Post Doctoral Research Associate in the group of Professor Steven V. Ley working on the synthesis of complex natural products and synthetic methodology.
College Lecturer and Fellow at Murray Edwards College.
Telephone number
01223 336698 (shared)
Zoe grew up on a farm in the small town of Warkworth, New Zealand. After completing her studies she moved to Auckland, New Zealand to attend the University of Auckland where she completed a Bachelor of Science in Medicinal Chemistry then a BSc (Hons) in Medicinal Chemistry under the supervision of Professor Margaret Brimble, working on the synthesis of anti-Helicobacter pylori compounds. She was then funded by a University of Auckland scholarship to carry out Ph.D. research with Professor Brimble into the synthesis of the extremophile natural product berkelic acid. Upon completion of her Ph.D. she was awarded a Newton International Fellowship from the Royal Society to move to the United Kingdom and join the research group of Professor Steven V. Ley in the Department of Chemistry, University of Cambridge. Upon completion of the two year Newton Fellowship, she was then employed as a Post-Doctoral Research Associate to continue working in the Ley group. While in Cambridge, she has been working on the total synthesis of the complex natural products azadirachtin and plantazolicins A and B, in the process developing novel chemistry. In October 2013 Zoe was appointed as a College Lecturer and Fellow at Murray Edwards College.

Teaching
Graduate Lecture Series – Reduction in Organic Chemistry (2 lectures) (2014, 2013)
Senior demonstrator Chemistry II laboratories (2014/2015)
Senior demonstrator Chemistry IB laboratories (2012/2013, 2013/2014)
College Lecturer at Murray Edwards College
Publications
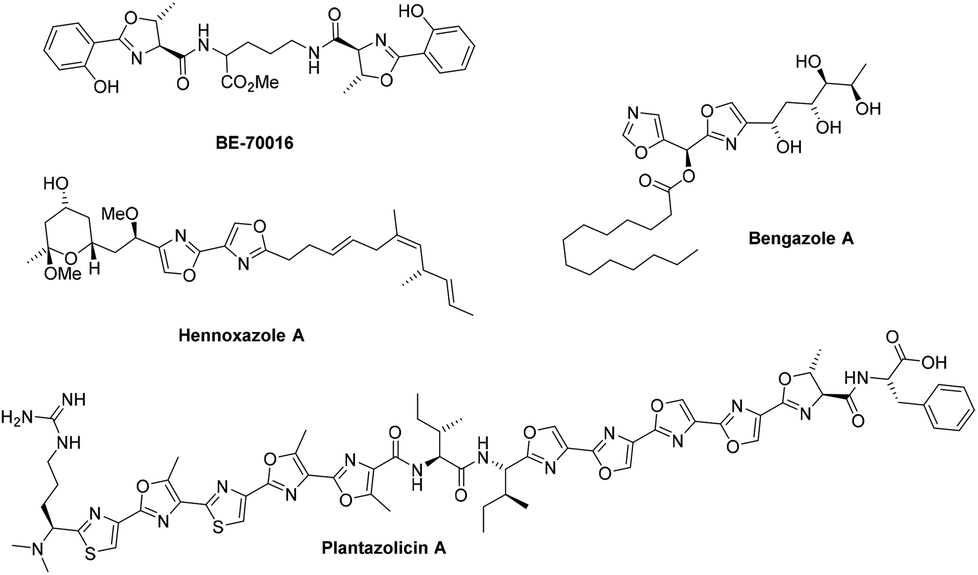
12. Zoe E. Wilson, Sabine Fenner and Steven V. Ley, “Total syntheses of linear poly-thiazole/oxazole plantazolicin A and its biosynthetic precursor plantazolicin B”, Angew. Chem. Int. Ed., 2015, 54, 1284 – 1288 DOI: 10.1002/anie.201410063R1
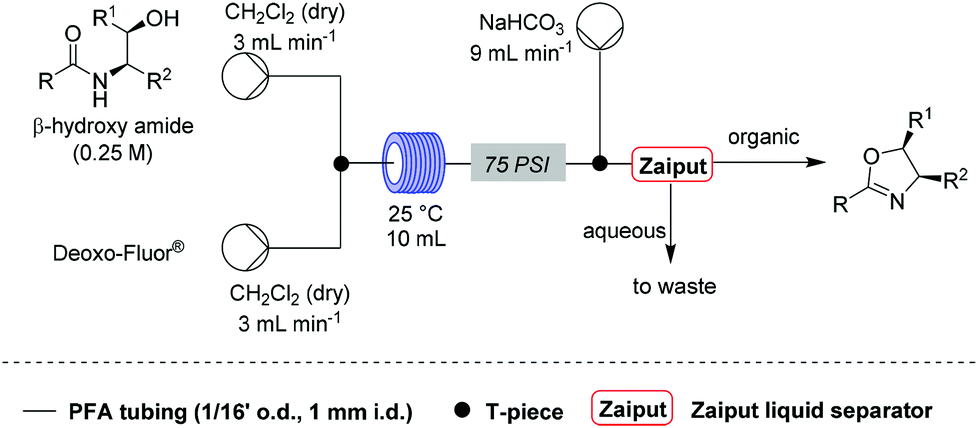
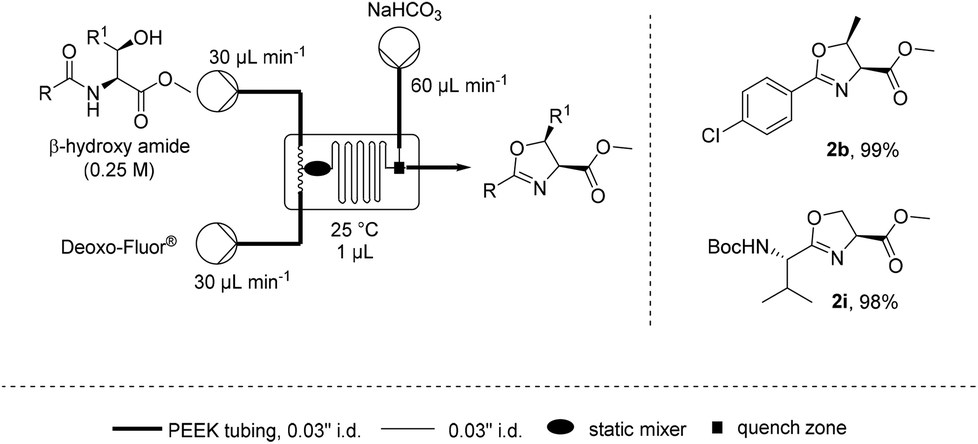
11. Steffen Glöckner, Duc N. Tran, Richard J. Ingham, Sabine Fenner, Zoe E. Wilson, Claudio Battilocchio and Steven V. Ley, “The rapid synthesis of oxazolines and their heterogeneous oxidation to oxazoles under flow conditions”, Org. Biomol. Chem.,2015, 13, 207–214, DOI: 10.1039/c4ob02105c
