













Regulatory Approval Pathways: EU vs US

Drug Authorization Procedures in the EU
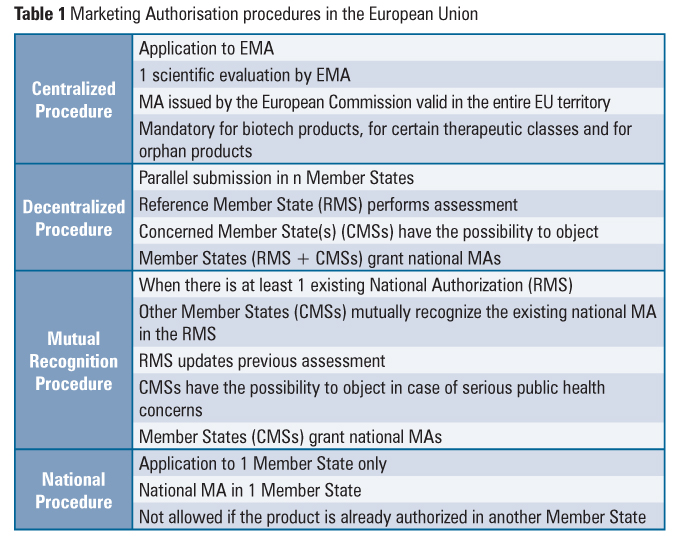
Sponsors have several options when seeking market approval for a new drug in Europe: a national authorization procedure, a decentralized procedure, a mutual recognition procedure and a centralized procedure. Depending on a product’s eligibility, each of these authorization routes offers different advantages and disadvantages to the sponsor, and these should be considered when setting up the market strategy of a product.
National Procedure
This procedure is used whenever a company wants to commercialize a product in only one EU Member State.
The National procedure is specific to each country. That is, each country within the EU has its own procedures for authorizing a marketing application for a new drug. Sponsors can find information regarding the requirements and procedure of each country on the websites of the regulatory agencies.
ADVANTAGES of National Procedure
There are some advantages in submitting a MAA through this procedure. First, it allows the sponsor to choose which country the company will submit to first. This is especially advantageous when the sponsor can’t afford to go through the centralized or decentralized procedure, due to lack of resources of distribution infrastructure for example. Choosing the country that the sponsor is most familiar with in regards to its regulation can also be an important factor. The national authorization procedure also allows the sponsor to, further down the line, get his drug approved through the mutual recognition procedure, seeing as one country already approved its drug. Overall, this procedure is less resource heavy than the others, and thus it is the cheapest and safest alternative for a sponsor.
DISADVANTAGES of National Procedure
The disadvantages are obvious, seeing as this procedure only allows the sponsor to commercialize in one single market, cutting potential revenue streams it could have by bringing the drug to more markets.
Centralized procedure
The centralized procedure is a Europe wide authorization procedure, conducted by EMA’s Committee for Human Medicinal Products (CHMP), an organization which has representatives of all Member states, EEA members, patient organizations and health professionals.
When a sponsor applies for drug approval through the Centralized Procedure, two member states are first selected, a rapporteur and a co-rapporteur. These two member states will be responsible for the creation of an evaluation report that will be assessed by the CHMP. First, a draft report is prepared and sent to the committee for review. The committee prepares a set of questions to send to the sponsor. After receiving a response, further discussions continue and a final evaluation report is arranged, containing a positive or negative opinion. This whole process can take up to 210 days. After the report is completed, it is sent to the European Commission in less than 15 days. The European Commission has the final say on the matter, granting the MA or not after evaluation of the CHMP’s report. The EC’s decision is applicable to all Member States of the European Union and EEA states – Iceland, Norway e Liechtenstein. After approval from the EC, the MA is valid for five years.
The centralized procedure, when it was introduced by Regulation (EEC) no 2309/93, followed the footsteps first established by Directive 87/22/EEC with its concertation procedure , and it was first made obligatory to products made from Recombinant DNA technology, controlled gene expression and monoclonal antibodies.
