Case studies
NUMBER 1
Application of the QbD Principles in the Development of the Casopitant Mesylate Manufacturing Process. Process Research Studies for the Definition of the Control Strategy of some Drug Substance-CQAs for Stages 2a, 2b, and 2c
Org. Process Res. Dev., 2010, 14 (4), pp 805–814

Casopitant was identified as a potent NK1 antagonist by GlaxoSmithKline (GSK). It was selected as part of a wide drug discovery programme within GSK for its potential activities on a number of therapeutic targets such as inflammatory bowel disease, overactive bladder, CNS disorders, and others.
The mesylate salt of casopitant was selected for full development. The manufacturing process to casopitant mesylate was developed and optimised by following a Quality by Design approach, whereby a control strategy was developed, underpinned by process understanding and risk analysis, for an enhanced level of quality assurance.
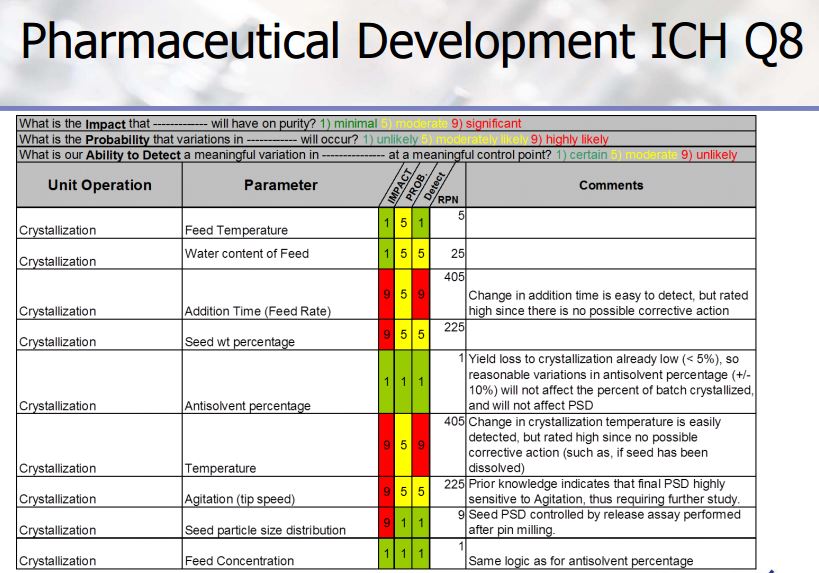
Quality process parameters and specifications levels for the Stages 2a, 2b, and 2c are the elements of the control strategy of the manufacturing process discussed in detail in this paper. The Design of Experiment approach has been extensively used to support the definition of the proven acceptable ranges for the process. The aim is to show the process development studies carried out to ensure quality control for the final drug substance.
(2R,4S)-4-(4-Acetyl-1-piperazinyl)-N-{(1R)-1-[3,5-bis(tri- fluoromethyl)phenyl]ethyl}-2-(4-fluoro-2-methylphenyl)-Nmethyl-1-piperidinecarboxamide Methanesulfonate Salt (Casopitant Mesylate 1). The solution of casopitant 2 was then diluted with EtOAc (overall solution of 2 in EtOAc was 4 L) and acetone (4.5 L) and was heated to the required temperature (from 39 °C). Then, neat methanesulfonic acid (0.12 L, 1.64 mol) was added, followed by a slurry of 2 (0.005 kg) in EtOAc (0.05 L) as seed. The obtained suspension was stirred for 1 h. Then, isooctane (3 L) was added in the required time (1 h), and the slurry was cooled to 20 °C in 2 h and aged 3 h. The suspension was filtered, and the solid was washed with EtOAc (3 × 4 L). The white solid was dried overnight under vacuum at 40 °C to give the desired casopitant mesylate 1 (0.94 kg, yield 48% mol with respect to 7). 1 H NMR (600 MHz, DMSO-d6): δ 9.57 (br s, 1H), 7.99 (br s, 1H), 7.68 (br s, 2H), 7.23 (m, 1H), 6.95 (dd, 1H), 6.82 (m, 1H), 5.31 (q, 1H), 4.45 (m, 1H), 4.20 (dd, 1H), 3.99 (m, 1H), 3.56 (m, 1H), 3.47 (m, 3H), 3.37 (m, 1H), 3.15 (m, 1H), 2.96 (m, 1H), 2.87 (m, 1H), 2.80 (t, 1H), 2.74 (s, 3H), 2.36 (s, 3H), 2.30 (s, 3H), 2.13 (m, 1H), 2.08 (m, 1H), 2.10 (s, 3H), 1.87 (m, 1H), 1.73 (m, 1H), 1.46 (d, 3H). MS: m/z 617 [MH]+, as free base.
