and Diastereoselective Synthesis of PSI-6130: A Clinically Efficacious Inhibitor of HCV NS5B Polymerase; J. Org. Chem., 2009, 74 (17), pp 6819–6824;Pharmasset, Inc.
Jeremy L. Clark, Laurent Hollecker, J. Christian Mason, Lieven J. Stuyver, Phillip M. Tharnish, Stefania Lostia, Tamara R. McBrayer, Raymond F. Schinazi, Kyoichi A. Watanabe, Michael J. Otto, Phillip A. Furman, Wojciech J. Stec, Steven E. Patterson, and Krzysztof W. Pankiewicz; Design, Synthesis, and Antiviral Activity of 2‘-Deoxy-2‘-fluoro-2‘-C-methylcytidine, a Potent Inhibitor of Hepatitis C Virus Replication; J. Med. Chem., 2005, 48 (17), pp 5504–5508; Pharmasset, Inc
- Harrison C. Patent battle lines drawn as sofosbuvir gains approval. , Nat Rev Drug Discov , Volume 13 , Issue 1 , 2013 Dec 31
- Traynor K. Sofosbuvir approved for chronic hepatitis C infection. , Am J Health Syst Pharm , Volume 71 , Issue 2 , 2014 Jan 15
- Benhamou Y. HCV F1/F2 patients: treat now or continue to wait. , Liver Int , Volume 34 Suppl 1 , 2014 Feb
- Asselah T. HCV direct-acting antiviral agents: the best interferon-free combinations. , Liver Int , Volume 34 Suppl 1 , 2014 Feb
- Marcellin P. Second-wave IFN-based triple therapy for HCV genotype 1 infection: simeprevir, faldaprevir and sofosbuvir. , Liver Int , Volume 34 Suppl 1 , 2014 Feb
- Corouge M. Treatment of hepatitis C virus genotype 3-infection. , Liver Int , Volume 34 Suppl 1 , 2014 Feb
- Osinusi A. IFNL4-ΔG Genotype is Associated with Slower Viral Clearance in Hepatitis C, Genotype-1 Patients Treated with Sofosbuvir and Ribavirin. , J Infect Dis , 2013 Dec 23
- Alavian SM. Sofosbuvir has come out of the magic box. , Hepat Mon , Volume 13 , Issue 12 , 2013 Dec 16
- Ferguson MC. Sofosbuvir with ribavirin is safe and effective in hepatitis C genotype 1 with unfavourable pretreatment characteristics. , Evid Based Med , 2013 Dec 12
- Hunt S. Minimal Impact of Sofosbuvir and Ribavirin on Health Related Quality of Life in Chronic Hepatitis C (CH-C). , J Hepatol , 2013 Dec 10
SOVALDI is the brand name for sofosbuvir, a nucleotide analog inhibitor of HCV NS5B polymerase.
The IUPAC name for sofosbuvir is (S)-Isopropyl 2-((S)-(((2R,3R,4R,5R)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-4-fluoro-3-hydroxy-4-methyltetrahydrofuran-2-yl)methoxy)-(phenoxy)phosphorylamino)propanoate. It has a molecular formula of C22H29FN3O9P and a molecular weight of 529.45. It has the following structural formula:
 |
Sofosbuvir is a white to off-white crystalline solid with a solubility of ≥ 2 mg/mL across the pH range of 2-7.7 at 37 oC and is slightly soluble in water.
SOVALDI tablets are for oral administration. Each tablet contains 400 mg of sofosbuvir. The tablets include the following inactive ingredients: colloidal silicon dioxide, croscarmellose sodium, magnesium stearate, mannitol, and microcrystalline cellulose. The tablets are film-coated with a coating material containing the following inactive ingredients: polyethylene glycol, polyvinyl alcohol, talc, titanium dioxide, and yellow iron oxide.
DO NOT FORGET TO CLICK
SEE………………….http://orgspectroscopyint.blogspot.in/2015/02/sofosbuvir-visited.html
J. Med. Chem. 2005, 48, 5504.
WO2008045419A1
CN201180017181

(WO2015139602) Sofosbuvir New Patent
In the literature (Journal of Medicinal Chemistry, 2005,48,5504) in order cytidine as a raw material, first selectively protected 3 ‘, 5′-hydroxyl group, and then oxidizing the 2′-hydroxyl to a carbonyl group, and the reaction of methyllithium get the 2’-hydroxyl compound, and then removing the protective group, use benzoyl protected 3 ‘, 5’-hydroxyl group, and then reacted with DAST fluorinated compound, followed by hydrolysis and aminolysis reaction products, such as the following Reaction Scheme. The method of route length, the need to use expensive silicon ether protecting group molecule relatively poor economy; conducting methylation time will generate a non-methyl enantiomer beta bits.

SOFOSBUVIR
NEW PATENT WO2015188782,
(WO2015188782) METHOD FOR PREPARING SOFOSBUVIR
CHIA TAI TIANQING PHARMACEUTICAL GROUP CO., LTD [CN/CN]; No. 8 Julong North Rd., Xinpu District Lianyungang, Jiangsu 222006 (CN)
Sofosbuvir synthesis routes currently used include the following two methods:
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015188782&redirectedID=true

![]()
