












A System of Equivalent Member States, a Coordinating Agency and a Centralized Institution
The regulatory system for supervision of pharmaceutical manufacturers and GMP inspection in the European Union is one of the most advanced in the world. Due to the globalization of pharmaceutical manufacture, it also affects industry, regulators and patients outside the European Union. This system, however, is often poorly understood beyond the EU borders.
What follows is an explanation of the EU system in order to increase awareness and facilitate cooperation on GMP between European Union regulators and those outside the European Union.

The European Union
The European Union includes 28 Member States located in Europe, which are: Austria, Belgium, Bulgaria, Croatia, Cyprus, Czech Republic, Denmark, Estonia, Finland, France, Germany, Greece, Hungary, Ireland, Italy, Latvia, Lithuania, Luxemburg, Malta, Netherlands, Poland, Portugal, Romania, Slovakia, Slovenia, Spain, Sweden, and United Kingdom. The EU total population is about 500 million people.

The European Union operates through a system of supranational independent institutions and intergovernmental negotiated decisions by its Member States. It is a legal entity and can negotiate international agreements on behalf of its Member States. The European Parliament, the Council of the European Union and the European Commission are the three main EU institutions. They produce through the “Ordinary Legislative Procedure” (formerly “co-decision”) the policies and laws that apply throughout the European Union.
The European Union has developed a single market through a standardized system of laws that apply in all its Member States. The same rules and harmonized procedures apply to all the 28 Member States regarding the authorization of medicines and the supervision of safety of medicines.

The EU Regulatory System for Medicines
The EU has developed a regulatory system based on a network of decentralized National Competent Authorities (NCAs) in the Member States, supported and coordinated by a centralized agency, theEuropean Medicines Agency (EMA).
![]()
The European Commission’s role is multifaceted and focuses on the following:
- Right of initiative: To propose new or amending legislation for the pharmaceutical sector
- Implementation: To adopt implementing measures as well as to ensure and monitor the correct application of EU law
- Risk management: To grant EU-wide marketing authorizations for centralized products or maximum residue limits on the basis of a scientific opinion of the EMA
- Supervisory authority: To oversee the activities of the EMA in compliance with the mandate of the EMA, EU law and the EU policy objectives
- Global outreach: To ensure appropriate collaboration with relevant international partners and to promote the EU system globally
The EMA was created in 1995 to coordinate the existing scientific resources in the EU Member States and is an interface for cooperation and coordination of Member States’ activities with respect to medicinal products. EMA scientific decisions are made through its scientific committees, whose members are chosen on the bases of their scientific expertise and are appointed by the Member States. One of the main roles of EMA is to mobilize scientific resources in the Member States, so that many of its scientific activities are carried out through a large network of scientific experts made available by the Member States.
The system for Marketing Authorisation (MA) of medicines, including the referral procedure, is an example of how the European Commission, the EMA and the Member States cooperate. The EU national, decentralized and mutual recognition MA procedures coexist with the centralized procedure (Table 1).
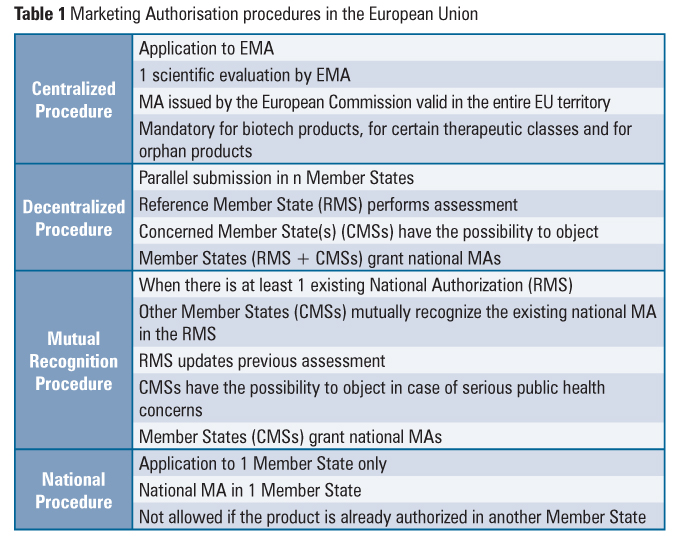
The referral procedure is an EU binding mechanism that ensures that the same measures are applied to products subject to national, decentralized and mutual recognition MA procedures. This procedure may be notably invoked when the conditions of authorizations need to be reviewed in the light of quality, safety and efficacy data (Union Interest Referral), when Member States have adopted different decisions regarding products that are authorized in at least two Member States (Divergent Decision Referral) or in the absence of agreement among Member States in the course of the mutual recognition or decentralized authorization procedures (Mutual Recognition and Decentralised Referral). This mechanism involves an opinion from the appropriate EMA committee and results in a decision of the European Commission that is binding for all Member States.
In order to provide for the same level of access to critical medicines to all the patients in the Union, the centralized procedure is mandatory for orphan products, biotechnological products, advanced-therapy products (gene therapy, somatic cell therapy and tissue engineering) and products intended for the treatment of critical therapeutic classes (HIV or AIDS, cancer, diabetes neurodegenerative diseases, auto-immune and other immune dysfunctions, and viral diseases). Veterinary medicines for use as growth or yield enhancers are also in the mandatory scope of the centralized procedure.