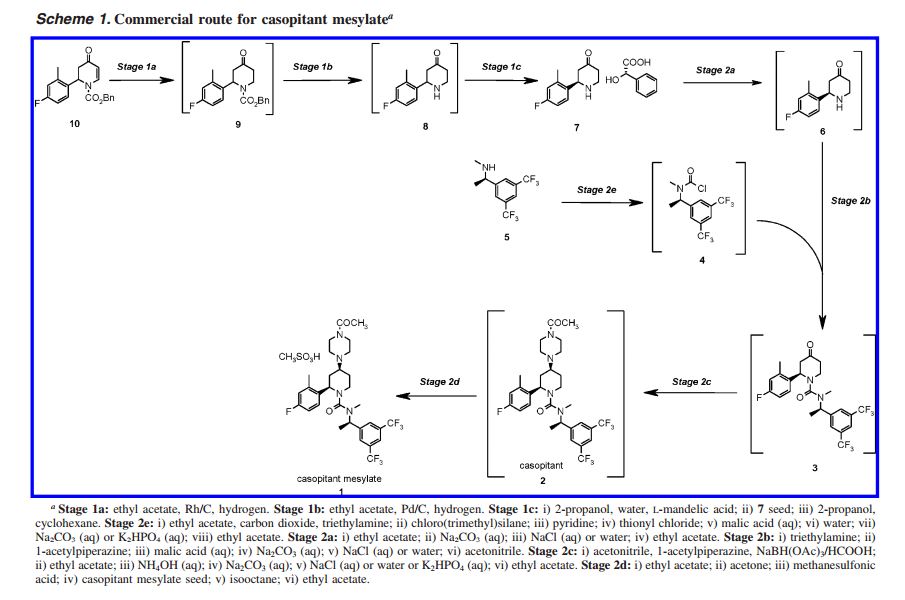
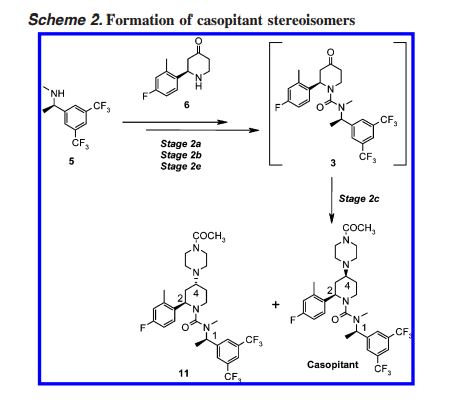
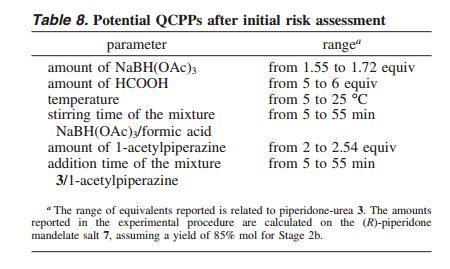




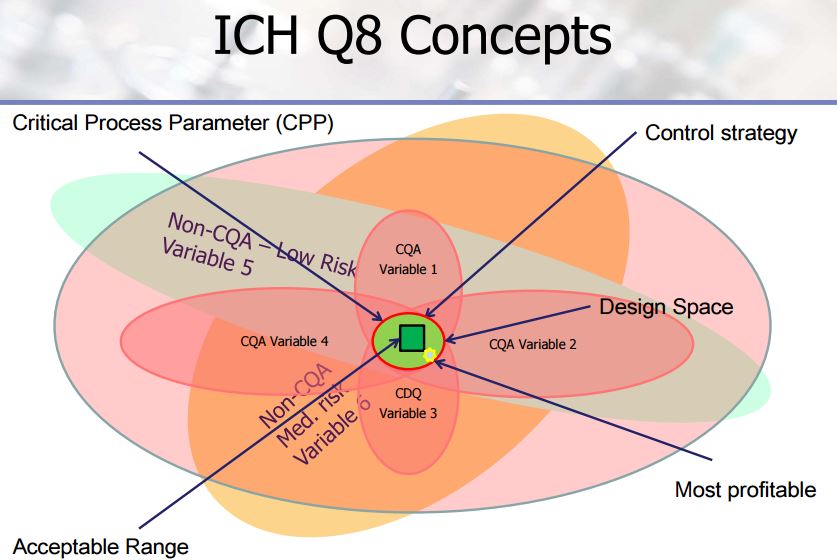
CASE STUDY 2
Development of a Control Strategy for a Defluorinated Analogue in the Manufacturing Process of Casopitant Mesylate
Org. Process Res. Dev., 2010, 14 (4), pp 832–839
DOI: 10.1021/op1000836
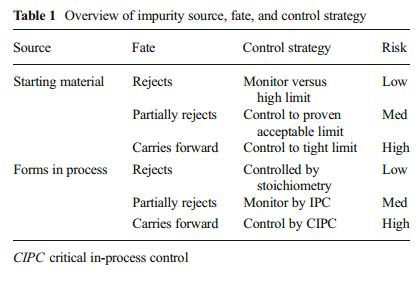
Casopitant mesylate was identified as part of the search for drugs with activity on the Central Nervous System (CNS) by GlaxoSmithKline. During late-phase development studies to develop the manufacturing process, a new impurity was found. This synthetic impurity, a defluorinated analogue of the drug substance, is discussed in detail to show the process development studies carried out to ensure quality control for the final drug substance following the principles of quality by design. The process understanding gained, in combination with risk analysis, allowed the development of a control strategy for enhanced level of quality assurance. This Control Strategy allows moving the control of this impurity to the point of origin instead of testing in the drug substance.







