












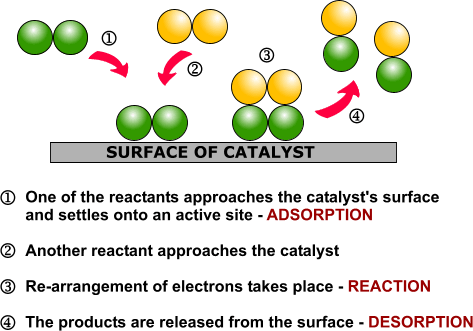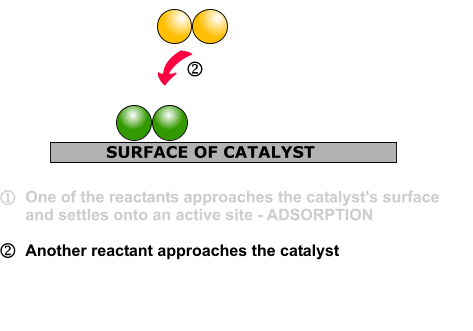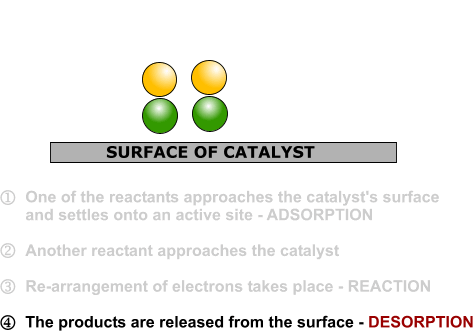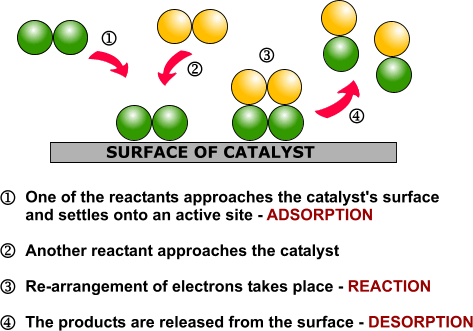
Heterogeneous catalysis and catalyst recycling
Heterogeneous catalysis is a type of catalysis in which the catalyst occupies a different phase from the reactants and products. This may refer to the physical phase — solid, liquid or gas — but also to immiscible fluids. Heterogeneous catalysts can be more easily recycled than homogeneous, but characterization of the catalyst and optimization of properties can be more difficult.


Heterogeneous catalysis is widely used in the synthesis of bulk and fine chemicals. In a general, small scale batch reaction, the catalyst, reactants, and solvent are stirred together until completion of the reaction, after which the bulk liquid is separated by filtration. The catalyst can then be collected for either recycling or disposal. In a continuous process, the catalyst can be fixed in space and the reaction mixture allowed to flow over it. The reaction and separation are thus combined in a single step, and the catalyst remains in the reactor for easy recycling. Beyond facilitating separation, thecatalyst may have improved lifetime due to decreased exposure to the environment, and reaction rates and turnover numbers can be enhanced through the use of high concentrations of a catalyst with continuous recycling. The benefits of flow are seemingly obvious, yet it has only recently become a widely adopted method for bench-scale synthesis.1



The most common application of continuous heterogeneous catalysis is in hydrogenation reactions,2 where the handling and separation of solid precious metal catalysts is not only tedious but hazardous under batch conditions. Moreover, the mixing between the three phases in a hydrogenation is generally quite poor. The use of a flow reactor gives a higher interfacial area between phases and thus more efficient reactions. For example, Ley and co-workers found that the hydrogenation of alkene 1 to 2 was challenging in batch, requiring multiple days at 80 bar of H2 (Scheme 1).3 Using a commercially available H-Cube® reactor, the reaction time was shortened to 4 hours, the pressure reduced to 60 bar, and manual separation and recycling of the catalyst from the reaction was unnecessary. The increased efficiency is due to a combination of improved mixing of the three phases, as well as the continuous recycling and high local concentration of the catalyst. The H-Cube offers a further safety advantage because it generates hydrogen gas on demand from water, obviating the need for a high pressure H2 tank.