10. Michael C. McLeod, Zoe E. Wilson and Margaret A. Brimble, “Formal synthesis of berkelic acid: a lesson in α-alkylation chemistry”, J. Org. Chem., 2012, 77, 1, 400–416, DOI: 10.1021/jo201988m
9. Michael C. McLeod, Margaret A. Brimble, Dominea C. K. Rathwell, Zoe E. Wilsonand Tsz-Ying Yuen, “Synthetic approaches to [5,6]-benzannulated spiroketal natural products”, Pure Appl. Chem., 2012, 84, 6, 1379-1390, DOI: 10.1351/PAC-CON-11-08-06
8. Michael C. McLeod, Zoe E. Wilson and Margaret A. Brimble, “An enantioselective formal synthesis of berkelic acid”, Org. Lett., 2011, 13, 19, 5382 – 5385, DOI: 10.1021/ol202265g
7. Zoe E. Wilson, Jonathan G. Hubert, Margaret A. Brimble, “A flexible approach to 6,5-benzannulated spiroketals”, Eur. J. Org. Chem., 2011, 3938-3945, DOI: 10.1002/ejoc.201100345
6. Jonathan Sperry, Yen-Cheng (William) Liu, Zoe E. Wilson, Jonathan G. Hubert, Margaret A. Brimble, “Synthesis of benzannulated spiroketals using an oxidative radical cyclization”, Synthesis, 2011, 9, 1383-1398, DOI: 10.1055/s-003001259981