CASE STUDY 3
Journal of Pharmaceutical Innovation
December 2013, Volume 8, Issue 4, pp 247–264
Impact of Quality by Design in Process Development on the Analytical Control Strategy for a Small-Molecule Drug Substance
Bryan C. Castle, Robert A. Forbes
DOI: 10.1007/s12247-013-9165-y
Cite this article as:
Castle, B.C. & Forbes, R.A. J Pharm Innov (2013) 8: 247. doi:10.1007/s12247-013-9165-y
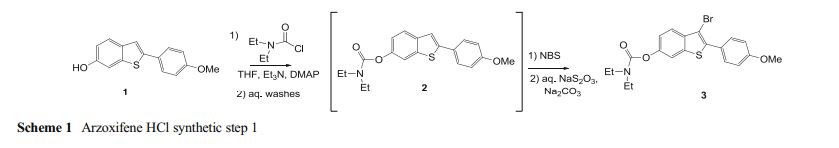
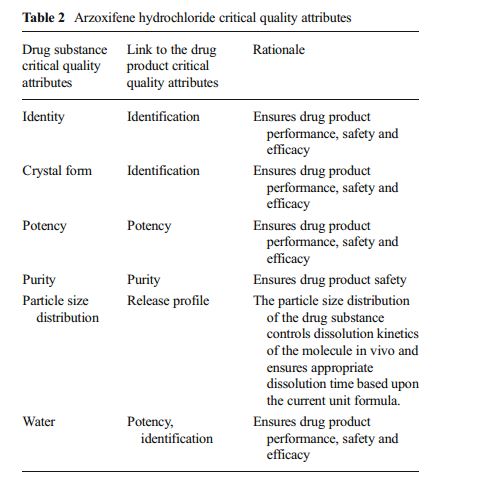
Development of the arzoxifene hydrochloride drug substance manufacturing process, first via a traditional approach and subsequently via an enhanced approach, provides an informative case study in quality by design (QbD). The primary focus of this paper is to illustrate the impact and advantages of QbD on the impurity control strategy. By operating the process at the extremes during design space studies, a larger collection of organic impurities and higher levels of typical impurities are observed in the intermediates. This enables a more thorough understanding of the ability of the process to purify the drug substance and results in a more complete and robust set of intermediate specifications as well as broader and more robust analytical methods. We demonstrate that, when each of the synthetic steps are operated within the design space, the byproduct impurities in the intermediates will not exceed levels found to be rejected in subsequent steps, ensuring that the drug substance will meet its critical quality attributes. Through the rigorous application of an enhanced process development approach, we have designed quality into the arzoxifene hydrochloride drug substance. As a result, real-time release of the intermediate batches is proposed to increase the process throughput and avoid the expense of nonvalue-added testing.
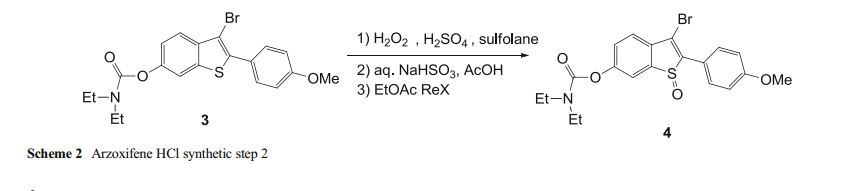
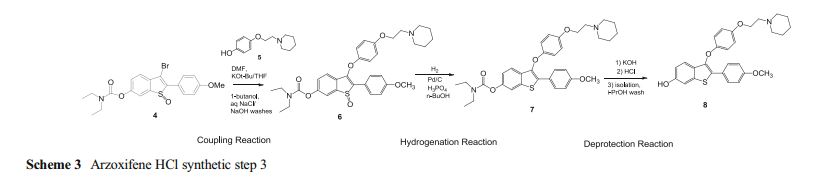



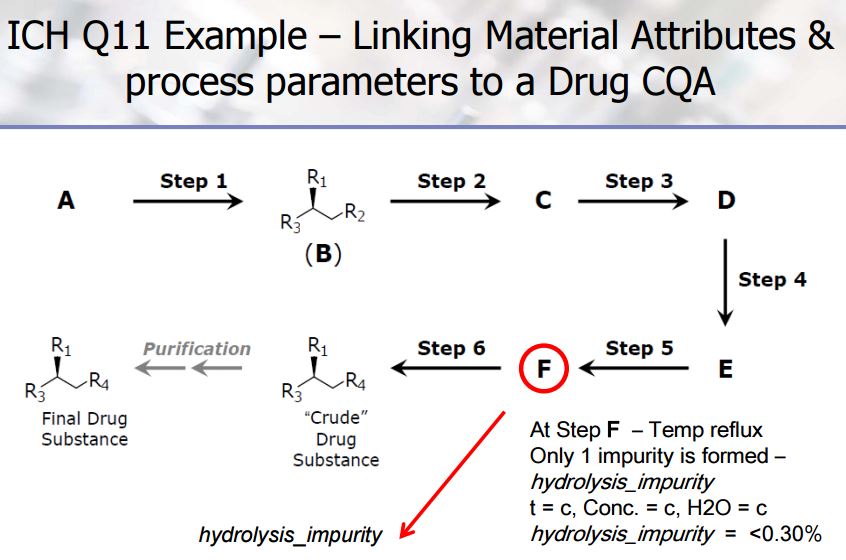


case study 4
Org. Process Res. Dev., 2012, 16 (4), pp 567–576
Modeling-Based Approach Towards Quality by Design for the Ibipinabant API Step
