












suramin
A polyanionic compound with an unknown mechanism of action. It is used parenterally in the treatment of African trypanosomiasis and it has been used clinically with diethylcarbamazine to kill the adult Onchocerca. (From AMA Drug Evaluations Annual, 1992, p1643) It has also been shown to have potent antineoplastic properties.
A polyanionic compound with an unknown mechanism of action. It is used parenterally in the treatment of African trypanosomiasis and it has been used clinically with diethylcarbamazine to kill the adult Onchocerca. (From AMA Drug Evaluations Annual, 1992, p1643) It has also been shown to have potent antineoplastic properties. Suramin is manufactured by Bayer in Germany as Germanin®.
Also known as: Naphuride, Germanin, Naganol, Belganyl, Fourneau, Farma, Antrypol, Suramine, Naganin
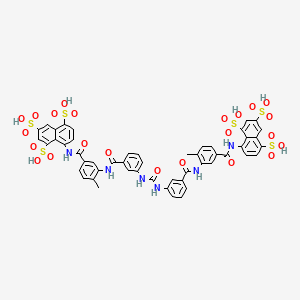
8,8′-{Carbonylbis[imino-3,1-phenylenecarbonylimino(4-methyl-3,1-phenylene)carbonylimino]}di(1,3,5-naphthalenetrisulfonic acid) …FREE FORM
8,8′-[Ureylenebis[m-phenylenecarbonylimino(4-methyl-m-phenylene)carbonylimino]]di(1,3,5-naphthalenetrisulfonic acid) hexasodium salt
CAS 145-63-1 FREE FORM
129-46-4 of hexa sodium
LAUNCHED 1940 BAYER
| Formula | C51H40N6O23S6 |
|---|---|
| Mol. mass | 1297.29 |
The molecular formula of suramin is C51H34N6O23S6. It is a symmetric molecule in the center of which lies urea, NH-CO-NH. Suramin contains eightbenzene rings, four of which are fused in pairs (naphthalene), four amide groups in addition to the one of urea and six sulfonate groups. When given as drug it usually contains six sodium ions that form a salt with the six sulfonate groups.
Suramin is a drug developed by Oskar Dressel and Richard Kothe of Bayer, Germany in 1916, and is still sold by Bayer under the brand nameGermanin.
Suramin sodium is a heparanase inhibitor that was first launched in 1940 by Bayer under the brand name Antrypol for the treatment of helminthic infection. It was later launched by Bayer for the treatment of trypanosomiasis (African sleeping sickness).
More recently, the product has entered early clinical development at Ohio State University for the treatment of platinum-pretreated patients with stage IIIB/IV non-small cell lung cancer, in combination with docetaxel or gemcitabine.
The National Cancer Institute (NCI) is conducting phase II clinical studies for the treatment of glioblastoma multiforme and for the treatment of adrenocortical carcinoma.
According to the National Cancer Institute there are no active clinical trials (as of April 1, 2008). Completed and closed clinical trials are listed here:[1]
In addition to Germanin, the National Cancer Institute also lists the following “Foreign brand names”: 309 F or 309 Fourneau,[1] Bayer 205, Moranyl, Naganin, Naganine.

It is used for treatment of human sleeping sickness caused by trypanosomes.[2]
It has been used in the treatment of onchocerciasis.[3]
It has been investigated as treatment for prostate cancer.[4]
Also, suramin as treatment for autism is being evaluated. [5]
Suramin is administered by a single weekly intravenous injection for six weeks. The dose per injection is 1 g.
The most frequent adverse reactions are nausea and vomiting. About 90% of patients will get an urticarial rash that disappears in a few days without needing to stop treatment. There is a greater than 50% chance of adrenal cortical damage, but only a smaller proportion will require lifelongcorticosteroid replacement. It is common for patients to get a tingling or crawling sensation of the skin with suramin. Suramin will cause clouding of the urine which is harmless: patients should be warned of this to avoid them becoming alarmed.
Kidney damage and exfoliative dermatitis occur less commonly.