5. Jonathan Sperry, Zoe E. Wilson, Dominea C. K. Rathwell and Margaret A. Brimble, “Isolation, biological activity and synthesis of benzannulated spiroketal natural products”, Nat. Prod. Rep., 2010, 27, 1117-1137, DOI: 10.1039/b911514p
4. Zoe E. Wilson and Margaret A. Brimble, “A flexible asymmetric synthesis of the tetracyclic core of berkelic acid using a novel Horner-Wadsworth-Emmons/oxa-Michael cascade”, Org. Biomol. Chem., 2010, 8, 1284-1286, DOI: 10.1039/B927219B
3. Zoe E. Wilson and Margaret A. Brimble, “Molecules derived from the extremes of life”, Nat. Prod. Rep., 2009, 26, 44–71, DOI: 10.1039/b800164m
Featured as an Instant insight article in Chemical Biology (“Life at the extremes”,Chemical Biology, 2008, 3, B95) and featured on the cover of the issue (Nat. Prod. Rep.,2009, 26, 1-2, DOI: 10.1039/B821737H)
2. Fiona J. Radcliff, John D. Fraser, Zoe E. Wilson, Amanda M. Heapy, James E. Robinson, Christina J. Bryant, Christopher L. Flowers, and Margaret A. Brimble, “Anti-Helicobacter pylori activity of derivatives of the phthalide-containing antibacterial agents spirolaxine methyl ether, CJ-12,954, CJ-13,013, CJ-13,102, CJ-13,104, CJ-13,108 and CJ-13,015”, Bioorg. Med. Chem., 2008, 16, 6179–6185, DOI: 10.1016/j.bmc.2008.04.037