This work presents a process modeling-based methodology towards quality by design that was applied throughout the development lifecycle of the ibipinabant API step. By combining mechanistic kinetic modeling with fundamental thermodynamics, the degradation of the API enantiomeric purity was described across a large multivariate process knowledge space. This knowledge space was then narrowed down to the process design space through risk assessment, target quality specifications, practical operating conditions for scale-up, and plant control capabilities. Subsequent analysis of process throughput and yield defined the target operating conditions and normal operating ranges for a specific pilot-plant implementation. Model predictions were verified via results obtained in the laboratory and at pilot-plant scale. Future efforts were focused on increasing fundamental process knowledge, improving model confidence, and using a risk-based approach to reevaluate the design space and selected operating conditions for the next scale-up campaign.
API process at the time of the first pilot-plant campaign

changed to

Process for the second pilot-plant implementation
Process parameter ranges and typical results from approximately 20 lab experiments conducted on the process shown in Scheme



Figure 3. Ishikawa diagram for the API step, highlighting factors that potentially affect the enantiomeric purity of the product. Factors shown in blue were accounted for in the sulfonylation reaction and distillative crystallization models. Factors shown in red were not included in the models
| process parameter |
min. value |
max. value |
# of “levels” |
| sulfonylation reaction model |
| temp. (°C) |
5 |
35 |
7 |
| 4-chlorobenzenesulfonyl chloride (equiv) |
1.0 |
1.2 |
6 |
| conc. (mL/g) |
5 |
10 |
6 |
| reaction time (h) |
2 |
5 |
4 |
| distillative crystallization model |
| pressure (mbar) |
300 |
1013 |
6 |
| residual 2(AP) |
0.05 |
2.0 |
6 |
| distillation time (h) |
8 |
48 |
4 |
| distillation end point (wt % EtOH) |
90 |
98 |
3 |


CASE STUDY FROM METTLER TOLEDO
Innovative Techniques
To Synthesize Breakthrough Molecules
SEE http://www.allfordrugs.com/qbd-presentations/
FOR PRESENTATION ON THIS
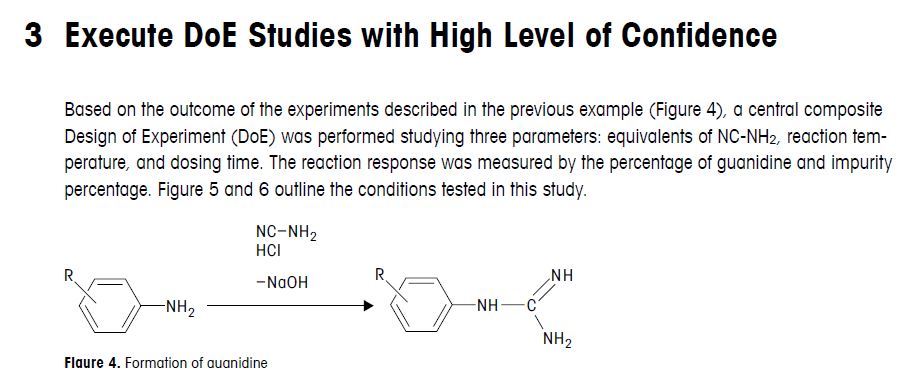
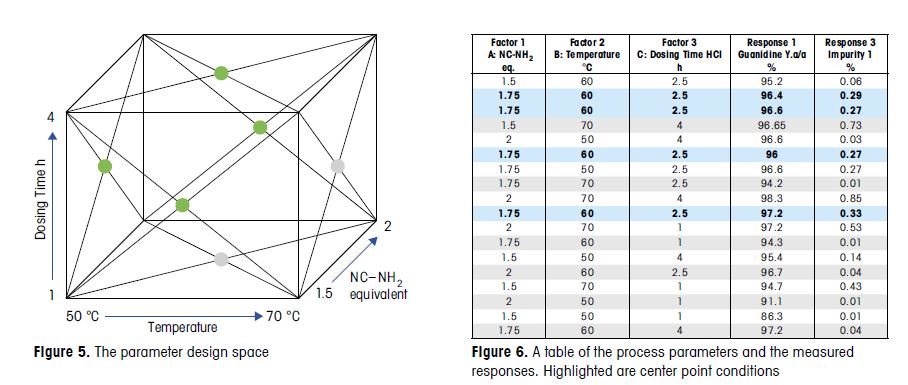
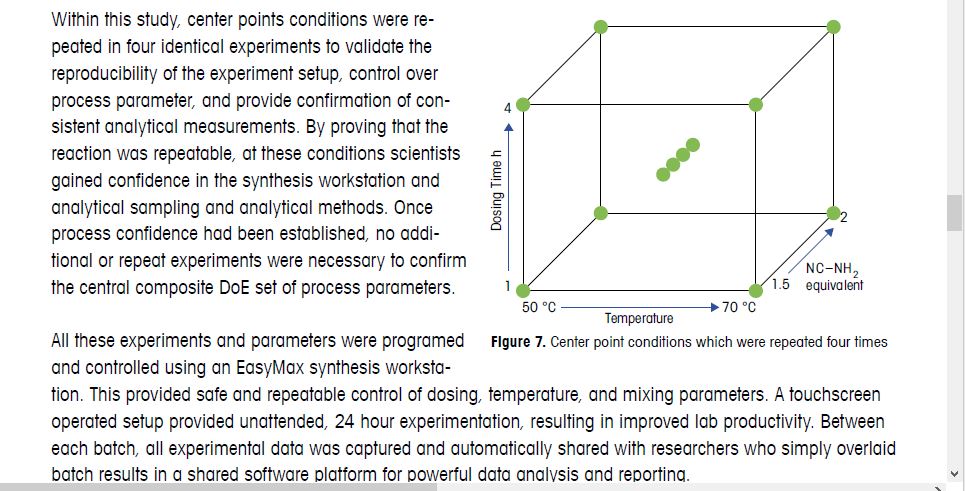
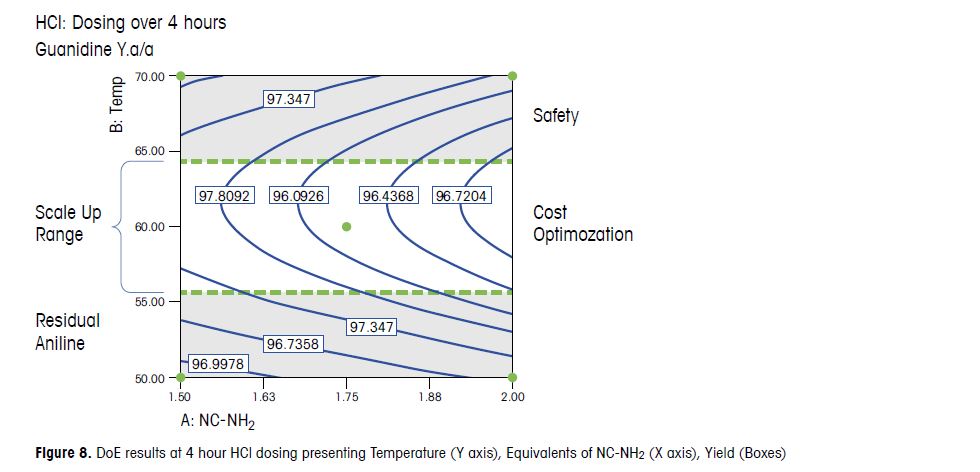
Based on the outcome of the DoE study, 1.75 equivalents of NC-NH2 with a 4 hour HCl dosing were chosen for
scale up. These conditions balanced process robustness with optimized yield, purity, and economic recovery.