Suramin has been applied clinically to HIV/AIDS patients resulting in a significant number of fatal occurrences and as a result the application of this molecule was abandoned for this condition. http://www.ncbi.nlm.nih.gov/pubmed/3548350
Suramin is also used in research as a broad-spectrum antagonist of P2 receptors[6][7] and agonist of Ryanodine receptors.[8]
suramin
Its effect on telomerase has been investigated.[9]
It may have some activity against RNA viruses.[10]
In addition to antagonism of P2 receptors, Suramin inhibits the acitivation of heterotrimeric G proteins in a variety of other GPCRs with varying potency. It prevents the association of heteromeric G proteins and therefore the receptors Guanine exchange functionality (GEF). With this blockade the GDP will not release from the Gα subunit so it can not be replaced by a GTP and become activated. This has the effect of blocking downstream G protein mediated signaling of various GPCR proteins including Rhodopsin, the A1 Adenosine receptor, and the D2 dopamine receptor.[11]
A polyanionic compound with an unknown mechanism of action. It is used parenterally in the treatment of African trypanosomiasis and it has been used clinically with diethylcarbamazine to kill the adult Onchocerca. (From AMA Drug Evaluations Annual, 1992, p1643) It has also been shown to have potent antineoplastic properties. Suramin is manufactured by Bayer in Germany as Germanin®.
|
8-1-2012
|
InCl3-catalysed synthesis of 2-aryl quinazolin-4(3H)-ones and 5-aryl pyrazolo[4,3-d]pyrimidin-7(6H)-ones and their evaluation as potential anticancer agents.
|
Bioorganic & medicinal chemistry letters
|
|
9-1-2012
|
Identification of a sirtuin 3 inhibitor that displays selectivity over sirtuin 1 and 2.
|
European journal of medicinal chemistry
|
|
1-1-2013
|
Inhibition of the human deacylase Sirtuin 5 by the indole GW5074.
|
Bioorganic & medicinal chemistry letters
|
|
5-9-2013
|
Discovery of thieno[3,2-d]pyrimidine-6-carboxamides as potent inhibitors of SIRT1, SIRT2, and SIRT3.
|
Journal of medicinal chemistry
|
- The formula of suramin was kept secret by Bayer for commercial reasons. But it was elucidated and published in 1924 by Fourneau and his team of the Pasteur Institute, and it is only on this date that its exact chemical composition was known. (E. Fourneau, J. and Th. Tréfouël and J. Vallée (1924). “Sur une nouvelle série de médicaments trypanocides”, C. R. Séances Acad. Sci. 178: 675.)
- Darsaud A, Chevrier C, Bourdon L, Dumas M, Buguet A, Bouteille B (January 2004). “Megazol combined with suramin improves a new diagnosis index of the early meningo-encephalitic phase of experimental African trypanosomiasis”. Trop. Med. Int. Health 9 (1): 83–91.doi:10.1046/j.1365-3156.2003.01154.x. PMID 14728611.
- Anderson J, Fuglsang H (July 1978). “Further studies on the treatment of ocular onchocerciasis with diethylcarbamazine and suramin”. Br J Ophthalmol 62 (7): 450–7.doi:10.1136/bjo.62.7.450. PMC 1043255. PMID 678497.
- Ahles TA, Herndon JE, Small EJ, et al. (November 2004). “Quality of life impact of three different doses of suramin in patients with metastatic hormone-refractory prostate carcinoma: results of Intergroup O159/Cancer and Leukemia Group B 9480”. Cancer 101 (10): 2202–8.doi:10.1002/cncr.20655. PMID 15484217.
- http://medicalxpress.com/news/2013-03-drug-treatment-autism-symptoms-mouse.html
- Abbracchio MP, Burnstock G, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, Knight GE, Fumagalli M, Gachet C, Jacobson KA, Weisman GA. (september 2006). “International Union of Pharmacology LVIII: update on the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy”. Pharmacol Rev. 58 (3): 281–341.doi:10.1124/pr.58.3.3. PMID 16968944.
- Khakh BS, Burnstock G, Kennedy C, King BF, North RA, Séguéla P, Voigt M, Humphrey PP. (march 2001). “International union of pharmacology. XXIV. Current status of the nomenclature and properties of P2X receptors and their subunits”. Pharmacol Rev. 53 (1): 107–118.PMID 11171941.
