Process Validation












http://www.lmi.no/media/2852497/process_validation_jarle_a_haugan_gehc.pdf

Process Validation and Regulatory Review
Drug Approval Strategies in the Age of Fast Track, Breakthrough Therapy and Accelerated Approval
To meaningfully discuss the process validation and regulatory approval strategies required for drugs that have been designated Fast Track, Breakthrough Therapy or Accelerated Approval drugs, we must first clarify these designations and briefly remind ourselves what the Process Validation guidance looks like. Then we will be able to clearly identify challenges and approaches to these barriers when working to bring a Fast Track, Accelerated Approval or Breakthrough Therapy drug to market.
Fast Track designation – Fast Track drugs treat serious conditions where there is an unmet medical need. Concluding that a condition is serious and that there is an unmet medical need most definitely leaves room for judgement, but generally speaking, the conditions these drugs treat are life-threatening, and the drug in question is expected to contribute to survival, daily functioning or the likelihood that a condition will advance to a very serious state. Fast Track drugs receive the benefit of more frequent meetings and communication with the FDA, and the drug qualifies for Accelerated Approval and rolling review of the Biologic License Application (BLA) or New Drug Application (NDA).
Breakthrough Therapy – Breakthrough Therapy status can be assigned to drugs that treat a serious condition when preliminary clinical data show significantly improved outcomes compared to treatments currently on the market. Breakthrough Therapies are eligible for: Fast Track designation benefits, extensive FDA guidance on effective drug development early in the development process and organizational commitment, including access to FDA senior managers.
Accelerated Approval – The FDA established accelerated approval regulations in 1992. Accelerated Approval could be given to drugs that met a serious unmet medical need, and approval was based on a surrogate endpoint. Fast forward to 2012 when Congress passed the Food and Drug Administration Safety Innovations Act (FDASIA). This amendment to the Federal Food, Drug, and Cosmetic Act (FD&C Act) allowed approval to be based on either a surrogate endpoint per the 1992 regulations or approval based on an intermediate clinical endpoint. For example, as a result of the 2012 legislation, a cancer drug could be approved based on the surrogate endpoint of increasing the probability of cancer to going into remission or the intermediate clinical endpoint of shrinking tumor size—an outcome that is strongly correlated with the ability to much more successfully treat cancer and induce remission.
These FDA designations are clearly designed to increase the availability and speed to market of drugs treating serious conditions where unmet medical needs exist. Given that nimbleness and speed has historically not been the pharmaceutical industry’s nor FDA’s strong suit—commercialization of a drug has historically taken on average 12 years and cost up to $2.5B (including expenditure outlays and opportunity costs). The ability for these designations to save both time and money is very attractive. However, given the slow-moving nature of the industry, changes in both mindset and approaches are needed by both drug innovators and regulators to validate processes and ensure drug quality within the faster-moving constructs.
Let’s now turn to the most recent Process Validation guidance so that we may juxtapose that system with the nimble needs of Fast Track Designation, Breakthrough Therapy and Accelerated Approval drugs—ultimately, making some observations regarding needed Process Validation and overall regulatory approval approaches as the industry moves towards accelerated development processes for an increasing number of drugs.

WHAT IS PROCESS VALIDATION?
According to the FDA’s 2011 Process Validation (PV) guidance, “For purposes of this guidance, process validation is defined as the collection and evaluation of data, from the process design stage through commercial production, which establishes scientific evidence that a process is capable of consistently delivering quality product. Process validation involves a series of activities taking place over the lifecycle of the product and process.”
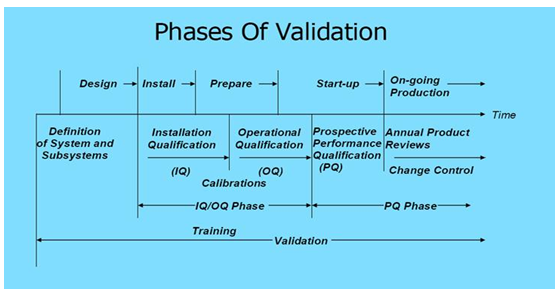
The Three Stages of Process Validation:
Stage 1: Process Design–manufacturing process is defined during this stage and is based on knowledge acquired through development and scale-up activities.
Stage 2: Process Qualification–process design is evaluated to determine if the process is capable of reproducible commercial manufacturing.
Stage 3: Continued Process Verification–ongoing assurance during manufacturing that the process is controlled and the outcome predictable.