Afterwards, Regulation (EC) No 726/2004 extended the scope of the procedure to include orphan medicinal products and new active substances for the treatment of acquired immune deficiency syndrome (HIV), cancer, neurodegenerative disorder or diabetes. It went into force in 20th November 2005.
Recital 8 and Point 3 of the Annex to Regulation (EC) No 726/2004 also established that, starting 20 May 2008, the centralized procedure would be obligatory for drug products containing new active substances for the treatment of autoimmune diseases and other immune dysfunctions and viral diseases.
Lastly, regulation EC No 1394/2007 made the procedure compulsory for Advanced Therapy Medicinal products, like gene therapy, tissue engineered and somatic cell therapy products.
Article 3(2) of Regulation (EC) No 726/2004 defines the optional scope of the centralized procedure. It states that the procedure can be followed optionally by medicines that contain a new active substance, or if the applicant shows that the therapeutic entity provides a significant therapeutic, scientific or technical innovation, and it would be in the best interest of public health if it was approved at a community level.
ADVANTAGES of Centralized Procedure
Products authorized through the centralized procedure are granted marketing authorizations that cover all EU member states and the EEA, a big, 500 million user market where the sponsor can potentially recoup the losses from drug development. The drug will be commercialized in all countries with a single, unique brand name.
The convenience of the centralized procedure is however accompanied by fees that are significantly higher than the national procedure’s.
DISADVANTAGES of Centralized Procedure
Also, it is also a very risky, all or nothing procedure. If the CHMP refuses an application, the drug is barred from sale in every EU country, whereas if the sponsor tried another authorization procedure, there was the possibility of getting approval in at least one country. Since the sponsor can’t choose the rapporteur countries like he can in other procedures, this also leaves him at a disadvantage.
Mutual Recognition Procedure
This procedure requires the drug to be already approved in a MS.
This procedure is based upon the principle that a marketing authorization and the evaluation in one Member State (the so-called reference Member State) ought to be recognized by the competent authorities of the other Member States (the so-called concerned Member States), that is, if a Member State concedes a national MA to a drug, other Member States can recognize the evaluation conducted by it and grant a MA for the drug themselves.
It’s also noteworthy to point out that both a Member State and the Sponsor can trigger the Mutual Recognition Procedure.
After the first marketing authorization in the Community is granted, the marketing authorization holder may request one or more Member State(s) to recognize an authorization approved by the reference Member State, by submitting an application in accordance with Article 28 of Directive 2001/83/EC.
Within 90 days of receipt of a valid application, the reference Member State will provide the assessment report together with the approved summary of product characteristics, labeling and package leaflet to the concerned Member States and to the marketing authorization holder.
Within 90 days of the receipt of these documents, the concerned Member States shall recognize the decision of the reference Member State and the approved summary of product characteristics, package leaflet and labeling by granting a MA.
If any country refuses to grant a MA by safety reasons, the matter will be taken to The Co-ordination Group for Mutual Recognition and Decentralized Procedures, which will attempt to make all member states reach a consensus in 60 days. If it fails, the request will be taken to the CHMP and treated like a centralized procedure.
Decentralized procedure
The decentralized procedure works in a similar way as the mutual recognition one, except here the medicinal product in question has not yet received a marketing authorization in any Member State at the time of application. Like the MRP, a reference member state is chosen, which will evaluate the MAA. The remaining member states then proceed to give their opinion on the evaluation. If all concerned member states agree on the evaluation by the reference member state, the drug will be approved and allowed for sale in those countries. If a member state disagrees, the Co-ordination Group for Mutual Recognition and Decentralized Procedures will, like in the MRP, play a referee role.
ADVANTAGES and DISADVANTAGES of MRP & Decentralized Procedure
Both the MRP and the decentralized procedure carry a set of advantages and disadvantages that sponsors ought to know before setting their product market strategy. Both of them allow a sponsor to avoid the need to go through different national procedures in each country. Moreover, they aren’t as risky as the centralized procedure, and, in the case of the MRP, the sponsor can choose the reference member state that will conduct the evaluation of the drug product (by first attaining a MA in that country). In both these procedures, fees have to be paid to all Member states who participate in the process, and, unlike the centralized procedure, the sponsor may have to attribute a different name for its drug product in different Member States., which may hurt brand awareness.