A fundamental aspect is that the legislation applicable to pharmaceuticals in the European Union is the same irrespective of the Member State or authorization route of the product, as it is developed at Union level. The same applies to the guidelines in use by assessors and inspectors for the assessment of MA applications and inspections, which are developed by EMA, in cooperation with Member States, through its scientific committees and working groups.
Clinical trials of Investigational Medicinal Products (IMPs) require authorization by each NCA and a favorable opinion by an ethics committee in which the clinical trial takes place and is granted in the form of a Clinical Trial Authorisation (CTA). The assessment for a CTA takes into account the holding of an appropriate authorization for each EU site of manufacture or importation.

The EU System for GMP Supervision of Manufacturers and Inspection
Any manufacturer, no matter where it is located, must comply with GMP if they are to supply products to the EU. There is a single system for GMP supervision of manufacturers which is valid throughout all the EU Member States; this includes authorized medicinal products for human or veterinary use placed on the market and IMPs used in clinical trials. The system is based on two main pillars, the authorization/registration of operators in the supply chain and inspection of those operators to ensure compliance with legal requirements, including compliance with GMP and the requirements in the MA or CTA.
Manufacturers and Importers of Medicinal Products*
Manufacturers and importers of medicinal products located in the EU need to be authorized to carry out their activities. This obligation also applies to manufacturers and importers of products only intended for export and IMPs. The competent authorities of each Member State are responsible for granting the authorizations for these activities occurring within their respective territory.
A condition for grant of a manufacturing or import authorization is that the manufacturers must comply with EU GMP. GMP principles and guidelines are set out in two Directives, one for medicines for human use and the other for medicines for veterinary use. More detailed guidelines have been developed through the work of the GMP and GDP Inspectors Working Group (GMDP IWG) and the European Commission and included in the EU GMP guide, published on the European Commission website.
Inspection of Manufacturers and Importers of Medicinal Products
Manufacturers and importers of medicinal products located in the European Union or manufacturers located in a third country are regularly inspected by an EU competent authority for compliance with EU GMP. The outcome of these inspections must be accepted by all other EU authorities. After every inspection a GMP certificate (positive outcome) or noncompliance report (negative outcome) must be issued by the inspecting authority and entered in the EudraGMDP database, which is accessible by regulators in other countries. Most of this information is also available to the general public.
Inspections of manufacturers are typically requested in order to grant or maintain a manufacturing or import authorization (EU sites) or in the context of assessment, approval and maintenance of an MA (typically sites outside the EU) or CTA. For example, EMA may request that an EU competent authority undertake a preapproval GMP inspection of a site included in a MA application through the Centralised procedure or that an EU competent authority undertake periodic repeated postauthorization surveillance inspections of sites named in centralized MAs, in order to verify ongoing compliance with GMP and that the requirements of the MA are being met.
According to EU legislation, the interval for repeated GMP inspection should be based on risk. As a result, a procedure outlining a risk-based model to frequency of inspections is included in theCompilation of European Union Procedures on Inspections and Exchange of Information.
Manufacturers and Importers of Active Substance**
Manufacturers, importers and distributors of active substance located in the European Union are required to comply with GMP and must be registered to the National Competent Authority of the Member State where they are located.
For active substances manufactured outside the EU and imported, each batch needs to be accompanied by a written confirmation issued by the competent authority of the country where it is produced, confirming, among other things, that GMP at least equivalent to that in place in the European Union has been applied to its manufacture. The competent authority of the exporting country also needs to confirm that any GMP noncompliance arising at the manufacturing site would be communicated to the European Union. The receipt of this noncompliance information is via the EMA.