Faster HCl dosing rates were avoided due to risk of accumulation. 60 °C was also chosen, higher temperatures
were avoided due to the safety risk and lower temperatures resulted in residual aniline. The process was
successfully introduced into production and multiple tons of material have been produced with this process,
without failure.
Next…………..
Application of On-Line NIR for Process Control during the Manufacture of Sitagliptin
George Zhou*, Shane Grosser, Lei Sun, Gabriel Graffius, Ganeshwar Prasad, Aaron Moment, Angela Spartalis,Paul Fernandez, John Higgins, Busolo Wabuyele, and Cindy Starbuck
Global Science, Technology and Commercialization, Merck Sharp & Dohme Corporation P.O. Box 2000, Rahway, New Jersey 07065, United States
Org. Process Res. Dev., 2016, 20 (3), pp 653–660
DOI: 10.1021/acs.oprd.5b00409
Publication Date (Web): February 12, 2016
Copyright © 2016 American Chemical Society
Abstract

The transamination-chemistry-based process for sitagliptin is a through-process, which challenges the crystallization of the active pharmaceutical ingredient (API) in a batch stream composed of multiple components. Risk-assessment-based design of experiment (DoE) studies of particle size distribution (PSD) and crystallization showed that the final API PSD strongly depends on the seeding-point temperature, which in turn relies on the solution composition. To determine the solution composition, near-infrared (NIR) methods had been developed with partial least squares (PLS) regression on spectra of simulated process samples whose compositions were made by spiking each pure component, either sitagliptin free base (FB), water, isopropyl alcohol (IPA), dimethyl sulfoxide (DMSO), or isopropyl acetate (IPAc), into the process stream according to a DoE. An additional update to the PLS models was made by incorporating the matrix difference between simulated samples in lab and factory batches. Overall, at temperatures of 20–35 °C, the NIR models provided a standard error of prediction (SEP) of less than 0.23 wt % for FB in 10.56–32.91 wt %, 0.22 wt % for DMSO in 3.77–19.18 wt %, 0.32 wt % for IPAc in 0.00–5.70 wt %, and 0.23 wt % for water in 11.20–28.58 wt %. After passing the performance qualification, these on-line NIR methods were successfully established and applied for the on-line analysis of production batches for compositions prior to the seeding point of sitagliptin crystallization.