- Wolner I, Kassack MU, Ullmann H, Karel A, Hohenegger M (October 2005). “Use-dependent inhibition of the skeletal muscle ryanodine receptor by the suramin analogue NF676”. Br. J. Pharmacol. 146 (4): 525–33. doi:10.1038/sj.bjp.0706359. PMC 1751178.PMID 16056233.
- Erguven M, Akev N, Ozdemir A, Karabulut E, Bilir A (August 2008). “The inhibitory effect of suramin on telomerase activity and spheroid growth of C6 glioma cells”. Med. Sci. Monit. 14(8): BR165–73. PMID 18667993.
- Mastrangelo E, Pezzullo M, Tarantino D, Petazzi R, Germani F, Kramer D, Robel I, Rohayem J, Bolognesi M, Milani M (2012) Structure-based inhibition of norovirus RNA-dependent RNA-polymerases. J Mol Biol
- Beindl W, Mitterauer T, Hohenegger M, Ijzerman AP, Nanoff C, Freissmuth M. (August 1996).“Inhibition of receptor/G protein coupling by suramin analogues”. ol. Pharmacology. 50 (2): 415–23. PMID 8700151.
- Drugs Fut 1986, 11(10): 860
- WO 2012159107
- WO 2012087336
- US 2011257109
- WO 2009022897
- WO 2009020613
- WO 2008094027
- EP 0486809
- US 5158940
- US 5173509
- WO 1993007864
- WO 1994008574
SURAMIN
- Suramin bound to proteins in the PDB
- Drug information
- Suramin, drug information by JBC Online
- Suramin in treating patients with recurrent bladder cancer
- National Cancer Institute
Enterovirus-71 (EV71) is one of the major causative reagents for hand-foot-and-mouth disease. In particular, EV71 causes severe central nervous system infections and leads to numerous dead cases. Although several inactivated whole-virus vaccines have entered in clinical trials, no antiviral agent has been provided for clinical therapy. In the present work, we screened our compound library and identified that suramin, which has been clinically used to treat variable diseases, could inhibit EV71 proliferation with an IC50 value of 40μM. We further revealed that suramin could block the attachment of EV71 to host cells to regulate the early stage of EV71 infection, as well as affected other steps of EV71 life cycle. Our results are helpful to understand the mechanism for EV71 life cycle and provide a potential for the usage of an approved drug, suramin, as the antiviral against EV71 infection.
- Suramin Hexasodium
- 129-46-4
Synonyms
- 309 F
- Antrypol
- BAY 205
- Bayer 205
- CI-1003
- EINECS 204-949-3
- Fourneau 309
- Germanin
- Moranyl
- Naganin
- Naganine
- Naganinum
- Naganol
- Naphuride sodium
- NF060
- NSC 34936
- SK 24728
- Sodium suramin
- Suramin Hexasodium
- Suramin sodium
- Suramina sodica
- Suramina sodica [INN-Spanish]
- Suramine sodique
- Suramine sodique [INN-French]
- Suramine sodium
- Suraminum natricum
- Suraminum natricum [INN-Latin]
- UNII-89521262IH
Suramin Sodium, is an anticancer agent with a wide variety of activities.
Recently suramin was shown to inhibit FSH binding to its receptor (Daugherty, R. L.; Cockett, A. T. K.; Schoen, S. R. and Sluss, P. M. “Suramin inhibits gonadotropon action in rat testis: implications for treatment of advanced prostate cancer” J. Urol. 1992, 147, 727-732).
This activity causes, at least in part, the decrease in testosterone production seen in rats and humans that were administered suramin(Danesi, R.; La Rocca, R. V.; Cooper, M. R.; Ricciardi, M. P.; Pellegrini, A.; Soldani, P.; Kragel, P. J.; Paparelli, A.; Del Tacca, M.; Myers, C. E, “Clinical and experimental evidence of inhibition of testosterone production by suramin.” J. Clin. Endocrinol. Metab. 1996, 81, 2238-2246).
Suramin is the only non-peptidic small molecule that has been reported to be an FSH receptor binding antagonist.