The requirement for the written confirmation can only be waived if the third country is included by the European Commission, after assessment, in a list of countries with an equivalent system of supervision and inspection or, exceptionally, in order to ensure availability of medicines in the EU market, if a GMP certificate for the site has been issued by an EU competent authority after inspection.
The requirement for written confirmation, introduced from July 2013 by Directive 2011/62/EU (the so called Falsified Medicines Directive), requires that authorities outside of the EU take responsibility for active substances manufactured in their territory, if exported to the EU. This requirement caused some debate before its implementation since there were concerns on its potential to cause shortages in the EU, if the exporting authorities were not willing or able to provide the written confirmations, which turned out not to be the case.
The increased dialogue and mutual understanding between the EU and the authorities of exporting countries was instrumental to ensure a smooth implementation of this requirement. It is a good example of the importance of regulatory cooperation in the current globalized manufacturing and supply environment to the benefit of all.
Inspection of Active Substance Manufacturers
The EU legislation places the responsibility for using active substances manufactured in compliance with GMP on the medicinal product manufacturer or the importer (in case the medicinal product is manufactured outside the European Union). The holder of the manufacturing authorization (medicinal product manufacturer in the European Union or EU importer) must verify the registration status of the manufacturer of the active substance and verify compliance by the manufacturer of active substance with GMP, by conducting audits at the manufacturing site. The holder of the manufacturing authorization shall verify compliance directly or they may use a third party acting under a contract.
Inspections of active substance manufacturers are carried out by EU competent authorities following a risk-based approach, or if there is suspicion of noncompliance.
Furthermore, every application for an MA must include a confirmation that the holder of the manufacturing authorization has verified compliance of the manufacturer of the active substance with principles and guidelines of GMP. The confirmation shall contain a reference to the date of the audit and a declaration by the Qualified Person that the outcome of the audit confirms that the manufacturing complies with GMP principles and guidelines.

Inspections of active substance manufacturers may also be organised by the European Directorate for the Quality of Medicines & Healthcare (EDQM) of the Council of Europe, on behalf of the EU. The Council of Europe has 47 members including all EU Member States and it has close cooperation with the EU. EDQM is responsible for developing and maintaining the European Pharmacopoeia.
EDQM issues Certificates of Suitability with the monographs of the European Pharmacopoeia (CEP) that can replace most of the data normally expected in EU MA dossiers for the active substance. In order to issue and maintain these certificates, EDQM runs its own inspection program of active substance manufacturers. Most of the inspections organised by the EDQM are carried out by inspectors from EU inspectorates.