Keys for Successful Validation Include:
• Gaining knowledge from the product and process development
• Understanding sources of variation in the production process
• Determining the presence of and degree of variation
• Understanding the impact of variation on the process and end product
• Controlling variation in a manner aligned with Critical Quality Attributes (CQA) and the risk a given attribute introduces to the process
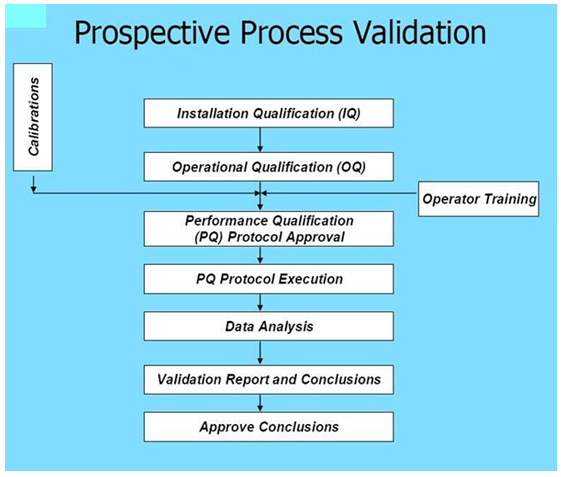
Process Qualification, a key component of Process Validation, should be based on overall level of product and process understanding, level of demonstrable control, data from lab, pilot and commercial batches, effect of scale and previous experience with similar products and processes. Process Qualification is generally recommended to be based on higher levels of sampling, additional testing and greater scrutiny of process performance than would be typical of routine commercial production.
As we will now explore, some of the demands of Process Qualification and overall Process Validation is severely challenged by the approaches required when bringing a Fast Track, Accelerated Approval or Breakthrough Therapy drug to market.
NOVEL APPROACHES NEEDED FOR ACCELERATED APPROVALS
Historically, it has taken an average of 12 years and, according to a Tufts Center for the Study of Drug Development (CSDD) report, including expenditures and opportunity costs, an average of ~$2.6 billion to bring a prescription drug to market. This paper will refrain from making editorial comments about this pharmaceutical industry fact; however, the undeniable reality is that the speed required at every point in the industry to develop Fast Track, Accelerated Approval or Breakthrough drugs is having a profound impact.
Approval of a Breakthrough drug, which of course is classified for Accelerated Approval, means manufacturers need to develop Chemistry, Manufacturing and Controls (CMC) data in about half the time of the traditional process. In addition, Breakthrough designation does not mean the innovator company can do less. In order to meet these accelerated timelines, they do need to start analytical methods creation and product and process characterization sooner, and handle the process differently. Validation of a process traditionally has called for sufficient data and an adequate number of runs to convince the manufacturer (and regulators) that the process works. As we will explore below, Breakthrough therapies are often in the market before the product is fully validated.
However, the guiding force behind these new approaches is that despite sharply reduced timeframes, manufacturers cannot compromise patient safety or product supply. Therefore, characterization of critical product and process attributes is typically required much earlier in the process.
Challenges and Realities of Process Validation and Regulatory Approval within the Accelerated Drug Paradigm:
• The collaboration and communication required between the FDA and innovator companies is extensive. Given limited FDA resources and extensive resources required by the organizations of innovator companies, is the growth of the Fast Track/Breakthrough Therapy/Accelerated Approval programs sustainable?
• New Drug Applications (NDA) for Breakthrough Therapies include less manufacturing information and data requiring alternative risk-mitigation approaches and often nontraditional statistical models.
• Both patient safety and product supply is at the forefront, without the data and historical knowledge traditionally used to address these concerns.
• The primary concerns for CMC reviewers include incomplete characterization of the drug, underdeveloped analytical methods and a lack of full understanding of a product’s Critical Quality Attributes (CQA) and associated risks.
• Process Validation will, in many cases, be incomplete at product launch.

THE CHANGED PARADIGM RESTORED TO ORDER (SORT OF)
The “restored order” for the approval of, and ultimate Process Validation for, Breakthrough/Accelerated Approval drugs will not look like anything we normally see. Again, all Breakthrough and Accelerated Approval drugs address very serious conditions and offer treatment where none currently exists, or offers benefits well above and beyond drug products currently on the market. Therefore, flexibility has been applied to segments of the traditional product review and approval process to speed the availability of treatments for these critical conditions.
Despite the flexibility in, and often changes to the product review and approval process, patient safety remains at the forefront, as well as the guarantee of consistent product supply.