1. Zoe E. Wilson, Amanda M. Heapy and Margaret A. Brimble, “Synthesis of indole analogues of the anti-Helicobacter pylori compounds CJ-13,015, CJ-13,102, CJ-13,104 and CJ-13,108”, Tetrahedron, 2007, 63, 5379–5385, DOI: 10.1016/j.tet.2007.04.067

















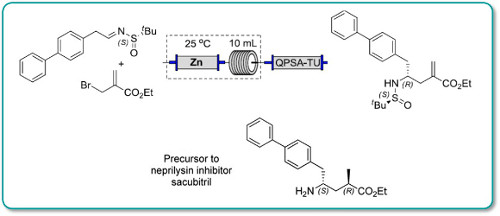


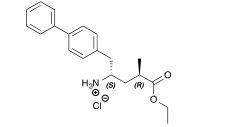
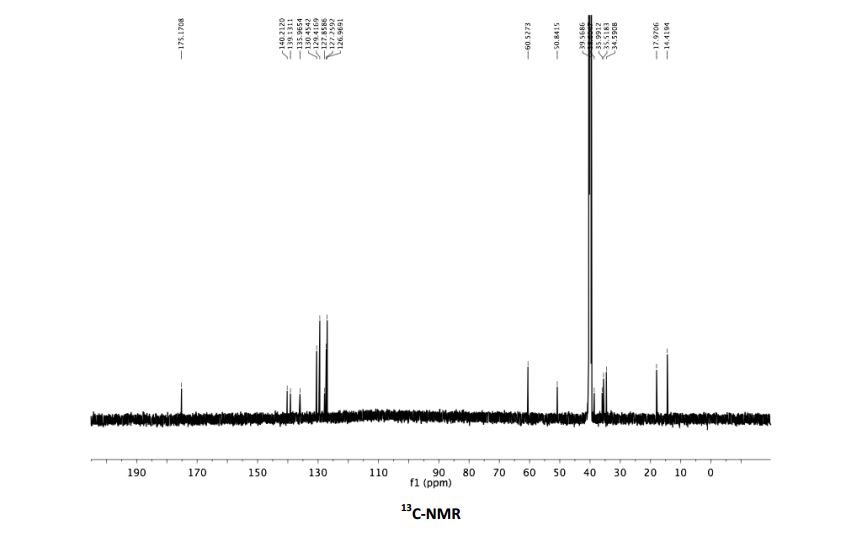
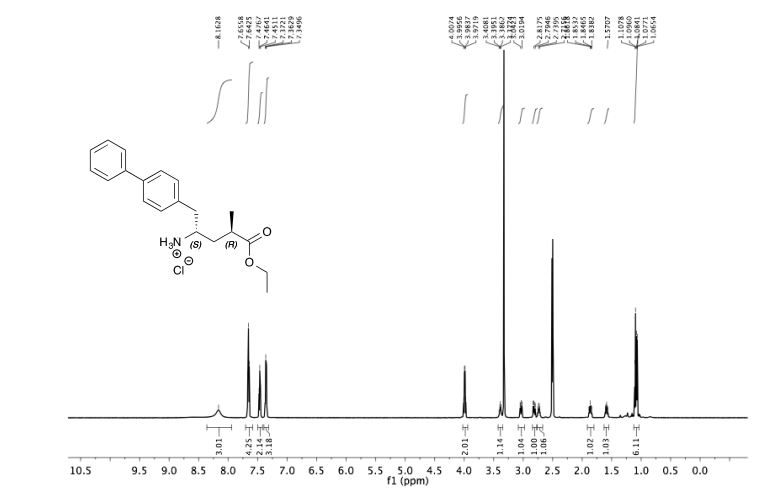





![[1860-5397-11-134-i11]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-11-134-i11.png?scale=2.0&max-width=1024&background=FFFFFF)




![[1860-5397-11-134-i8]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-11-134-i8.png?scale=2.0&max-width=1024&background=FFFFFF)



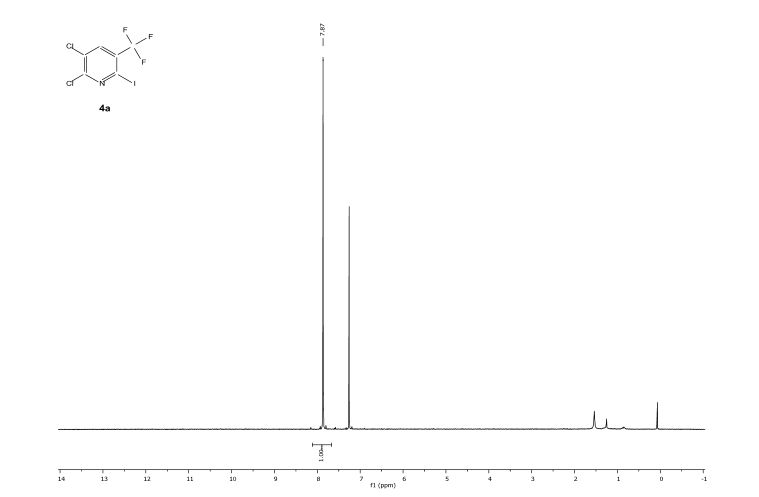
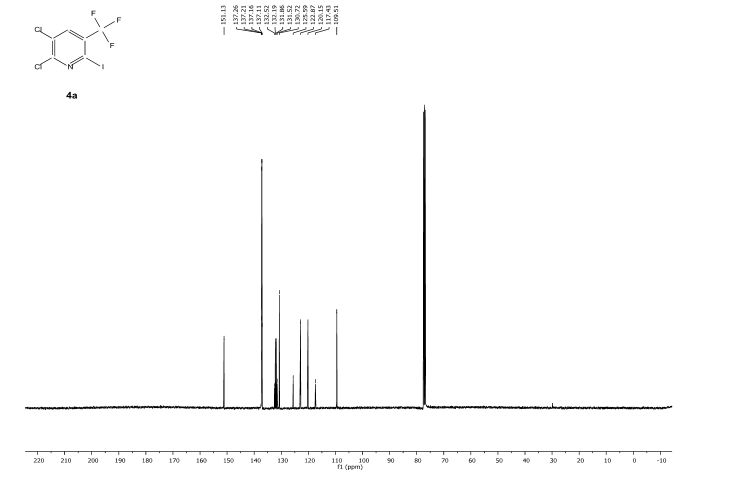




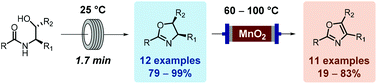
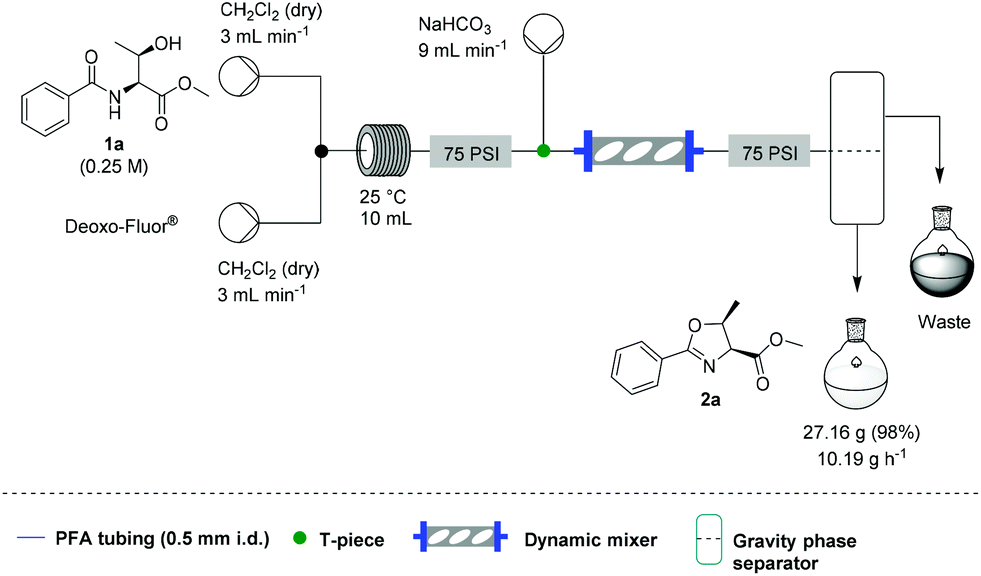
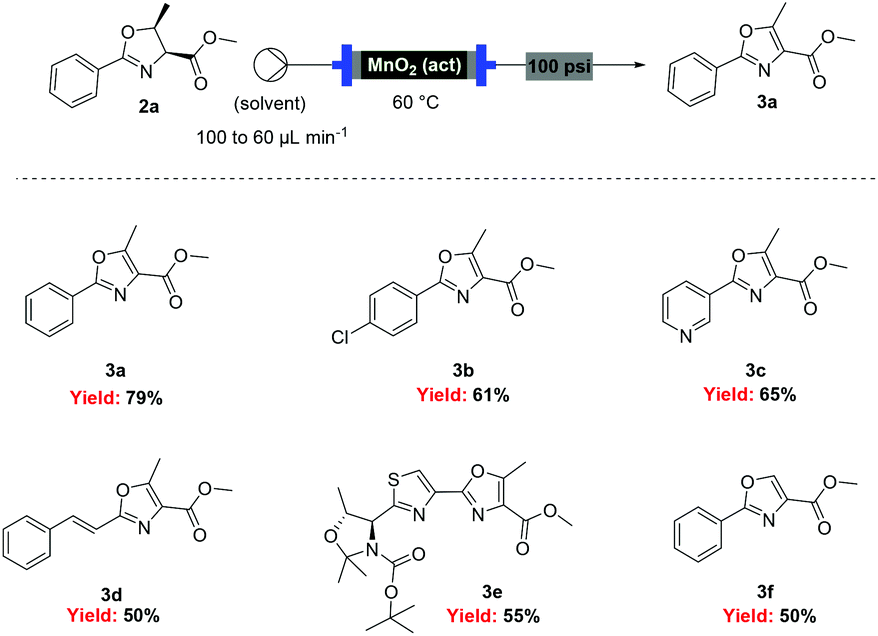
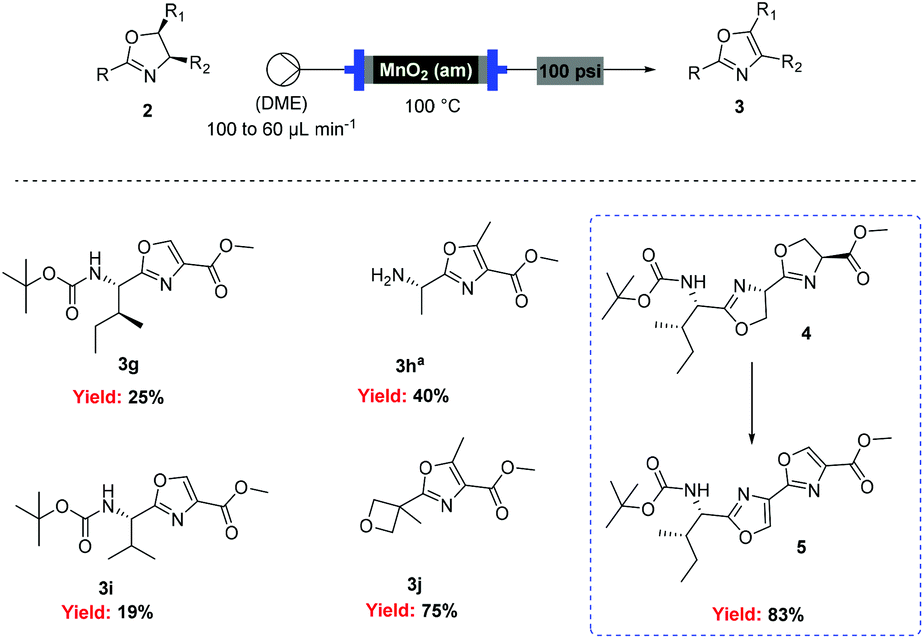
![[thin space (1/6-em)]](http://www.rsc.org/images/entities/char_2009.gif) Deprotection was observed.