http://pubs.acs.org/doi/abs/10.1021/acs.oprd.5b00409?journalCode=oprdfk
Next…………..
www.soci.org/~/media/Files/…/2011/…/Jake_Janey_Presentation.ashx
Nov 2, 2011 – 9 Steps, 52% overall yield, >100Kg of sitagliptin prepared ….. FDA filings requires “Quality by Design”: A way to allow process changes within.
A PRESENTATION
A PRESENTATION
https://www.pmda.go.jp/files/000213677.pdf
Aug 11, 2016 – QbD assessment experience in Japan … Number of Approved Products with QbD … Active Ingredient : Sitagliptin Phosphate Hydrate.
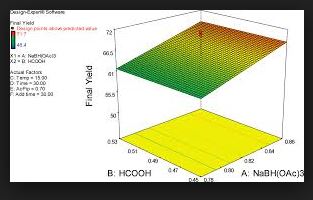
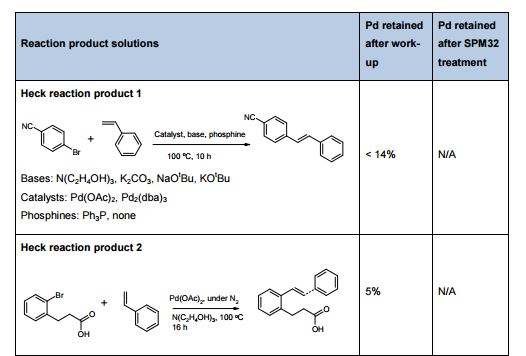
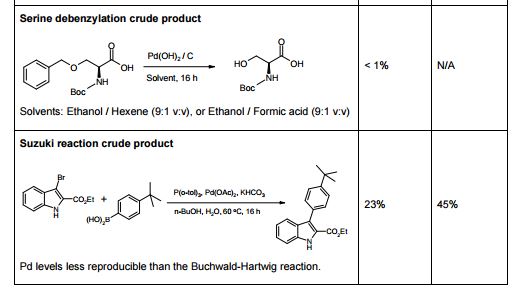
Statistical DoE Approach to the Removal of Palladium from Active Pharmaceutical Ingredients (APIs) by Functionalized Silica Adsorbents

The influence of four parameters (temperature, scavenging time, amount of scavenger, and concentration of palladium in the solution) on the efficiency of Pd removal from a cross-coupling reaction, using a commercially available Pd scavenger, SPM32, was studied. The DoE-based method employed yielded more information than is readily attainable from standard adsorption isotherms and kinetics experiments. The optimal regime of scavenging was identified; intuitive and nonintuitive effects of temperature, scavenging time, and scavenger amounts were highlighted; and a mathematical model quantifying predicted Pd removal from the synthetic intermediate was built.
link http://pubs.acs.org/doi/abs/10.1021/op5000336
Statistical DoE Approach to the Removal of Palladium from Active Pharmaceutical Ingredients (APIs) by Functionalized Silica Adsorbents
PhosphonicS Ltd., 44c Western Avenue, Milton Park, Abingdon, OX14 4RU, United Kingdom
Org. Process Res. Dev., 2014, 18 (5), pp 626–635
DOI: 10.1021/op5000336
Publication Date (Web): April 14, 2014
Copyright © 2014 American Chemical Society
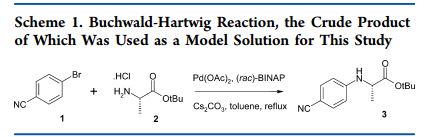
Preparation of tert-butyl 2-[(4-cyanophenyl)amino]propanoate (3).
4-Bromobenzonitrile (18.20 g, 100 mmol), L-alanine tert-butyl ester hydrochloride (21.73 g, 120 mmol), ±BINAP (1.25 g, 2 mmol) and cesium carbonate (48.87 g, 150 mmol) were added to a 3-neck round bottom flask containing a magnetic stirrer. Toluene (167 mL) was added and a reflux condenser, thermometer and a rubber septum were attached. Argon gas was bubbled through as the heterogeneous mixture was warmed to reflux temperature with slow agitation from the magnetic stirrer. Palladium acetate (0.45 g, 2 mmol) was added quickly through one of the side-arm joints and de-gassing was continued for 5 min. The reaction mixture was kept under argon at reflux and the disappearance of 4-bromobenzonitrile was monitored by GC-MS. After 16-24 h, the reaction mixture was filtered through a sinter funnel, washed with toluene and then filtered through a nylon membrane (Sigma Aldrich catalogue no. Z290793, 0.45 µm pore size) and washed with toluene. This crude reaction mixture was used in the DoE matrix without further purification.