The MRP often sees disagreements between member states, holding up the procedure and causing delays. In these occasions, a lengthy dispute solving mechanism has to be employed, costing both time and money to the sponsor
The decentralized procedure avoids some of the potential disputes between member states by engaging each of the member states the applicant wishes to apply to at the time the first marketing authorization is made. Disputes are this less common in the decentralized procedure than in the MRP. Lastly, the decentralized procedure is faster than the MRP. The first can take up to 210 days to complete its two steps. The MRP, on the other hand, a national MA is first needed, which can take up to 210 days, alongside the update period of the MA license before the MRP procedure starts proper, which can take more 180 days. The take home message is that there is no one-size fits all in regards to drug authorization procedures. Each one of the four available has different advantages and disadvantages, which have to be carefully weighed out by the sponsor.


Drug Approval Process for the US
http://www.jpsr.pharmainfo.in/Documents/Volumes/vol5issue06/jpsr05061302.pdf
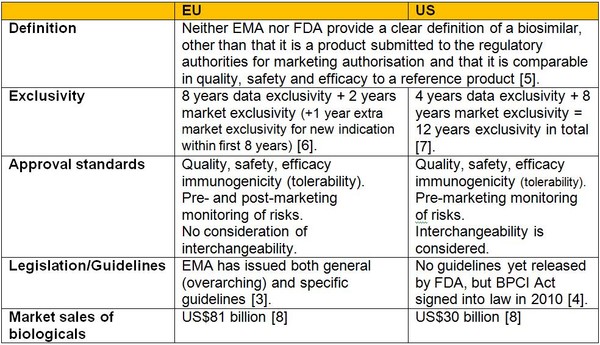
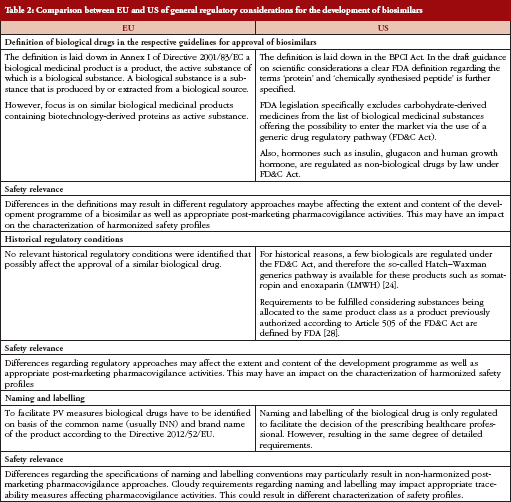
Types of Applications Submitted to the US FDA for New Medicines/Treatments
Investigational New Drug (IND) – Federal law requires that a drug be the subject of an approved marketing application before it is transported or distributed across state lines.
New Drug Application (NDA) – When the sponsor of a new drug believes that enough evidence on the drug’s safety and effectiveness has been obtained to meet FDA’s requirements for marketing approval, the sponsor submits a new drug application (NDA) to FDA. The application must contain data from specific technical viewpoints for review, including chemistry, pharmacology, medical, biopharmaceutics, and statistics. If the NDA is approved, the product may be marketed in the United States.
Biologic License Application (BLA) – Biological products are approved for marketing under the provisions of the Public Health Service Act. The Act requires a firm who manufactures a biologic for sale in interstate commerce to hold a license for the product. A biologics license application is a submission that contains specific information on the manufacturing processes, chemistry, pharmacology, clinical pharmacology and the medical effects of the biologic product. If the information provided meets FDA requirements, the application is approved and a license is issued allowing the firm to market the product.
US Drug Approval Process
If an IND drug survives the clinical trials (phase 1-3), an NDA is submitted to the FDA. An NDA contains all the preclinical and clinical information obtained during the testing phase. The application contains information on the chemical makeup and manufacturing process, pharmacology and toxicity of the compound, human pharmacokinetics, results of the clinical trials, and proposed labeling. An NDA can include experience with the medication from outside the United States as well as external studies related to the drug.