Suramin is 8,8′ – (carbonylbis(imino-3,1-phenylenecarbonylimino (4-methyl-3,1-phenylene) carbonylimino)) bis-1,3 ,5-naphthalenetrisulfonic acid (GB Patent No. 224849). This polyanionic compound has been used for many decades as a prophylactic and therapeutic agent for try- panosomiasis. It was subsequently shown that suramin is able to block the activity of a variety of proteins like cellular and viral enzymes and growth factors (Mitsuya, M. et al. Science 226 : 172 (1984), Hosang, M. J. Cell. Biochem. 29 : 265 (1985), De Clercq, E. Cancer Lett. 8 : 9 (1979)).
|
5-32-1977
|
Complement inhibitors
|
|
|
5-25-1977
|
Aromatic amidines as antiviral agents in animals
|
|
|
5-4-1977
|
Complement inhibitors
|
|
|
5-4-1977
|
Complement inhibitors
|
|
|
4-27-1977
|
Cyclodextrin sulfate salts as complement inhibitors
|
|
|
4-20-1977
|
Ureylenebis methyl-phenylene-carbonyl-bis-dihydro-2-oxo-naphthoxazine disultonic acids
|
|
|
3-30-1977
|
Water treatment for controlling the growth of algae employing biguanides
|
|
|
3-2-1977
|
Isoxazole substituted nitroimidazoles
|
|
|
2-16-1977
|
Amidophenyl-azo-naphthalenesulfonic complement inhibitors and method of use thereof
|
|
|
2-9-1977
|
Complement inhibitors
|
|
2-10-2011
|
MODULATION OF HUMAN MAST CELL ACTIVATION MODULATION OF HUMAN MAST CELL ACTIVATION
|
|
|
11-18-2010
|
Admixtures for inorganic binders based on a hydrogenated disaccharide, inorganic binders containing these admixtures and process for their preparation Admixtures for inorganic binders based on a hydrogenated disaccharide, inorganic binders containing these admixtures and process for their preparation
|
|
|
10-28-2010
|
THERAPEUTIC INHIBITORS OF VASCULAR SMOOTH MUSCLE CELLS
|
|
|
9-9-2010
|
APPARATUS FOR USING ELECTROPORATION MEDIATED DELIVERY OF DRUGS AND GENES
|
|
|
4-8-2010
|
PREPARATION AND USE OF SULFATED OLIGOSACCHARIDES
|
|
|
10-29-2009
|
THERAPEUTIC INHIBITOR OF VASCULAR SMOOTH MUSCLE CELLS THERAPEUTIC INHIBITOR OF VASCULAR SMOOTH MUSCLE CELLS
|
|
|
8-20-2009
|
METHOD OF MAKING MINERAL FIBRES METHOD OF MAKING MINERAL FIBRES
|
|
|
6-25-2009
|
OXYGEN-FUEL BOOST REFORMER PROCESS AND APPARATUS
|
|
|
4-23-2009
|
METHODS OF TREATING VASCULAR DISEASE WITH TNF ANTAGONISTS METHODS OF TREATING VASCULAR DISEASE WITH TNF ANTAGONISTS
|
|
|
3-26-2009
|
COPOLYMER COMPOSITIONS FOR ORAL DELIVERY
|
|
5-3-1978
|
1,3,5- Or 1,3,6-naphthalenetriyltris(sulfonylimino)aryl acids and salts
|
|
|
3-22-1978
|
Nitroimidazoles
|
|
|
2-15-1978
|
Treatment of rheumatoid arthritis and related diseases
|
|
|
1-4-1978
|
AROMATIC AMIDINES AS ANTIVIRAL AGENTS IN ANIMALS
|
|
|
1-4-1978
|
Malto-dextrin poly(H-)sulfates
|
|
|
12-14-1977
|
Disazo compounds useful as complement inhibitors
|
|
|
12-7-1977
|
Bis-substituted naphthalene-azo phenyleneazo-stilbene-disulfonic and naphthalene-sulfonic acid
|
|
|
9-28-1977
|
UREIDOPHENYLENEBIS(CARBONYLIMINO)DINAPHTHALENETRISULFONIC ACID COMPOUNDS
|
|
|
9-21-1977
|
Substituted bisnaphthylazo diphenyl ureido complement inhibitors
|
|
|
9-7-1977
|
Substituted-hydroxy-naphthalenedisulfonic acid compounds
|
|
1-12-1977
|
Complement inhibitors
|
|
|
12-22-1976
|
Complement inhibitors
|
|
|
10-13-1976
|
Complement inhibitors
|
| EP0183352A2 * | Sep 27, 1985 | Jun 4, 1986 | THE UNITED STATES OF AMERICA as represented by the Secretary United States Department of Commerce | Use of suramin for clinical treatment of infection with any of the members of the family of human-t-cell leukemia (htvl) viruses including lymphadenopathy virus (lav) |