Approaches for Successfully Handling the Approval and Validation of Accelerated Approval Drugs:
• Open and transparent communication with the FDA is essential throughout the entire approval and post-market process. The pharmaceutical company mindset of not wanting to learn certain information for fear of needing to revalidate based on those discoveries has no place in this new reality. New information will be learned pre- and post-launch, and plenty of amendments will need to be filed.
• Given the compressed development timeframes, less stability data will be available at submission. Additional data will be submitted via amendments during the review cycle, and in some cases, post-market.
• Launch commercial process with limited experience and optimize post-approval–the classic three runs is not the guiding force within this construct. The level of flexibility regulators will extend is determined for each specific product. Factors taken into consideration include: riskiness of product characteristics, seriousness of the condition and medical need, complexity of manufacturing processes, state of the innovator’s quality system and merits of the innovator’s risk-based quality assessment including Critical Quality Attributes (CQA).
• Novel statistical models and approaches will need to be applied in many cases. Representative samples and assays for these models will likely need to be acquired from sources, like prior knowledge and use of comparability protocols. Also, determination of the appropriate use of stability data from representative pilot scale lots will be required.
• Manufacturers should freely acknowledge where data is limited, demonstrate that the missing data pose no risk to patient safety or product supply and outline post-market strategy for acquiring the missing data. Conversations with the FDA are clearly required for successful outcomes.
• Focus on patient safety and reliable supply of quality product at launch, not process optimization. In addition, begin critical product attributes and process characterization work much earlier than a typical pharmaceutical development process. In many cases, consider broader product quality ranges for non-Critical Quality Attributes until further manufacturing experience is acquired post-approval.
Enhance analytical methods and understanding to offset more limited process understanding and to support future comparability work. Extremely important, involve commercial Quality Control representatives in the development assay design.
• Again, CMC activities that may be incomplete at launch include: Process Validation, stability studies on commercial product, manufacturing scale/tech transfer data and complete control system data.
• A post-approval product lifecycle management plan is a must, and it needs to be included in the filing to support deferred CMC activities.
Fast Track, Breakthrough Therapy and Accelerated Approval drugs have profoundly changed the thinking and approach to Process Validation and other CMC activities.
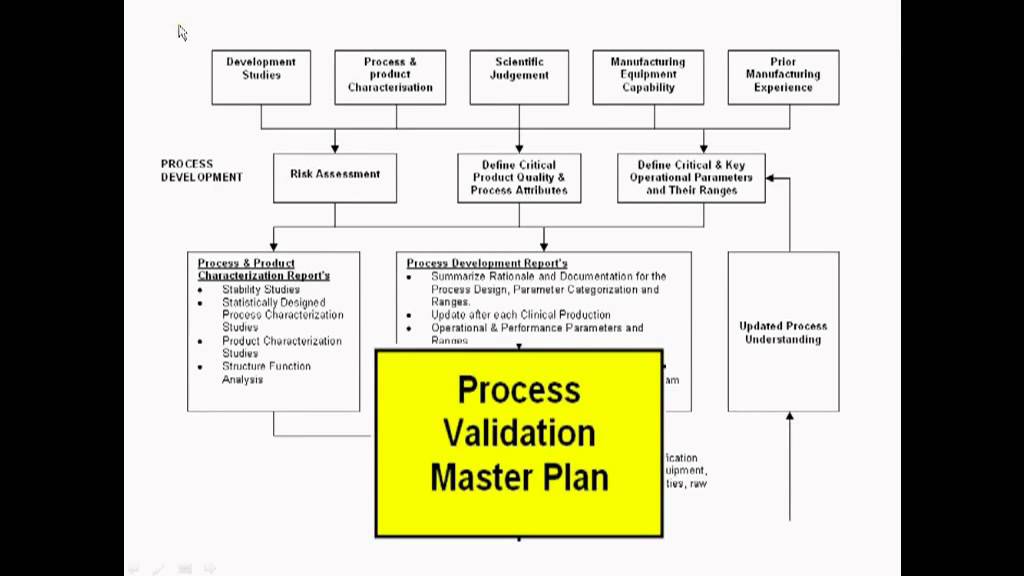
Sources:
Joseph A. DiMasia, Henry G. Grabowskib, Ronald W. Hansenc, “Innovation in the Pharmaceutical Industry: New Estimates of R&D costs,” Tufts Center for the Study of Drug Development, Tufts UniversityJ. Wechsler, “Breakthrough Drugs Raise Development and Production Challenges,” Pharmaceutical Technology 39 (7) 2015.Earl S. Dye, PhD, “CMC/GMP Considerations for Accelerated Development and Launch of Breakthrough Therapy Products,” Roche“Guidance for Industry Expedited Programs for Serious Conditions – Drugs and Biologics,” U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER), May 2014 ProceduralAnthony Mire-Sluis, Michelle Frazier, Kimberly May, Emanuela Lacana, Nancy Green, Earl Dye, Stephan Krause, Emily Shacter, Ilona Reischl, Rohini Deshpande and Joe Kutza, “Accelerated Product Development: Leveraging Industry and Regulator Knowledge to Bring Products to Patients Quickly,” BioProcess International, December 2014
Daniel Alsmeyer and Ajay Pazhayattil, Apotex Inc., “A Case for Stage 3 Continued Process Verification,” Pharmaceutical Manufacturing, May 2014