After receiving an NDA, the FDA completes an independent review and makes its recommendations. The Prescription Drug User Fee Act of 1992 (PDUFA) was designed to help shorten the review time. This act allowed the agency to collect user fees from pharmaceutical companies as financial support to enhance the review process. The 1992 Prescription Drug User Fee Act (PDUFA) established a two-tiered system – Standard Review and Priority Review.
Standard Review is applied to a drug that offers at most, only minor improvement over existing marketed therapies. The 2002 amendments to PDUFA set a 10 month goal for a standard review.
Priority Review designation is given to drugs that offer major advances in treatment, or provide a treatment where none existed. The goal for completing a Priority Review is six months.
If during the review the FDA staff feels there is a need for additional information or corrections, they will make a written request to the applicant. During the review process it is not unusual for the FDA to interact with the applicant staff.
The following four FDA programs are intended to facilitate and expedite development and review of new drugs to address unmet medical need in the treatment of a serious or life-threatening3 condition: fast track designation, breakthrough therapy designation, accelerated approval, and priority review designation.
Drug development in the fast lane: FDA approaches to expedited approval.
Fast track designation applies to the drug (either alone or in combination with other drugs) and the specific use for which it is being studied. The term drugrefers to the combination of two or more drugs if the combination is the subject of the fast track designation or request. Where appropriate, FDA may grant designation to the development of a new use of an approved drug.
- Serious Condition
- Demonstrating the Potential to Address Unmet Medical Need
The type of information needed to demonstrate the potential of a drug to address an unmet medical need will depend on the stage of drug development at which fast track designation is requested. Early in development, evidence of activity in a nonclinical model, a mechanistic rationale, or pharmacologic data could be used to demonstrate such potential. Later in development, available clinical data should demonstrate the potential to address an unmet medical need.
BREAKTHROUGH Therapy Designation
Section 506(a) of the FD&C Act provides for designation of a drug as a breakthrough therapy “. . . if the drug is intended, alone or in combination with 1 or more other drugs, to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on 1 or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development.” It is important to recognize that the standard for breakthrough therapy designation is not the same as the standard for drug approval. The clinical evidence needed to support breakthrough designation is preliminary. In contrast, as is the case for all drugs, FDA will review the full data submitted to support approval of drugs designated as breakthrough therapies to determine whether the drugs are safe and effective for their intended use before they are approved for marketing.
ACCELERATED APPROVAL
The accelerated approval provisions of FDASIA in section 506(c) of the FD&C Act provide that FDA may grant accelerated approval to:
. . . a product for a serious or life-threatening disease or condition . . . upon a determination that the product has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments.
For drugs granted accelerated approval, post marketing confirmatory trials have been required to verify and describe the anticipated effect on IMM or other clinical benefit
Post marketing surveillance is important, because even the most well-designed phase 3 studies might not uncover every problem that could become apparent once a product is widely used. Furthermore, the new product might be more widely used by groups that might not have been well studied in the clinical trials, such as elderly patients. A crucial element in this process is that physicians report any untoward complications. The FDA has set up a medical reporting program called Medwatch to track serious adverse events (1-800-FDA-1088). The manufacturer must report adverse drug reactions at quarterly intervals for the first 3 years after approval, including a special report for any serious and unexpected adverse reactions
Regulatory Links for the US FDA Guidances
Guidance for Industry -Expedited Programs for Serious Conditions – Drugs and Biologics, May 2014
http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/default.htm
Good Review Practice: Refuse to File, available on the Internet at http://www.fda.gov/downloads/aboutfda/centersoffices/officeofmedicalproductsandtobacco/cder/manualofpoliciesprocedures/ucm370948.htm and CBER SOPP 8404, Refusal to File Procedures for Biologic License Applications (August 27, 2007), available on the Internet athttp://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/ProceduresSOPPs/ucm073474.htm.
