
cilnidipine
西尼地平
CAS 132203-70-4
- (E) – (±) 1 ,4 a dihydro-2 ,6 – dimethyl-4 – (3 – nitrophenyl) -3,5 – pyridinedicarboxylic acid, 2 – methoxy- ethyl butylester 3 – phenyl – 2 – propenyl ester FRC-8653 Cinalong
- More FRC 8653 1,4-Dihydro-2 ,6-dimethyl-4-(3-nitrophenyl) 3 ,5-pyridinedicarboxylic acid 2-methoxyethyl (2E)-3-phenyl-2-propenyl ester
- Molecular formula:C 27 H 28 N 2 O 7
- Molecular Weight:492.52
CAS Name: 1,4-Dihydro-2,6-dimethyl-4-(3-nitrophenyl)-3,5-pyridinedicarboxylic acid 2-methoxyethyl (2E)-3-phenyl-2-propenyl ester
Additional Names: (±)-(E)-cinnamyl 2-methoxyethyl 1,4-dihydro-2,6-dimethyl-4-(m-nitrophenyl)-3,5-pyridinedicarboxylate
Manufacturers’ Codes: FRC-8653
Trademarks: Atelec (Morishita); Cinalong (Fujirebio); Siscard (Boehringer, Ing.)
Molecular Formula: C27H28N2O7
Molecular Weight: 492.52
Percent Composition: C 65.84%, H 5.73%, N 5.69%, O 22.74%
Properties: Crystals from methanol, mp 115.5-116.6°. LD50 in male, female mice, rats (mg/kg): ³5000, ³5000, ³5000, 4412 orally;³5000 all species s.c.; 1845, 2353, 441, 426 i.p. (Wada).
Melting point: mp 115.5-116.6°
Toxicity data: LD50 in male, female mice, rats (mg/kg): ³5000, ³5000, ³5000, 4412 orally; ³5000 all species s.c.; 1845, 2353, 441, 426 i.p. (Wada)
Antihypertensive; Dihydropyridine Derivatives; Calcium Channel Blocker; Dihydropyridine Derivatives.

Cilnidipine (INN) is a calcium channel blocker. It is sold as Atelec in Japan, asCilaheart, Cilacar in India, and under various other trade names in East Asian countries.
Cilnidipine is a dual blocker of L-type voltage-gated calcium channels in vascular smooth muscle and N-type calcium channels in sympathetic nerve terminals that supply blood vessels. However, the clinical benefits of cilnidipine and underlying mechanisms are incompletely understood.
Clinidipine is the novel calcium antagonist accompanied with L-type and N-type calcium channel blocking function. It was jointly developed by Fuji Viscera Pharmaceutical Company, Japan and Ajinomoto, Japan and approved to come into market for the first time and used for high blood pressure treatment in 1995. in india j b chemicals & pharmaceuticals ltd and ncube pharmaceutical develope a market of cilnidipine.
Hypertension is one of the most common cardiovascular disease states, which is defined as a blood pressure greater than or equal to 140/90 mm Hg. Recently, patients with adult disease such as hypertension have rapidly increased. Particularly, since damages due to hypertension may cause acute heart disease or myocardial infarction, etc., there is continued demand for the development of more effective antihypertensive agent.
Meanwhile, antihypertensive agents developed so far can be classified into Angiotensin II Receptor Blocker (ARB), Angiotensin-Converting Enzyme Inhibitor (ACEI) or Calcium Chanel Blocker (CCB) according to the mechanism of actions. Particularly, ARB or CCB drugs manifest more excellent blood pressure lowering effect, and thus they are more frequently used.
However, these drugs have a limit in blood pressure lowering effects, and if each of these drugs is administered in an amount greater than or equal to a specific amount, various side-effects may be caused. Therefore, there have been many attempts in recent years to obtain more excellent blood pressure lowering effect by combination therapy or combined preparation which combines or mixes two or more drugs.
Particularly, since side-effect due to each drug is directly related to the amount or dose of a single drug, there have been active attempts to combine or mix two or more drugs thereby obtaining more excellent blood pressure lowering effect through synergism of the two or more drugs while reducing the amount or dose of each single drug.