Overcoming The Challenge Of The 2011 FDA Process Validation Guidance

A paradigm shift is underway in process validation. The FDA revised the guidance to industry for process validation in January 2011. This guidance defines process validation as “the collection and evaluation of data, from the process design stage through commercial production which establishes scientific evidence that a process is capable of consistently delivering quality product.” The new emphasis is on “scientific” rather than “documented” evidence. [1]
This new approach is aimed at increasing process understanding and control. The consequences of ineffective process validation are significant as evidenced by increased cost of compliance: Adverse events, warning letters, delays in product approval and launch, plant remediation, field alerts, recalls and product shortages.
Implications For Industry
Pharmaceutical manufacturers must now make deliberate decisions about which statistical tools and analyses are appropriate for their products and processes. For example, the traditional “rule of three” for the number of product batches required to validate a process has been challenged. The use of statistical tools are now encouraged not only to determine the number of batches required to validate a process but throughout the process life cycle to demonstrate product integrity, safety, and efficacy.
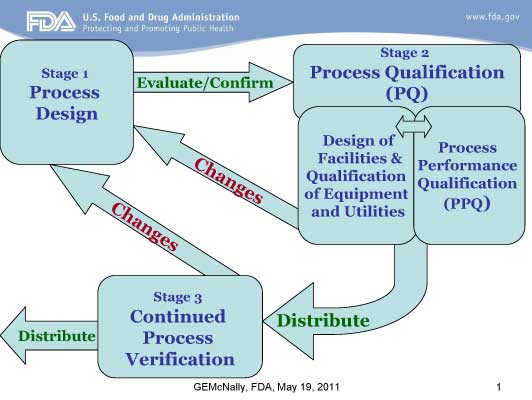
The larger paradigm shift is that the former approach to validation focused only on what is now called Stage 2: Process Qualification. The new guidance looks at the life cycle beginning with Stage 1: Process Design and ending with Stage 3: Continued Process Verification, in addition to the existing Stage 2.
This has produced a vexing problem for many pharmaceutical executives, especially problematic for midsize to small firms. The standards and expectations are being raised at the same time that drug products are becoming more sophisticated. Historical formulations were generally simpler; now products have complex release mechanisms making it more challenging to demonstrate equivalent bioavailability. Risk assessment is now integral to the validation process and must consider supply chain not under direct control, such as global raw material sources. There is also pressure to drive costs down and reduce production cycles.
New questions are raised: How is an understanding of process design demonstrated? What variables must be considered? What are the inputs and outputs? How are the new risk assessments accomplished? How are statistically valid results demonstrated? Is the internal statistical staff current with FDA guidance?
Some of the specific technical statistical skills that would be required to fully cover the new FDA Guidance include Design of Experiments (DOE), sample size determination, linear regression and correlation, nonlinear regression, Analysis of Variance (ANOVA), and measurement system analysis, to name a few. In addition to competency with the statistical tools, the ability to interpret data and integrate the science of the process with the specific statistical tools is critical. The higher the level of physical-chemical knowledge and pharmaceutical science knowledge coupled with a broad base of statistical skills, the better the outcome.
The benefits of statistical analysis are numerous (think “Cheaper/Better/Faster”): accelerated innovation cycle time, reduced total cost of process and product, improved compliance levels, enhanced quality and safety, reduced rework and recalls and improved patient safety and efficacy.
Practical Statistical Applications
The 2011 Guidance promotes a life-cycle approach to process validation that includes scientifically sound design practices, robust qualification, and process verification. The lifecycle concept links product and process development, qualification of the commercial manufacturing process, and maintenance of the process in a state of control during commercial production and throughout the ownership of the product by the manufacturer.