Deprotection was observed.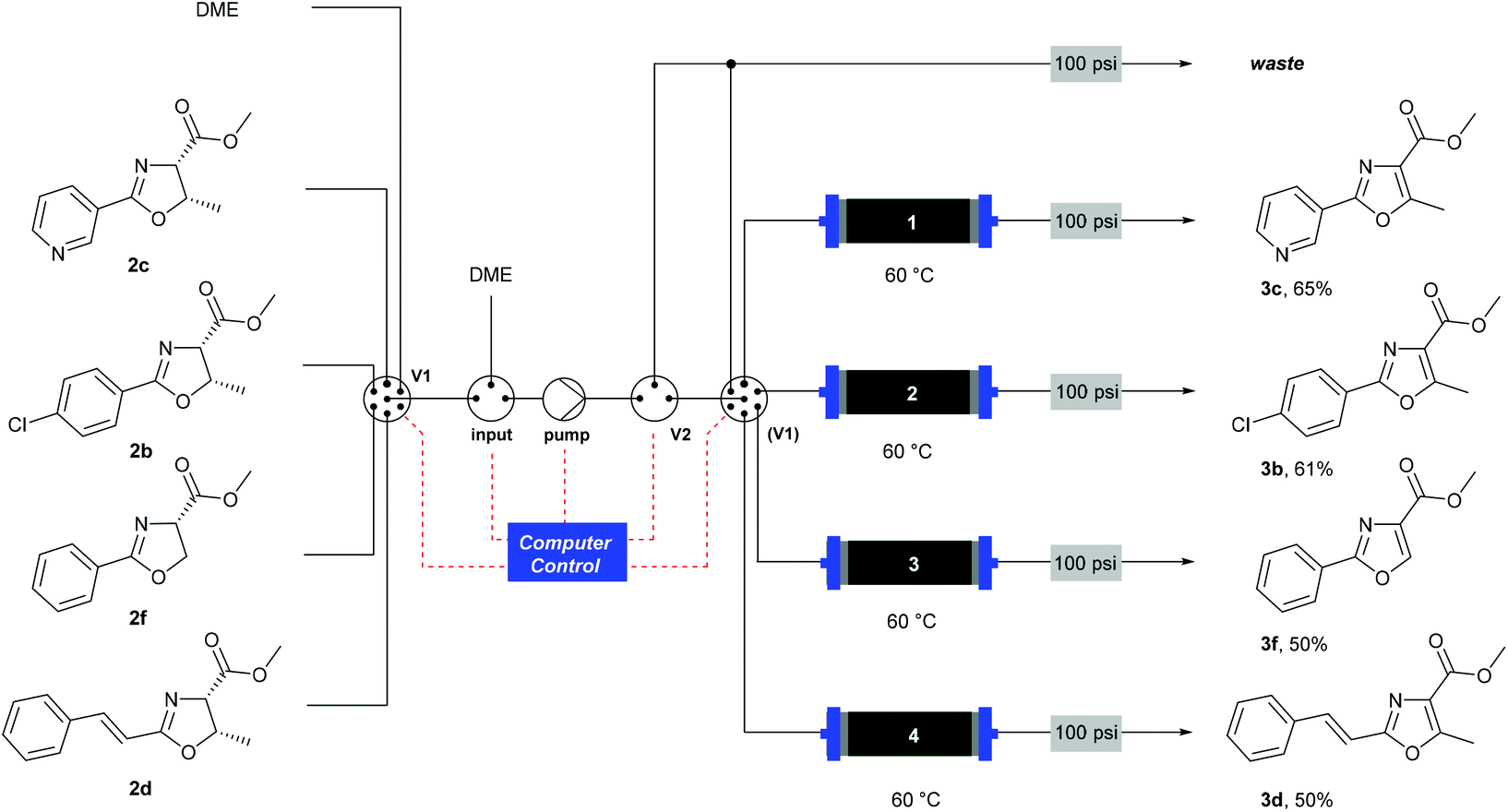
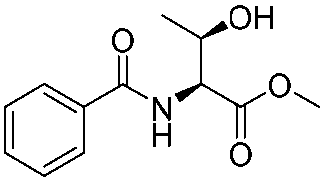
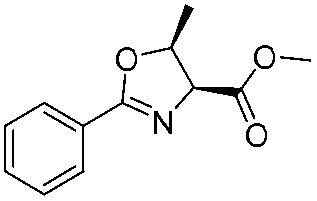
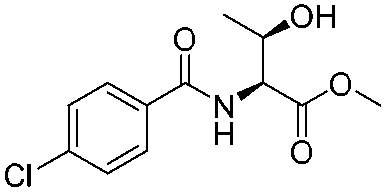
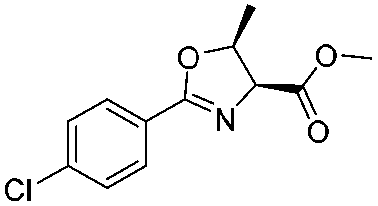
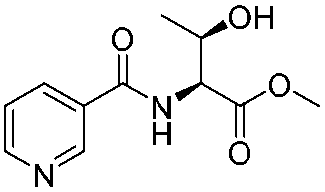
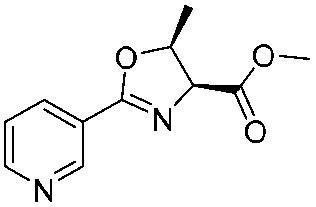
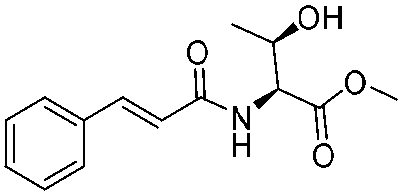
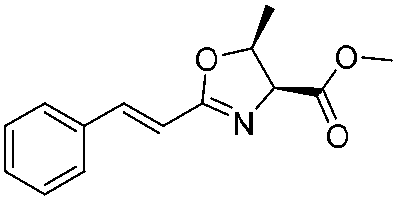
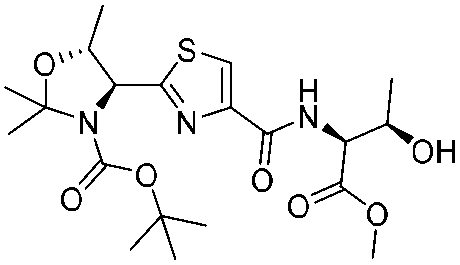
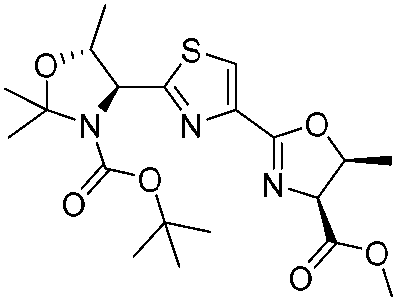