The Supervisory Authority
As inspections are carried out by inspectorates of Member States, in order to avoid duplication it is necessary to identify the Member State responsible for supervision and inspection of any manufacturing sites involved in production of active substances and medicines for the EU market. This is achieved through the identification of one or more Supervisory Authority (SA); the SA is the NCA in the EU responsible for the GMP supervision of the site, including granting the manufacturing or import authorization and GMP inspection.
If the manufacturing site is in the EU, the SA is the NCA of the Member State where the site is located. In cases where the manufacturing site is outside the EU, the SA is the NCA of the Member State in which the importer of the product(s) is located. Where products from a manufacturing site located in a country outside the EU are imported in more than one Member State, there may be more than one SA, which cooperate in the supervision of the manufacturing site.
The Qualified Person & Batch Certification Prior to Release
An important feature of the supervision system in place in Europe is the role of Qualified Person (QP). In order to obtain an authorization, EU manufacturers and importers must have at their disposal the services of at least one Qualified Person. The Qualified Person must take responsibility for securing that each batch of medicinal product, manufactured or imported, has been manufactured in accordance with EU GMP, and must certify compliance with GMP and with the relevant MA(s). A batch may only be released by a manufacturer or importer for distribution in the EU after certification by the QP. Member States are empowered to take administrative and disciplinary measures against QPs if they have failed to fulfil their obligations.
Furthermore, imported batches need to undergo a full retest in the EU to ensure the quality of the product in accordance with the MA specification. There-testing requirement is waived if there is an operational Mutual Recognition Agreement in place between the EU and the exporting country.
Consequences of Noncompliance with EU GMP
The discovery of serious GMP noncompliance may have implications not only for the Member State which carries out the inspection but also other, possibly all, Member States as well as international authorities should the active substance or product be supplied to them. A mechanism that ensures a coordinated approach for protection of public and/or animal health is taken throughout the European Union has been developed and is published in the Compilation of European Union Procedures. The objective of this procedure is to achieve a coordinated and harmonized assessment and proportionate supervisory actions to balance the protection of patients and minimize supply disruptions whilst ensuring maximum efficiency and avoiding full parallel reviews on a national level across the European Union.
European legislation provides that manufacturer and import authorizations may be suspended or not granted as a result of noncompliance with GMP. Also, existing MAs for the products affected can be varied (e.g., to delete a certain manufacturing site), not granted or revoked. Urgent measures include prohibition of manufacture, importation or supply, and/or withdrawal of all, or of specific batches from the market.
EudraGMDP
EudraGMDP is a publicly accessible Union database which is a repository of, among other things, manufacturing and import authorizations, GMP certificates and non-compliance reports. After every GMP inspection carried out by an EU competent authority, a GMP certificate (positive outcome) or a noncompliance report (negative outcome) is issued by the inspecting authority and entered in the EudraGMDP database.
The database includes a planning module (only accessible to the relevant regulators) for coordination of inspections planned by EU authorities in countries outside the European Union. Data are entered into the planning module in order to facilitate exchange of information between competent authorities and reduce duplication and ensure the best use of inspectional resources. EMA and EU authorities recognize the global nature of modern pharmaceutical supply chains and the need for close collaboration and cooperation with regulatory authorities outside the European Union and therefore work is ongoing to extend the use of the EudraGMDP database planning module to include exchange of information on inspections planned by authorities outside the European Union.
Overview of Inspection Activities
The chart below shows a summary of the inspections carried out by EEA competent authorities in 2014. Domestic inspections are inspections carried out by EEA competent authorities within the EEA territory. Foreign inspections are inspections carried out by EEA competent authorities outside the EEA. The data are extracted from EudraGMDP.

Ensuring and Maintaining Equivalence among Member States Inspectorates
In order to ensure the functioning of the EU system for GMP supervision of manufacturers and inspections described above, it is necessary to ensure that all the National inspectorates in the Member States are equivalent as regards the level of supervision they are able to provide. A number of measures are put in place to ensure that this is the case, summarized below.
Legislation
The pharmaceutical legislation is developed at EU level, mainly in the form of Regulations and Directives. Both are applicable to all the Member States, the difference being that Regulations are directly applicable to the entire EU territory while Directives have to be transposed into national legislation, in a timeframe established in the Directive itself, usually 18 months.
The EU legal framework for medicinal products is intended to ensure a high level of public health protection and to promote the functioning of the EU internal market. The system is also designed to encourage innovation. It is a large body of legislation that ensures extensive harmonization within the European Union, including GMP and inspections. The pharmaceutical legislation is published in the Official Journal of the European Union.
The EU GMP guide
A single GMP guide is in use in the European Union. The guide is referenced in the EU legislation (Directives 2001/83/EC for human products, 2001/82/EC for veterinary products and in clinical trial legislation) and has long since replaced any previously existing national GMP guide. The EU GMP guide provides the standards and requirements used by EU inspectors for any GMP inspections, both in or outside of the European Union.
The guide is subdivided into tree parts and 19 annexes dealing with specific types of manufacture. Part 1 is the GMP for finished products, Part 2 GMP for active substances and Part 3 includes GMP-related documents. The EU GMP guide is harmonized with the PIC/S GMP guidelines on an ongoing basis. EU GMP Part 2 reflects the EU’s agreement to the ICH Q7 guidelines and forms the basis of the detailed guidelines.