Experimental results
NMR δC (62.9 MHz, CDCl3): 172.7, 150.0, 133.7, 120.4, 112.7, 99.3, 82.3, 51.7, 28.0, 18.5 ppm.
NMR δH (250 MHz, CDCl3): 7.39 (2H, d, J = 8.8 Hz, Ph), 6.52 (2H, d, J = 8.8 Hz, Ph), 4.85 (1H, d, J = 7.4 Hz, NH), 4.01 (1H, quintet, J = 7.4 Hz, CH(Me), 1.43 (9H, s, tBu), 1.42 (3H, d, J = 7.4 Hz, Me).
GC/MS: GC method used: hold at 50 °C for 4 min; increase temperature from 50 to 280 °C at 30 °C / min; hold at 280 °C for 5 min. Peaks were recorded and identified as follows: 4-Bromobenzonitrile: 10.05 min. Molecular peak observed at m/z = 181 for the 79Br isotope, m/z = 183 for the 81Br isotope L-alanine tert-butyl ester hydrochloride: not observed. Retention time less than 5 min, peak lost within the solvent front. Product: 13.64 min. Molecular peak observed at m/z = 246, main fragment at m/z = 146 (M – CO2 t Bu) Side product (not identified or quantified, minor peak): 14.56 min.
Corresponding Author *E-mail: jan.recho@phosphonics.com.
WILL BE UPDATED WITH MORE, WATCH OUT

/////////
| Name |
Explanation |
| Active Pharmaceutical Ingredient (API) |
An active pharmaceutical ingredient (API) is a substance used in a finished pharmaceutical product, intended to furnish pharmacological activity or to otherwise have direct effect in the diagnosis, cure, mitigation, treatment or prevention of disease, or to have direct effect in restoring, correcting or modifying physiological functions in human beings. |
| Annual Product Reviews (APR) |
The Annual Product Reviews (APR) include all data necessary for evaluation of the quality standards of each drug product to determine the need for changes in drug product specifications or manufacturing or control procedures. The APR is required by the U.S. Code of Federal Regulations. |
| ANVISA |
The Brazilian Health Surveillance Agency (in Portuguese, Agência Nacional de Vigilância Sanitária) is a governmental regulatory body in Brazil. Similar to the FDA in the United States, it oversees the approval of drugs and other health products and regulates cosmetics, food products, and other health-related industries. |
| Biologic License Application (BLA) |
The Biologics License Application (BLA) is a request for permission to introduce, or deliver for introduction, a biologic product into commerce in the U.S. |
| CFDA |
The China Food and Drug Administration is similar to the FDA in the United States and is responsible for regulating food and drug safety. |
| cGMP |
Current Good Manufacturing Practices govern the design, monitoring, and control of manufacturing facilities and processes and are enforced by the US FDA. Compliance with these regulations helps safeguard a drug’s identity, strength, quality, and purity. |
| COFEPRIS |
The Federal Commission for Protection against Sanitary Risks (in Spanish, Comisión Federal para la Protección contra Riesgos Sanitarios) is a government agency in Mexico. It regulates food safety, drugs, medical devices, organ transplants, and environmental protection. |
| Common Technical Document (CTD) |
The Common Technical Document (CTD) is the mandatory common format for new drug applications in the EU and Japan, and the U.S. The CTD assembles all the Quality, Safety and Efficacy information necessary for a drug application. |
| European Medicines Agency (EMA) |
The European Medicines Agency (EMA) is a decentralised agency of the European Union (EU), located in London. It began operating in 1995. The Agency is responsible for the scientific evaluation, supervision and safety monitoring of medicines developed by pharmaceutical companies for use in the EU. |
| Food and Drug Administration (FDA) |
The Food and Drug Administration (FDA) is an agency within the U.S. Department of Health and Human Services. The FDA is responsible for the approval of new pharmaceutical products for sale in the U.S. and performs audits at the companies participating in the manufacture of pharmaceuticals to ensure that they comply with regulations. |
| Human growth hormone |
A growth hormone (GH or HGH) is a peptide hormone produced by the pituitary gland that stimulates growth in children and adolescents. It is involved in several body processes, including cell reproduction and regeneration, regulation of body fluids, and metabolism. It can be produced by the body (ie, somatotropin) or genetically engineered (ie, somatropin). |
| In-Process Control (IPC) |
In-Process Controls (IPC) are checks performed during production in order to monitor and if necessary to adjust the process to ensure that the product conforms its specification. |
| Interferons (INFs) |
Interferons are proteins produced by the body as part of the immune response. They are classified as cytokines, proteins that signal other cells to trigger action. For example, a cell infected by a virus will release interferons to stimulate the defenses of nearby cells. |
| Interleukins |
Interleukins are proteins produced by cells as an inflammatory response. Most interleukins help leukocytes communicate with and direct the division and differentiation of other cells. |
| Investigational Medicinal Product Dossier (IMPD) |
The Investigational Medicinal Product Dossier (IMPD) is the basis for approval of clinical trials by the competent authorities in the EU. The IMPD includes summaries of information related to the quality, manufacture and control of the Investigational Medicinal Product, data from non-clinical studies and from its clinical use. |