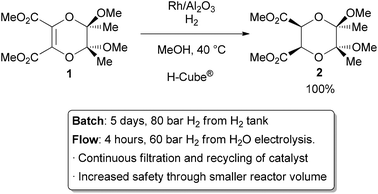 |
||
| Scheme 1 Hydrogenation with an immobilized heterogeneous catalyst. | ||
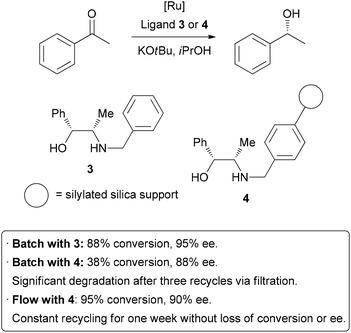
Homogeneous catalysis has many advantages over heterogeneous catalysis, such as increased activity and selectivity, and mechanisms of action that are more easily understood. Unfortunately, the difficulty associated with separating homogeneous catalysts from the product is a significant hindrance to their large scale application. In an attempt to combine the high activity of homogeneous catalysis with the practical advantageous of heterogeneous catalysis, there has been much research into immobilizing homogeneous catalysts on solid supports.4 This is generally achieved by linking thecatalyst to the surface of an insoluble solid such as silica or polymer beads. As was the case in batch hydrogenation reactions, the process of separating and purifying the catalyst is inefficient, potentially dangerous, and may lead to degradation and loss of material. Performing these reactions in a flow system can help overcome these problems.5 A highly efficient example has been demonstrated by van Leeuwen and co-workers, who sought to immobilize a catalyst used in transfer hydrogenation reactions (Scheme 2).6Their test reaction was the asymmetric reduction of acetophenone; homogeneousreduction with ruthenium and ligand 3 provided 88% conversion and 95% enantioselectivity. The ligand was then covalently linked to silica gel through the benzyl group to form 4. Using this heterogenized system under batch conditions, conversion dropped to 38% on the same time scale, and a slight decrease in enantioselectivity occurred. A reduction in activity of a catalyst upon immobilization is common, so highly efficient recycling is required. Unfortunately, when attempting to re-use the catalyst after filtration, significant degradation and leaching occurred. The catalyst was then packed in a glass column for application in flow chemistry. After a short optimization of flow rate, 95% conversion and 90% ee were obtained. Importantly, the reaction could be run continuously for up to one week without significant degradation in conversion or enantioselectivity. The physical isolation of catalyst species on the solid support is suggested to contribute to the long catalystlifetime. Interestingly, the basic potassium tert-butoxide additive was only required initially to activate the catalyst, and the reaction could subsequently be run without additional base, allowing the product to be isolated completely free of additives. It is important to note, on top of the decreased activity due to modification, that leaching from cleavage off the solid support and the increased cost of the catalyst due to derivatization are all potential downsides of immobilization of catalysts. In some instances, a seemingly heterogeneous catalyst has been shown to leach active homogeneous species into solution.7 However, as can be seen above, robust systems can be developed which do combine the best features of both homogeneous and heterogeneous catalysis.
 |
||
| Scheme 7 Immobilization of a homogeneous catalyst on a solid support. | ||
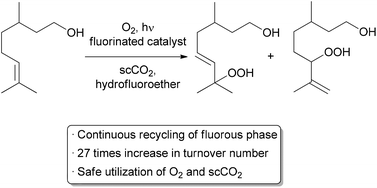
Another important method for recycling expensive catalysts is through the use of liquid–liquid biphasic conditions where the catalyst and reactants can be separated by extraction upon completion of the reaction. Such processes have already been utilized on the medium and large scale in a continuous or semi-continuous fashion.8,9 Recycling on a small scale is typically done through batch liquid–liquid extractions, but examples using continuous methods are increasing.10-13 A recent automated small scale recycling of a biphasic catalyst system was demonstrated by the George group in the continuous oxidation of citronellol (Scheme 3).14A highly fluorinated porphyrin was used as the photocatalyst, and a combination of hydrofluoroether (HFE) and scCO2 was used as the solvent. Under high pressure flow conditions, a single phase was observed. Depressurization occurred after the reactor, resulting in two phases – the organic product in one, and the catalyst and HFE in the other. The denser, catalyst-containing fluorous phase was continuously pumped back through the reactor. With this method, the catalyst was recycled 10 times while maintaining 75% of its catalytic activity, giving an increase in TON of approximately 27-fold compared to previous batch conditions. Some leaching of the fluorinated catalyst into the organic product was observed, accounting for the decreased activity over time.