For example, US 20040198789 discloses a pharmaceutical composition for lowering blood pressure combining lercanidipine, one of CCB, and valsartan, irbesartan or olmesartan, one of ARB, etc. In addition, a combined preparation composition which combines or mixes various blood pressure lowering drugs or combination therapy thereof has been disclosed.
cilnidipine Compared with other calcium antagonists, clinidipine can act on the N-type calcium-channel that existing sympathetic nerve end besides acting on L-type calcium-channel that similar to most of the calcium antagonists. Due to its N-type calcium-channel blocking properties, it has more advantages compared to conventional calcium-channel blockers. It has lower incidence of Pedal edema, one of the major adverse effects of other calcium channel blockers. Cilnidipine has similar blood pressure lowering efficacy as compared to amlodipine. One of the distinct property of cilnidipine from amlodipine is that it does not cause reflex tachycardia.
In recent years, cardiovascular disease has become common, the incidence increased year by year, about a patient of hypertension in China. 3-1. 500 million, complications caused by hypertension gradually increased, and more and more young patients with hypertension technology. In recent years, antihypertensive drugs also have great development, the main first-line diuretic drug decompression 3 – blockers, calcium channel blockers, angiotensin-converting enzyme inhibitors, ar blockers and vascular angiotensin II (Ang II) receptor antagonist.
In the anti-hypertensive drugs, calcium antagonists are following a – blockers after another rapidly developing cardiovascular drugs, has been widely used in clinical hypertension, angina and other diseases, in cardiovascular drugs in the world, ranked first.
Cilnidipine for the long duration of the calcium channel blockers, direct relaxation of vascular smooth muscle, dilation of peripheral arteries, the peripheral resistance decreased, with lower blood pressure, heart rate without causing a reflex effect.
Cilnidipine is a dihydropyridine CCB as well as an antihypertensive. Cilnidipinehas L- and N-calcium channel blocking actions. Though many of the dihydropyridine CCBs may cause an increase in heart rate while being effective for lowering blood pressure, it has been confirmed that cilnidipine does not increase the heart rate and has a stable hypotensive effect. (Takahiro Shiokoshi, “Medical Consultation & New Remedies” vol. 41, No. 6, p. 475-481)

- http://www.mcyy.com.cn/e-product2.asp
- Löhn M, Muzzulini U, Essin K, et al. (May 2002). “Cilnidipine is a novel slow-acting blocker of vascular L-type calcium channels that does not target protein kinase C”. J. Hypertens. 20 (5): 885–93. PMID 12011649.

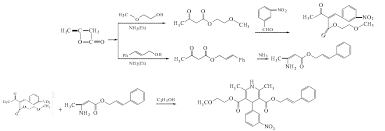
Cilnidipine (CAS NO.: 132203-70-4), with its systematic name of (+-)-(E)-Cinnamyl 2-methoxyethyl 1,4-dihydro-2,6-dimethyl-4-(m-nitrophenyl)-3,5-pyridinedicarboxylate, could be produced through many synthetic methods.
Following is one of the synthesis routes: By cyclization of 2-(3-nitrobenzylidene)acetocetic acid cinnamyl ester (I) with 2-aminocrotonic acid 2-methoxyethyl ester (II) by heating at 120 °C.
MORE
NMR
CARBOHYDRATE POLYMERS 90 PG 1719-1724 , YR2012
Numerous peaks were found in the spectrum of cilnidipine: 2.3555 (3H, s, CH3), 2.3886(3H, s, CH3), 3.2843(CD3OD), 3.3292(3H, s, OCH3), 3.5255–3.5623(2H, m, CH3OCH2CH2 ), 4.1224–4.1597(2H, m, CH3OCH2CH2 ), 4.6695–4.7293(2H, m, CH2 CH CH ), 4.8844(D2O), 5.1576(1H, s, CH), 6.2609(1H, dt, CH2 CH CH ), 6.5518(1H, d, CH2 CH CH ), 7.2488–7.3657(6H, m, ArH), 7.7002(1H, dd, ArH), 7.9805(1H, dd, ArH), 8.1548(1H, s, ArH)


References:
Dihydropyridine calcium channel blocker. Prepn: T. Kutsuma et al., EP 161877; eidem, US 4672068(1985, 1987 both to Fujirebio).