Regulatory Links for the EU:
Directive 2001/20/EC of the European Parliament and of the Council of 4 April2001 on the approximation of the laws, regulations and administrative provisions of the MS relating to the implementation of good clinical practice in the conduct of clinical trials on medicinal products for human use. http://eur-lex.europa.eu/LexUriServ/LexUriServ.douri=OJ:L:2001:121:0034:0044:en:PDF
Detailed guidance on the request to the competent authorities for authorization of a clinical trial on a medicinal product for human use, the notification of substantial amendments and the declaration of the end of the trial (CT-1) (2010/C 82/01) http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:C:2010:082:0001:0019:
EFPIA: Status of the implementation of the European Union Clinical Trials
Directive at member state level, Circular N° 12.784 , June 2008
Klingmann I et al. Impact on Clinical Research of European Legislation. Final report, February 2009http://www.efgcp.be/downloads/icrel_docs/Final_report_ICREL.pdf
Assessment of the functioning of the “Clinical Trials Directive” 2001/20/EC, Public Consultation Paper, ENTR/F/2/SF D(2009) 32674http://ec.europa.eu/enterprise/sectors/pharmaceuticals/files/clinicaltrials/docs/2009_ 10_09_public-consultation-paper.pdf
Report of the multidisciplinary workshop on “A single CTA in multinational clinical trials – dream or option?”, Brussels, Belgium, 7 July 2009http://www.efgcp.be/Conference_details.asp?id=265&L1=10&L2=2&TimeRef=2
Clinical Trials Facilitation Groups, Guidance document for a VoluntaryHarmonization Procedure (VHP) for the assessment of multinational Clinical Trial Applications, Version 2 ; Doc.ref.: CTFG/VHP/2010/Rev1, March 2010 http://www.hma.eu/uploads/media/VHP_version_2_March_2010.pdf
European Commission Enterprise Directorate-General. Detailed guidance on the application format and documentation to be submitted in an application for an Ethics Committee opinion on the clinical trial on medicinal products for human use (ENTR/CT2), Revision 1, February 2006http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/vol-10/12_ec_guideline_200 60216.pdf
The EFGCP Report on The Procedure for the Ethical Review of Protocols forClinical Research Projects in Europe, Update April 2010http://www.efgcp.be/EFGCPReports.asp?L1=5&L2=1
European Commission-European Medicines Agency Conference on the Operation of the Clinical Trials Directive (Directive 2001/20/EC) and Perspectives for the Future, Report on the Conference held on 3 October 2007 at the EMEA, London, Doc. ref.: EMEA/565466/2007http://www.eortc.be/services/doc/EUCTD/EC-EMEA_report_CT_20071003.pdf
Assessment of the functioning of the “Clinical Trials Directive” 2001/20/EC,Summary of responses to the public consultation paper, SANCO/C/8/SF/dn D(2010) 380240http://ec.europa.eu/enterprise/sectors/pharmaceuticals/files/clinicaltrials/2010_03_30_summary_responses.pdf
Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community Code relating to Medicinal Products for Human Use, as amendedhttp://ec.europa.eu/enterprise/pharmaceuticals/eudralex/vol-1/dir_2001_83/dir_2001 _83_de.pdf
Responses to the Public consultation paper “Assessment of the functioning of the ‘Clinical Trials Directive’ 2001/20/EC”, March 2010http://ec.europa.eu/enterprise/sectors/pharmaceuticals/human-use/clinicaltrials/ developments/responses_2010-02_en.htm
Regulation (EC) No 1394/2007 of the European Parliament and of the Council of 13 November 2007 on advanced therapy medicinal products and amending Directive 2001/83/EC and Regulation (EC) No 726/2004 http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2007:324:0121:0137:
Commission Directive 2005/28/EC of 8 April 2005 laying down principles and detailed guidelines for good clinical practice as regards investigational medicinal products for human use, as well as the requirements for authorization of the manufacturing or importation of such products http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2005:091:0013:0019:
European Commission, Impact Assessment, 2010 Roadmaps “Legislative proposal on a Regulation/Directive amending the Clinical Trials Directive 2001/20/EC”, Version 2, 23/03/2010http://ec.europa.eu/governance/impact/planned_ia/docs/47_sanco_clinical_trials_directive_en.pdf
//////////Regulatory Approval Pathways, EU vs US