The Compilation of European Union Procedures on Inspections and Exchange of Information
The Compilation of European Union Procedures on Inspections and Exchange of Information (CoUPs) is a collection of procedures for GMP and Good Distribution Practice (GDP) inspectorates, applicable to all the inspectorates in the European Union. It provides a tool to facilitate cooperation between EU Member States and a means to achieve harmonization. The CoUP covers, among other things, the basis for national procedures that form part of the national inspectorates’ quality systems, how quality defects and noncompliance are handled and how GMP and GDP inspections are carried out and reported.
The contents of the CoUP are constantly updated, developed and agreed, under the coordination of the EMA, by representatives of the Inspectorates of each Member State, including those supervising the manufacture and import of veterinary medicinal products only. Once agreed, they are adopted by the European Commission and then published on its behalf by the EMA.
Common Union formats for manufacturing and import authorizations, GMP certificates and for statements of non-compliance with GMP have been agreed and published in the compilation and implemented by EU competent authorities in order to enhance communication, collaboration and co-operation between authorities. This common format enables Member States to enter manufacturing, importing and distribution authorizations in the Union database, EudraGMDP.
![]()
The GMP/GDP Inspectors Working Group
The GMP/GDP Inspectors Working Group (GMDP IWG) is a group of senior inspectors appointed by all the EEA competent authorities which meets at EMA premises four times a year. It is chaired by EMA and a European Commission representative attends the meetings, as well as observers from the European EDQM, accession countries (countries which have applied to be part of the EU but have not joined yet) and MRA partners. Representatives from other international authorities can be invited on a case-by-case basis.
![]()
The group is a forum for harmonization and discussion of common issues which are taken by the inspectors back to their NCA for implementation. Any new or amended text of the EU GMP guide is developed by this group, with the European Commission responsible for the final adoption. The GMDP IWG also maintains the CoUP and oversees, on behalf of the Heads of Medicines Agencies (HMAs) the Joint Audit Programme.
Training
The GMDP IWG organises training for EEA inspectors and inspectors from accession countries, aimed at raising the technical capability of the inspectors, ensuring common understanding of issues related to GMP and harmonization. In addition, EMA has signed a partnership agreement with PIC/S on cooperation on training for GMP inspectors, which recognizes the role that PIC/S plays in this area and avoids duplication of effort.
Ensuring Equivalence before Joining the EU
Becoming a member of the European Union is a complex procedure and there are strict conditions for EU membership to ensure that new members are admitted only when they are fully able to take on the obligations of membership, including compliance with all the EU’s standards and rules. For the purpose of accession negotiations, these are divided into 35 different policy fields(chapters).
For acceding to the EU, a candidate country must implement the EU rules and regulations in all areas. The length of the membership negotiations can vary and depends on the time needed to complete the necessary reforms and the alignment with EU law. The candidates are supported financially, administratively and technically during this preaccession period.
In order to ensure that new Member States joining the European Union have reached the same level as the other members before the date of accession, a number of measures are put in place. These include:
- The European Commission checks compliance with the EU legislation (including pharmaceutical legislation)
- Through the TAIEX program, financed by the European Commission, technical support may be provided
- Accession countries are invited as observers to EU meetings (including the GMDP IWG)
- Specific training on EU procedures is organized
Auditing Member States
Auditing is an important part of the measures put in place in order to oversee the equivalence of Member States. There are a number of contexts in which Member States NCAs and/or inspectorates can be audited.
The Joint Audit Program (JAP) of the EU NCAs’ GMP inspectorates is an internal audit program under the Heads of Medicines Agencies (HMA) and is run on behalf of HMA by the GMDP IWG. JAP aims at achieving and maintaining equivalence between Member States’ national inspectorates responsible for GMP. It was established in October 2000 and is an important part of the quality system adopted by all GMP inspectorates in the EU.
JAP auditors are senior GMP inspectors, further qualified for auditing inspectorates through specific training. A list of qualified JAP auditors is maintained by the Compliance Group, which is a subgroup of the GMDP IWG. JAP auditors also provide technical advice and support to accession countries before they become EU Member States.
EU inspectorates are audited through the JAP onsite, at intervals established through a risk-based approach (typically every five to six years). Mutual Recognition Agreement and other international partners are invited on a case-by-case basis to join JAP audits of EU Member States inspectorates as observers.
Audits are also organized in the framework of the Pharmaceutical Inspection Convention and Pharmaceutical Inspection Co-operation Scheme (jointly referred to as PIC/S) and Mutual Recognition Agreement (MRA) (see International Cooperation Activities below). Since most of the EU authorities and all MRA partners are member of PIC/S, synergies between the various audit schemes are used in order to avoid duplication.