| Investigational New Drug (IND) |
An Investigational New Drug application is provided to the FDA to obtain permission to test a new drug in humans in Phase I – III clinical studies. The IND is reviewed by the FDA to ensure that study participants will not be placed at unreasonable risk. |
| Marketing Authorization Application (MAA) |
The Marketing Authorization Application (MAA) is a common document used as the basis for a marketing application across all European markets, plus Australia, New Zealand, South Africa, and Israel. This application is based on a full review of all quality, safety, and efficacy data, including clinical study reports. |
| Master batch records |
These general manufacturing instructions, which are required by cGMP, are the bases for a precise, detailed description of a pharmaceutical manufacturing process. They ensure that all proper ingredients are included, each process step is completed, and the process is controlled. |
| Medicines and Healthcare Products Regulatory Agency (MHRA) |
The Medicines and Healthcare products Regulatory Agency (MHRA) regulates medicines, medical devices and blood components for transfusion in the UK. MHRA is an executive agency, sponsored by the Department of Health. |
| MFDS |
The Ministry of Food and Drug Safety (formerly the Korean Food & Drug Administration) is a government agency that oversees the safety and efficacy of drugs and medical devices in South Korea. |
| Monoclonal antibodies |
Monoclonal antibodies are antibodies made in a laboratory from identical immune cells that are clones of a single cell. They are distinct from polyclonal antibodies, which are made from different immune cells. |
| NDA |
A New Drug Application (NDA) is the vehicle submitted to the FDA by drug companies in order to gain approval to market a new product. Safety and efficacy data, proposed package labeling, and the drug’s manufacturing methods are typically included in an NDA. |
| New Drug Application (NDA) |
The New Drug Application (NDA) is the vehicle through which drug sponsors formally propose that the FDA approve a new chemical pharmaceutical for sale and marketing in the U.S. |
| Oligonucleotides |
These short nucleic acid chains (made up of DNA or RNA molecules) are used in genetic testing, research, and forensics. |
| Parenteral |
Parenteral medicine is taken or administered in a manner other than through the digestive tract. Intravenous and intramuscular injections are two examples. |
| Peptide hormones |
Peptide hormones are proteins secreted by organs such as the pituitary gland, thyroid, and adrenal glands. Examples include follicle-stimulating hormone (FSH) and luteinizing hormone. Similar to other proteins, peptide hormones are synthesized in cells from amino acids. |
| PMDA |
The Pharmaceuticals Medical Devices Agency is an independent administrative agency that works with the Ministry of Health, Labour and Welfare to oversee the safety and quality of drugs and medical devices in Japan. |
| Process Analytical Technology (PAT) |
These analytical tools help monitor and control the manufacturing process, including accommodating for variability in material and equipment, in order to ensure consistent quality. |
| Product Quality Reviews (PQR) |
The Product Quality Reviews (PQR) of all authorized medicinal products, is conducted with the objective of verifying the consistency of the existing process, the appropriateness of current specifications for both starting materials and finished product, to highlight any trends and to identify product and process improvements. The PQR is required by the EU GMP Guideline. |
| Quality by Design (QbD) |
This concept involves a holistic, proactive, science- and risk-based approach to the development and manufacturing of drugs. At the heart of QbD is the idea that quality is achieved through in-depth understanding of the product and the process by which it is developed and manufactured. |
| Restricted Access Barrier System (RABS) |
This advanced aseptic processing system provides an enclosed environment that reduces the risk of contamination to the product, containers, closures, and product contact surfaces. As a result, it can be used in many applications in a fill-finish area. |
| Scale-up |
Scale-up involves taking a small-scale manufacturing system developed in the laboratory to a commercially viable, robust production process. |
| Six Sigma |
Six Sigma is a set of quality management methods, techniques, and tools used to improve manufacturing, transactional, and other business processes. The goal is to enhance quality (as well as employee morale and profits) by identifying and eliminating the cause of errors and process variations. |
| Target Product Profile (TPP) |
This key strategic document summarizes the features of an intended drug product. Characteristics may include the dosage form, route of administration, dosage strength, pharmacokinetics, and drug product quality criteria. |
| TFDA |
The Taiwan Food & Drug Administration is a governmental body devoted to enhancing food safety and drug quality in that country. |
“ALL FOR DRUGS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This is a compilation for educational purposes only. P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
50,248 total views, 2 views today