| EP0205077A2 * | Jun 3, 1986 | Dec 17, 1986 | Bayer Ag | Suramin sodium for use as an immunostimulant |
| EP0515523A1 * | Feb 13, 1991 | Dec 2, 1992 | THE UNITED STATES OF AMERICA as represented by the Secretary United States Department of Commerce | Use of suramin to treat rheumatologic diseases |
| EP0755254A1 * | Mar 24, 1995 | Jan 29, 1997 | The Trustees Of The University Of Pennsylvania | Prevention and treatment of ischemia-reperfusion and endotoxin-related injury using adenosine and purino receptor antagonists |
| EP1460087A1 * | Feb 17, 1997 | Sep 22, 2004 | The Kennedy Institute Of Rheumatology | Methods of treating vascular disease with TNF antagonists |
| EP1940376A2 * | Oct 3, 2006 | Jul 9, 2008 | Rottapharm S.P.A. | Use of neboglamine in the treatment of toxicodependency |
| EP1945204A2 * | Oct 27, 2006 | Jul 23, 2008 | Brane Discovery S.R.L. | V-atpase inhibitors for use in the treatment of septic shock |
| US5453444 * | Oct 6, 1994 | Sep 26, 1995 | Otsuka Pharmaceutical Co., Ltd. | Method to mitigate or eliminate weight loss |
| US5534539 * | Jun 12, 1995 | Jul 9, 1996 | Farmitalia Carlo Erba S.R.L. | Biologically active ureido derivatives useful as anit-metastic agenst |
| US5596105 * | Jan 13, 1995 | Jan 21, 1997 | Farmitalia Carlo Erba S.R.L. | Therapeutically active naphthalenesulfonic pyrrolecarboxamido derivatives |
| US7476693 | Mar 26, 2003 | Jan 13, 2009 | Eastern Virginia Medical School | Suramin and derivatives thereof as topical microbicide and contraceptive |
| US7608262 | Feb 16, 1996 | Oct 27, 2009 | The Kennedy Institute Of Rheumatology | Methods of preventing or treating thrombosis with tumor necrosis factor antagonists |
| US8552064 | Dec 19, 2008 | Oct 8, 2013 | Eastern Virginia Medical School | Suramin and derivatives thereof as topical microbicide and contraceptive |
| WO1994008574A1 * | Oct 12, 1993 | Apr 28, 1994 | Otsuka America Pharmaceutical | Treatment of cachexia and inhibition of il-6 activity |
| WO1994010990A1 * | Nov 12, 1993 | May 26, 1994 | British Bio Technology | Inhibition of tnf production |
| WO1997030088A2 * | Feb 17, 1997 | Aug 21, 1997 | Kennedy Inst Of Rheumatology | Methods of treating vascular disease with tnf antagonists |
| WO2004113920A1 * | Jun 18, 2004 | Dec 29, 2004 | Babon Jeff James | Screening method for substances binding to merozoite surface protein-1/42 |
| WO2008138943A2 * | May 14, 2008 | Nov 20, 2008 | Mara Galli | Prophylactic and therapeutic use of sirtuin inhibitors in tnf-alpha mediated pathologies |
| WO2009137471A2 * | May 5, 2009 | Nov 12, 2009 | University Of Miami | Azo dye related small molecule modulators of protein-protein interactions |
| WO2010016628A1 * | Jul 10, 2009 | Feb 11, 2010 | Sammy Opiyo | Conjugated suramin amino compounds for medical conditions |
| WO2012159107A1 * | May 21, 2012 | Nov 22, 2012 | Rhode Island Hospital | Inhibition of renal fibrosis |

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family





 11.25 M in NH3)], Na2CO3 [15% w/w solution (4 L)]. More EtOAc (4 L) was added, and the organic layer was washed with water (4 L). The organic phase was then concentrated to 2.5 L; again fresh EtOAc (4 L) was added, and the solution was concentrated to 2.5 L to give a solution of casopitant 2.