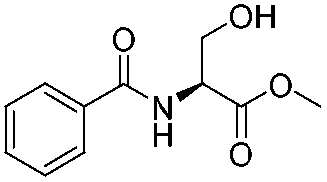
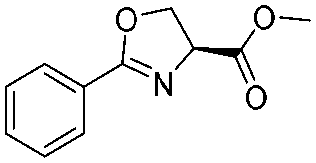
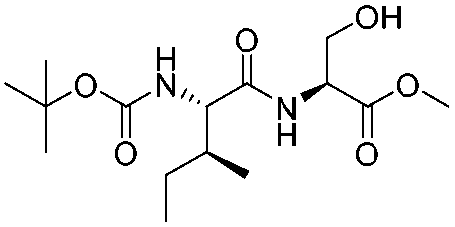
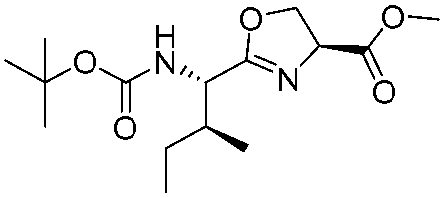
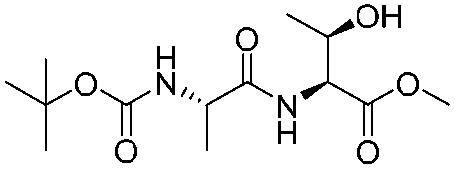
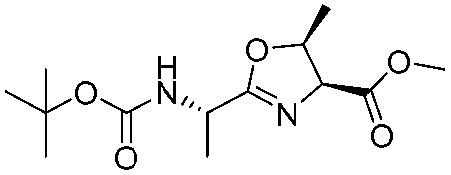
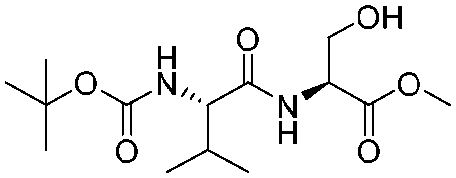
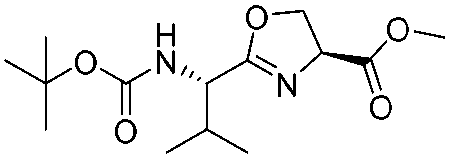
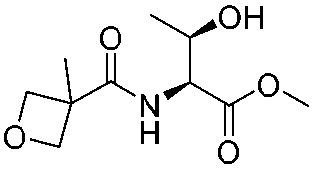
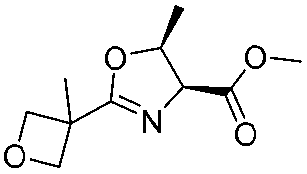
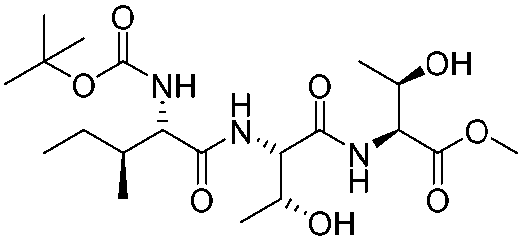
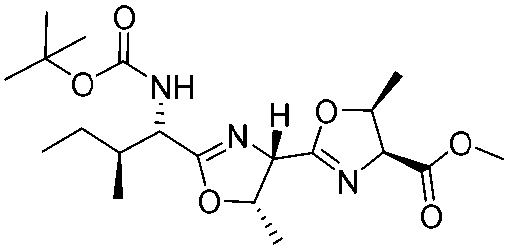