 |
||
| Scheme 3 Automated recycling of a biphasic catalyst system. | ||
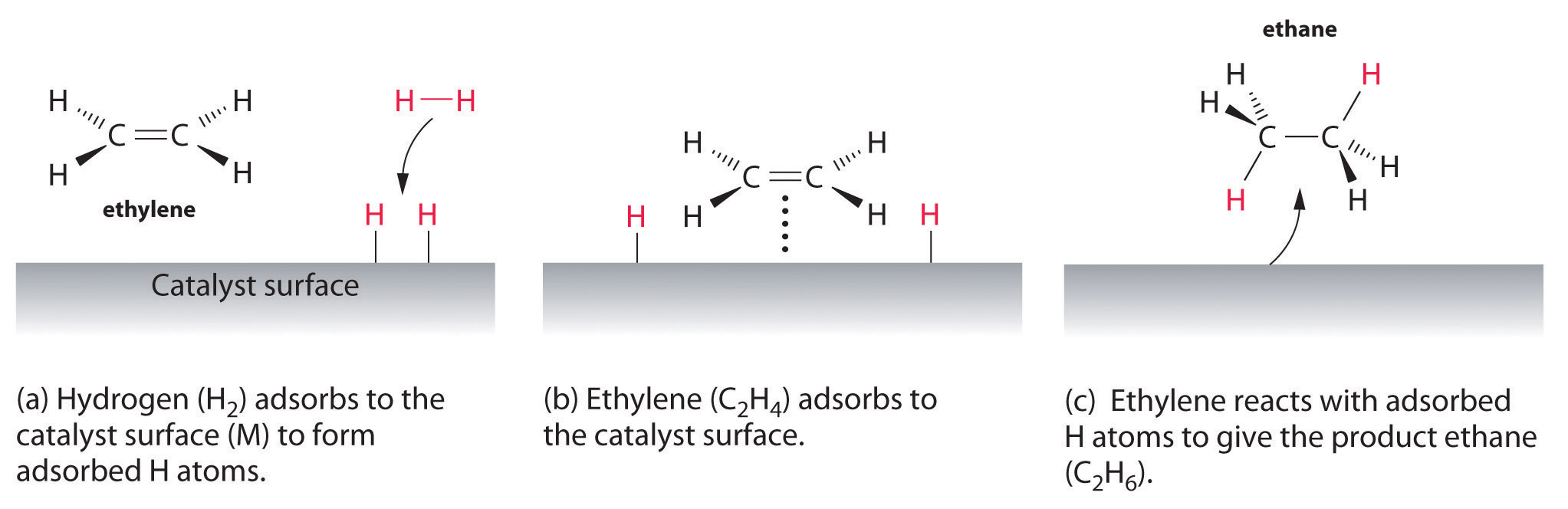
| Examples of heterogeneous catalysisThe hydrogenation of a carbon-carbon double bondThe simplest example of this is the reaction between ethene and hydrogen in the presence of a nickel catalyst.
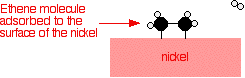
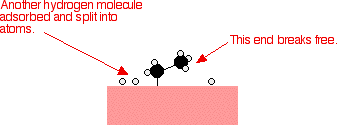
Hydrogen molecules are also adsorbed on to the surface of the nickel. When this happens, the hydrogen molecules are broken into atoms. These can move around on the surface of the nickel.
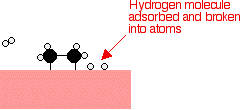
If a hydrogen atom diffuses close to one of the bonded carbons, the bond between the carbon and the nickel is replaced by one between the carbon and hydrogen.
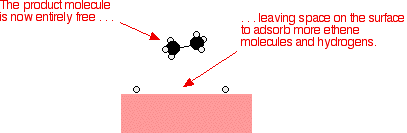
That end of the original ethene now breaks free of the surface, and eventually the same thing will happen at the other end.
As before, one of the hydrogen atoms forms a bond with the carbon, and that end also breaks free. There is now space on the surface of the nickel for new reactant molecules to go through the whole process again.
Catalytic converters Catalytic converters change poisonous molecules like carbon monoxide and various nitrogen oxides in car exhausts into more harmless molecules like carbon dioxide and nitrogen. They use expensive metals like platinum, palladium and rhodium as the heterogeneous catalyst. The metals are deposited as thin layers onto a ceramic honeycomb. This maximises the surface area and keeps the amount of metal used to a minimum. Taking the reaction between carbon monoxide and nitrogen monoxide as typical:
|
|
| Catalytic converters can be affected by catalyst poisoning. This happens when something which isn’t a part of the reaction gets very strongly adsorbed onto the surface of the catalyst, preventing the normal reactants from reaching it.Lead is a familiar catalyst poison for catalytic converters. It coats the honeycomb of expensive metals and stops it working.In the past, lead compounds were added to petrol (gasoline) to make it burn more smoothly in the engine. But you can’t use a catalytic converter if you are using leaded fuel. So catalytic converters have not only helped remove poisonous gases like carbon monoxide and nitrogen oxides, but have also forced the removal of poisonous lead compounds from petrol.
The use of vanadium(V) oxide in the Contact Process During the Contact Process for manufacturing sulphuric acid, sulphur dioxide has to be converted into sulphur trioxide. This is done by passing sulphur dioxide and oxygen over a solid vanadium(V) oxide catalyst.
|
|
| This example is slightly different from the previous ones because the gases actually react with the surface of the catalyst, temporarily changing it. It is a good example of the ability of transition metals and their compounds to act as catalysts because of their ability to change their oxidation state. | |
| The sulphur dioxide is oxidised to sulphur trioxide by the vanadium(V) oxide. In the process, the vanadium(V) oxide is reduced to vanadium(IV) oxide. |
|