11.25 M in NH3)], Na2CO3 [15% w/w solution (4 L)]. More EtOAc (4 L) was added, and the organic layer was washed with water (4 L). The organic phase was then concentrated to 2.5 L; again fresh EtOAc (4 L) was added, and the solution was concentrated to 2.5 L to give a solution of casopitant 2.




















 approved by the FDA")



























 Cod liver oil has been a popular supplement for many years and naturally contains very high levels of vitamin A and vitamin D. Cod liver oil provides 10001IU (1667% DV) per 100 gram serving, or 1360IU (340% DV) in a single tablespoon.
Cod liver oil has been a popular supplement for many years and naturally contains very high levels of vitamin A and vitamin D. Cod liver oil provides 10001IU (1667% DV) per 100 gram serving, or 1360IU (340% DV) in a single tablespoon. Various types of fish are high in vitamin D. Typically raw fish contains more vitamin D than cooked, and fatty cuts will contain more than lean cuts. Further, fish canned in oil will have more vitamin D than those canned in water. Raw fish is typically eaten in the form of sushi. Raw Atlantic Herring provides the most vitamin D with 1628IU (271% DV) per 100 gram serving, 2996IU (499% DV) per fillet, and 456IU (76% DV) per ounce. It is followed by Pickled Herring with 680IU (113% DV) per 100g serving, Canned Salmon (127% DV), Raw Mackerel (60% DV), Oil Packed Sardines (45% DV), Canned Mackerel (42% DV), and oil packed Tuna (39% DV).
Various types of fish are high in vitamin D. Typically raw fish contains more vitamin D than cooked, and fatty cuts will contain more than lean cuts. Further, fish canned in oil will have more vitamin D than those canned in water. Raw fish is typically eaten in the form of sushi. Raw Atlantic Herring provides the most vitamin D with 1628IU (271% DV) per 100 gram serving, 2996IU (499% DV) per fillet, and 456IU (76% DV) per ounce. It is followed by Pickled Herring with 680IU (113% DV) per 100g serving, Canned Salmon (127% DV), Raw Mackerel (60% DV), Oil Packed Sardines (45% DV), Canned Mackerel (42% DV), and oil packed Tuna (39% DV). A breakfast staple in the Americas, most commercial cereals are fortified with the essential vitamins and nutrients. Exercise caution and check food labels when purchasing cereals, be sure to pick products that have little or no refined sugars, and no partially hydrogenated oils! Fortified cereals can provide up to 342IU (57% DV) per 100 gram serving (~2 cups), and even more if combined with fortified dairy products or fortified soy milk. Products vary widely so be sure to check the nutrition label before buying.
A breakfast staple in the Americas, most commercial cereals are fortified with the essential vitamins and nutrients. Exercise caution and check food labels when purchasing cereals, be sure to pick products that have little or no refined sugars, and no partially hydrogenated oils! Fortified cereals can provide up to 342IU (57% DV) per 100 gram serving (~2 cups), and even more if combined with fortified dairy products or fortified soy milk. Products vary widely so be sure to check the nutrition label before buying. In addition to vitamin D, Oysters are a great source of vitamin b12, zinc, iron, manganese, selenium, and copper. Oysters are also high in cholesterol and should be eaten in moderation by people at risk of heart disease or stroke. Raw wild caught Eastern Oysters provide 320IU (80% DV) per 100 gram serving, 269IU (67% DV) in six medium oysters.