Pharmacology: K. Ikeda et al., Oyo Yakuri 44, 433 (1992).
Mechanism of action study: M. Hosonoet al., J. Pharmacobio-Dyn. 15, 547 (1992).
LC-MS determn in plasma: K. Hatada et al., J. Chromatogr. 583, 116 (1992). Clinical study: M. Ishii, Jpn. Pharmacol. Ther. 21, 59 (1993).
Acute toxicity study: S. Wada et al., Yakuri to Chiryo 20, Suppl. 7, S1683 (1992), C.A. 118, 32711 (1992).
U.S Patent No. 4,572,909 discloses amlodipine; U.S Patent No. 4,446,325 discloses aranidipine; U.S Patent No. 4,772,596 discloses azelnidipine; U.S Patent No. 4,220,649 discloses barnidipine; U.S Patent No. 4,448,964 discloses benidipine; U.S Patent No. 5,856,346 discloses clevidipine; U.S Patent No. 4,466,972 discloses isradipine; U.S Patent No. 4,885,284 discloses efonidipine; and U.S Patent No. 4,264,61 1 discloses felodipine.
U.S Patent No. 5,399,578 discloses Valsartan; European Patent No. 0 502 314 discloses Telmisartan; U.S Patent No. 5,138,069 discloses Losartan; U.S Patent No. 5,270,317 discloses Irbesartan; U.S Patent No. 5,583,141 and 5,736,555 discloses Azilsartan; U.S Patent No. 5,196,444 discloses Candesartan; U.S Patent No. 5,616,599 discloses Olmesartan; and U.S Patent No. 5,185,351 discloses Eprosartan.
U.S Patent No. 4,374,829 discloses enalapril; U.S Patent No. 4,587,258 discloses ramipril; U.S Patent No. 4,344,949 discloses quinapril; U.S Patent No. 4,508,729 discloses perindopril; U.S Patent No. 4,374,829 discloses lisinopril; U.S Patent No. 4,410,520 discloses benazepril; U.S Patent No. 4,508,727 discloses imidapril; U.S Patent No. 4,316,906 discloses zofenopril; U.S Patent Nos. 4,046,889 and 4,105,776 discloses captopril; and U.S Patent No. 4,337,201 discloses fosinopril.
-
Planar chemical structures of these calcium blockers of formula (I) are shown below.
-
Amlodipine is 2-(2-aminoethoxymethyl)-4-(2-chlorophenyl)-3-ethoxycarbonyl-5-methoxycarbonyl-6-methyl-1,4-dihydropyridine disclosed in USP 4,572,909, Japanese patent publication No. Sho 58-167569 and the like.
-
Aranidipine is 3-(2-oxopropoxycarbonyl)-2,6-dimethyl-5-methoxycarbonyl-4-(2-nitrophenyl)-1,4-dihydropyridine disclosed in USP 4,446,325 and the like.
-
Azelnidipine is 2-amino-3-(1-diphenylmethyl-3-azetidinyloxycarbonyl)-5-isopropoxycarbonyl-6-methyl-4-(3-nitrophenyl)-1,4-dihydropyridine disclosed in USP 4,772,596, Japanese patent publication No. Sho 63-253082 and the like.
-
Barnidipine is 3-(1-benzyl-3-pyrrolidinyloxycarbonyl)-2,6-dimethyl-5-methoxycarbonyl-4-(3-nitrophenyl)-1,4-dihydropyridine disclosed in USP 4,220,649, Japanese patent publication No. Sho 55-301 and the like.
-
Benidipine is 3-(1-benzyl-3-piperidinyloxycarbonyl)-2,6-dimethyl-5-methoxycarbonyl-4-(3-nitrophenyl)-1,4-dihydropyridine and is described in the specifications of U.S. Patent No. 4,501,748, Japanese patent publication No. Sho 59-70667 and the like.