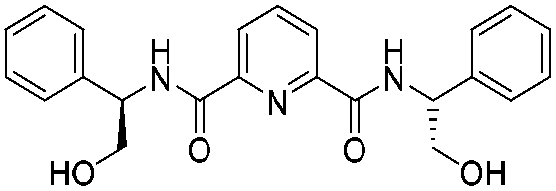
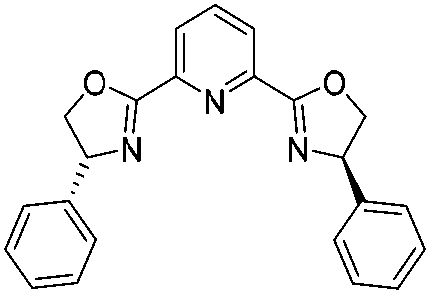


![[small nu, Greek, tilde]](http://www.rsc.org/images/entities/i_char_e0e1.gif) (cm−1) = 2953, 1736, 1645, 1603, 1580, 1496, 1450, 1384, 1349, 1244, 1197, 1174, 1067, 1045, 1001, 973, 934, 904, 886, 851, 778, 695;
(cm−1) = 2953, 1736, 1645, 1603, 1580, 1496, 1450, 1384, 1349, 1244, 1197, 1174, 1067, 1045, 1001, 973, 934, 904, 886, 851, 778, 695; 



 LinkedIn
LinkedIn Facebook
Facebook Twitter
Twitter GooglePlus
GooglePlus