BEMA Audits
The Benchmarking of European Medicines Agencies (BEMA) is an internal EU program managed by the Heads of Medicines Agencies, based on assessment of the systems and processes in individual agencies against a set of indicators in four main areas:
- Management systems
- Assessment of marketing authorization applications
- Pharmacovigilance (drug safety) activities
- Inspection services
The assessment identifies strengths and best practices in agencies and any opportunity for improvement. The program has concluded its third cycle in 2015.
International Cooperation Activities
The European Union and its Member States are involved in several bilateral and multilateral cooperation activities with international partners in the GMP area. The main advantage is that international cooperation allows, by relying on information received from trusted international authorities, to reallocate foreign inspections towards areas more at risk. It thus optimizes available inspection resources.
PIC/S
The Pharmaceutical Inspection Convention and Pharmaceutical Inspection Co-operation Scheme (jointly referred to as PIC/S) aims at harmonizing inspection procedures worldwide by developing common standards in the field of GMP and by providing training opportunities to inspectors. It also aims at facilitating cooperation and networking between competent authorities, regional and international organisations, thus increasing mutual confidence. Most EU Member States are members of PIC/S while EMA is participating in PIC/S activities as a partner organization.
Mutual Recognition Agreements
Mutual Recognition Agreements (MRAs) are official agreements on the mutual recognition of assessment of conformity of regulated products which are negotiated and signed at EU level. MRAs concluded by the European Union include pharmaceuticals and cover GMP. Consequently, inspection results carried out by MRA partners in their territory are recognized by EU Member States and vice versa and retesting upon importation into the European Union is not needed in the QP batch certification process. The MRA scope can cover both human and veterinary products, finished products, active substances and Investigational Medicinal Products, but there are differences in scope between the various MRAs.
Currently, the European Union has operational MRAs in place with Australia, Canada, Japan, New Zealand and Switzerland. The EU also has in place an Agreement on Conformity Assessment and Acceptance of industrial products (ACAA), which includes GMP, with Israel. An ACAA is a specific type of MRA; the main practical difference is that in the ACAA case results of inspections carried out outside the territory of the agreement partners are mutually recognized as well, in addition to inspections carried out in the partners’ territory. An MRA between the European Union and the United States was signed in 1999; at the time of this writing it is operational only toward rapid alerts.
International Coalition of Medicines Regulatory Authorities
The European Commission, EMA and some EU Member States (France, Germany, Ireland, Italy, Spain and UK) participate to the activities of the International Coalition of Medicines Regulatory Authorities (ICMRA). ICMRA is a recent initiative started by Heads of Medicines Agencies worldwide, which aims at providing global strategic coordination and direction on areas that are common to many regulatory authorities’ missions worldwide, and which builds on existing arrangements such as those of PIC/S. The ICMRA has the objective to establish synergies and to foster global cooperation among regulators and GMP is one of the ICMRA main areas of interest.
Other International Cooperation Activities
In addition to MRAs, the European Union is involved in several less formalized cooperation schemes on GMP with international partners and/or in areas not covered by an MRA.
The API international cooperation project has as main objectives the sharing of information on inspection planning, policy and inspection reports and joint inspections on manufacturers located outside the participating countries. It includes the following participants: the EMA and all EU member States, the European EDQM, the U.S. FDA, the Australian Therapeutic Goods Administration (TGA) and WHO.
Several bilateral pilots and programs between EMA and FDA were also developed during the last ten years with the view to increase collaboration on domestic and third country GMP inspections.
This less formal form of cooperation in the last years has allowed the building of confidence among cooperating countries and regions, mainly through joint inspections and exchange of information, and is opening new possibilities of mutual reliance on inspection results. In this perspective, it is worth noting that the European Union has identified the recognition of GMP inspections carried out in the European Union and the United States and in third countries as a main objective for the pharmaceutical sector in the context of the negotiations of the Transatlantic Trade and Investment Partnership (TTIP).

Disclaimer: The views expressed in this article are those of the authors and may not be understood or quoted as being made on behalf of or reflecting the position of the Agencies or Institutions with which the authors are affiliated.
Notes
*The term “Medicinal Product” in the European Union approximately corresponds to the term “Drug Product” in the United States. Sometimes the term “Finished Product” is used instead.
**The term “Active Substance” in the European Union corresponds to drug “Drug Substance” in the United States.
Tags: EMA , Europe , inspections , GMP , EC , European Commission , European regulations , PIC/S , GMP regulation