In addition to vitamin D, Oysters are a great source of vitamin b12, zinc, iron, manganese, selenium, and copper. Oysters are also high in cholesterol and should be eaten in moderation by people at risk of heart disease or stroke. Raw wild caught Eastern Oysters provide 320IU (80% DV) per 100 gram serving, 269IU (67% DV) in six medium oysters. Caviar is a common ingredient in sushi and more affordable than people think. Caviar provides 232IU (58% DV) of vitamin D per 100 gram serving, or 37.1IU (9% DV) per teaspoon.
Caviar is a common ingredient in sushi and more affordable than people think. Caviar provides 232IU (58% DV) of vitamin D per 100 gram serving, or 37.1IU (9% DV) per teaspoon. Fortified soy products are often fortified with both vitamin D and calcium. Fortified Tofu can provide up to 157IU (39% DV) of vitamin D per 100 gram serving, or 44IU (11% DV) per ounce. Fortified Soy Milk can provide up to 49IU (12% DV) of vitamin D per 100 gram serving, 119IU (30% DV) per cup. Amounts of vitamin D vary widely between products, so be sure to check nutrition facts for vitamin D content.
Fortified soy products are often fortified with both vitamin D and calcium. Fortified Tofu can provide up to 157IU (39% DV) of vitamin D per 100 gram serving, or 44IU (11% DV) per ounce. Fortified Soy Milk can provide up to 49IU (12% DV) of vitamin D per 100 gram serving, 119IU (30% DV) per cup. Amounts of vitamin D vary widely between products, so be sure to check nutrition facts for vitamin D content. Salami, Ham, and Sausages are a good source of vitamin b12, and copper. Unfortunately, they are also high in cholesterol and sodium, and so should be limited by people at risk of hypertension, heart attack, and stroke. Salami provides 62.0IU (16% DV) of vitamin D per 100 gram serving, or 16.7IU (4% DV) per ounce (3 slices). It is followed by Bologna Pork 56IU (9% DV) per 100 grams, and Bratwurst 44IU (7% DV) per 100 gram serving.
Salami, Ham, and Sausages are a good source of vitamin b12, and copper. Unfortunately, they are also high in cholesterol and sodium, and so should be limited by people at risk of hypertension, heart attack, and stroke. Salami provides 62.0IU (16% DV) of vitamin D per 100 gram serving, or 16.7IU (4% DV) per ounce (3 slices). It is followed by Bologna Pork 56IU (9% DV) per 100 grams, and Bratwurst 44IU (7% DV) per 100 gram serving. Dairy products are already high in calcium, so it makes sense to fortify them with vitamin D. Milk can provide up to 52.0IU (13% DV) of vitamin D per 100 gram serving, 127IU (32% DV) per cup. Cheese can provide up to 6.6IU (2% DV) in a cubic inch, and butter provides 7.8IU (2% DV) in a single tablespoon. Check nutrition labels for exact amounts.
Dairy products are already high in calcium, so it makes sense to fortify them with vitamin D. Milk can provide up to 52.0IU (13% DV) of vitamin D per 100 gram serving, 127IU (32% DV) per cup. Cheese can provide up to 6.6IU (2% DV) in a cubic inch, and butter provides 7.8IU (2% DV) in a single tablespoon. Check nutrition labels for exact amounts. In addition to vitamin D, eggs are a good source of vitamin B12, and protein. Eggs provide 37.0IU (9% DV) of vitamin D per 100 gram serving, or 17.0IU (4% DV) in a large fried egg.
In addition to vitamin D, eggs are a good source of vitamin B12, and protein. Eggs provide 37.0IU (9% DV) of vitamin D per 100 gram serving, or 17.0IU (4% DV) in a large fried egg. More than just a high vitamin D food, mushrooms also provide Vitamin B5 (Pantothenic Acid) and copper. Lightly cooked white button mushrooms provide the most vitamin D with 27.0IU (7% DV) per 100 gram serving, or 7.6IU (2% DV) per ounce.
More than just a high vitamin D food, mushrooms also provide Vitamin B5 (Pantothenic Acid) and copper. Lightly cooked white button mushrooms provide the most vitamin D with 27.0IU (7% DV) per 100 gram serving, or 7.6IU (2% DV) per ounce.

 LinkedIn
LinkedIn Facebook
Facebook Twitter
Twitter GooglePlus
GooglePlus