- C. G. Frost and L. Mutton, Green Chem., 2010, 12, 1687–1703 .
- M. Irfan, T. N. Glasnov and C. O. Kappe, ChemSusChem, 2011, 4, 300–316
- C. F. Carter, I. R. Baxendale, M. O’Brien, J. P. V. Pavey and S. V. Ley, Org. Biomol. Chem., 2009, 7, 4594–4597 .
- P. McMorn and G. J. Hutchings, Chem. Soc. Rev., 2004, 33, 108–122.
- S. Ceylan and A. Kirschning, in Recoverable and Recyclable Catalysts, ed. M. Benaglia, John Wiley & Sons Ltd, 2009, pp. 379–410 .
- A. J. Sandee, D. G. I. Petra, J. N. H. Reek, P. C. J. Kamer and P. W. N. M. Van Leeuwen, Chem.–Eur. J., 2001, 7, 1202–1208
- M. Pagliaro, V. Pandarus, R. Ciriminna, F. Belénd and P. D. Cerà, ChemCatChem, 2012, 4, 432–445 .
- C. W. Kohlpaintner, R. W. Fischer and B. Cornils, Appl. Catal., A, 2001, 221, 219–225
- W. A. Herrmann, C. W. Kohlpaintner, H. Bahrmann and W. Konkol, J. Mol. Catal., 1992, 73, 191
- A. B. Theberge, G. Whyte, M. Frenzel, L. M. Fidalgo, R. C. R. Wootton and W. T. S. Huck, Chem. Commun., 2009, 6225–6227 .
- A. Yoshida, X. Hao and J. Nishikido, Green Chem., 2003, 5, 554–557 .
- E. Perperi, Y. Huang, P. Angeli, G. Manos, C. R. Mathison, D. J. Cole-Hamilton, D. J. Adams and E. G. Hope, Dalton Trans., 2004, 2062–2064 .
- S. Liu, T. Fukuyama, M. Sato and I. Ryu, Org. Process Res. Dev., 2004, 8, 477–481
- T. Fukuyama, M. T. Rahman, M. Sato and I. Ryu, Synlett, 2008, 151–163
- J. F. B. Hall, X. Han, M. Poliakoff, R. A. Bourne and M. W. George, Chem. Commun., 2012, 48, 3073–3075 .
- R. A. Bourne, X. Han, M. Poliakoff and M. W. George, Angew. Chem., Int. Ed., 2009, 48, 5322








//////