-
Cilnidipine is 2,6-dimethyl-5-(2-methoxyethoxycarbonyl)-4-(3-nitrophenyl)-3-(3-phenyl-2-propenyloxycarbonyl)-1,4-dihydropyridine disclosed in USP 4,672,068, Japanese patent publication No. Sho 60-233058 and the like.
-
Efonidipine is 3-[2-(N-benzyl-N-phenylamino)ethoxycarbonyl]-2,6-dimethyl-5-(5,5-dimethyl-1,3,2-dioxa-2-phosphonyl)-4-(3-nitrophenyl)-1,4-dihydropyridine disclosed in USP 4,885,284, Japanese patent publication No. Sho 60-69089 and the like.
-
Elgodipine is 2,6-dimethyl-5-isopropoxycarbonyl-4-(2,3-methylenedioxyphenyl)-3-[2-[N-methyl-N-(4-fluorophenylmethyl)amino]ethoxycarbonyl]-1,4-dihydropyridine disclosed in USP 4,952,592, Japanese patent publication No. Hei 1-294675 and the like.
-
Felodipine is 3-ethoxycarbonyl-4-(2,3-dichlorophenyl)-2,6-dimethyl-5-methoxycarbonyl-1,4-dihydropyridine disclosed in USP 4,264,611, Japanese patent publication No. Sho 55-9083 and the like.
-
Falnidipine is 2,6-dimethyl-5-methoxycarbonyl-4-(2-nitrophenyl)-3-(2-tetrahydrofurylmethoxycarbonyl)-1,4-dihydropyridine disclosed in USP 4,656,181, Japanese patent publication (kohyo) No. Sho 60-500255 and the like.
-
Lemildipine is 2-carbamoyloxymethyl-4-(2,3-dichlorophenyl)-3-isopropoxycarbonyl-5-methoxycarbonyl-6-methyl-1,4-dihydropyridine disclosed in Japanese patent publication No. Sho 59-152373 and the like.
-
Manidipine is 2,6-dimethyl-3-[2-(4-diphenylmethyl-1-piperazinyl)ethoxycarbonyl]-5-methoxycarbonyl-4-(3-nitrophenyl)-1,4-dihydropyridine disclosed in USP 4,892,875, Japanese patent publication No. Sho 58-201765 and the like.
-
Nicardipine is 2,6-dimethyl-3-[2-(N-benzyl-N-methylamino)ethoxycarbonyl]-5-methoxycarbonyl-4-(3-nitrophenyl)-1,4-dihydropyridine disclosed in USP 3,985,758, Japanese patent publication No. Sho 49-108082 and the like.
-
Nifedipine is 2,6-dimethyl-3,5-dimethoxycarbonyl-4-(2-nitrophenyl)-1,4-dihydropyridine disclosed in USP 3,485,847 and the like.
-
Nilvadipine is 2-cyano-5-isopropoxycarbonyl-3-methoxycarbonyl-6-methyl-4-(3-nitrophenyl)-1,4-dihydropyridine disclosed in USP 4,338,322, Japanese patent publication No. Sho 52-5777 and the like.
-
Nisoldipine is 2,6-dimethyl-3-isobutoxycarbonyl-5-methoxycarbonyl-4-(3-nitrophenyl)-1,4-dihydropyridine disclosed in USP 4,154,839, Japanese patent publication No. Sho 52-59161 and the like.
-
Nitrendipine is 3-ethoxycarbonyl-2,6-dimethyl-5-methoxycarbonyl-4-(3-nitrophenyl)-1,4-dihydropyridine disclosed in USP 3,799,934, Japanese patent publication (after examination) No. Sho 55-27054 and the like.
-
Pranidipine is 2,6-dimethyl-5-methoxycarbonyl-4-(3-nitrophenyl)-3-(3-phenyl-2-propen-1 -yloxycarbonyl)-1,4-dihydropyridine disclosed in USP 5,034,395, Japanese patent publication No. Sho 60-120861 and the like.



