Stage 1- Process Design: The commercial manufacturing process is defined during this stage based on knowledge gained through development and scale-up activities. The objective of this stage is to demonstrate understanding and robustness of the design space. Statistical applications include:
- Risk assessment / cause and effect matrix to identify potential variables that impact the process and any risks in the system
- DOE to explore process variables
- Sample size determination to specify the number of samples needed to detect significant differences in the variables
- Multiple regression analysis to determine the significant variables and define the operating regions of the variables
- Development of the control strategy to document the operating ranges for the variables to be controlled
- Analytical method validation to obtain an adequate mechanism for measuring the in-process and release parameters
- Analysis to ensure the measurement system is repeatable and reproducible
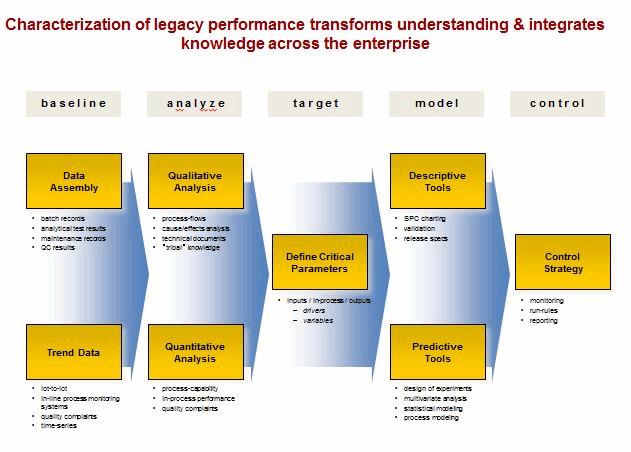
Stage 2- Process Qualification: During this stage, the process design is evaluated to determine if the process is capable of reproducible commercial manufacturing. The objective of this stage is to demonstrate process capability and repeatability. Statistical applications include:
- Sample size determination
- ANOVA to detect inter/intra batch variability
- Development of specifications
- Process capability demonstration
- Refinement of the control strategy to the operator level
- Stability analysis for determination of shelf-life
- Statistical documentation to support to the CMC filing
Stage 3 – Continued Process Verification (CPV): Ongoing assurance is gained during routine production that the process remains in a state of control. The objective of this stage is to demonstrate a stable and effective process. Statistical applications include:
- Customer complaint analysis for ongoing monitoring
- Setting up and ongoing use of control charts
- Ongoing monitoring of process capability
- Implementation of measurements and controls
- Integration of Annual Product Review (APR) statistics and CPV
Legacy Products
The three stages of the pharmaceutical life cycle as described above are applied to new products as they are developed and introduced. However, most products currently in operation are legacy products and may not have had the benefit of the new guidance.
Legacy products and processes are vulnerable because knowledge can be fragmented, often caused by: step-wise development cycles, multiple tech transfers, compartmentalized data, incompatible information systems, ineffective supplier relations and aggressive cost reduction. Statistical tools can be used to characterize legacy products and processes in the existing facilities and prior to product transfer to other manufacturing operations.
The solution is within the organization. Objectives include: defining a continuum of drivers and variables, embedding process understanding, building an early warning and signaling process, and developing and adjusting process know-how. Statistical tools must be integrated into the fabric of the manufacturing operation for addressing deviations and managing change control.
Problematic for many pharmaceutical operations is a lack of fundamental understanding of their processes and products. When a deviation occurs, this lack of understanding makes it difficult to determine the root cause of the deviation. The investigator has no baseline to examine for sources of variation or causal events. Lack of root cause determination for deviations is a common FDA citation.
Organizing For Success
An increase in the use of statistics is inevitable. There is no single statistician that can possibly maintain all of the required skills at a high enough level to meet the overwhelming challenges of the new process validation guidance. There are alternative ways to address this challenge:
- Insource statistical skills
- Outsource statistical skills
- Hybrid approach
Insourcing completely requires a large staff, however complete coverage of statistical needs can be achieved. This is appropriate for large firms that can leverage these skills. However, the disadvantage is in the cost of maintaining this staff. Even large firms may face pressure to reduce the cost of analytics and may seek some level of outsourcing. In addition, a “next generation” gap could be forming as statisticians face retirement and replacements are strictly from academia without industry knowledge.
Outsourcing completely is appropriate for very small firms at the startup stage. However, this approach creates a dependency that may put the firm at continuity risk.
A Hybrid approach leverages the statistical competence of the existing internal staff while reaching out to fill the remaining skill gaps. This begins with the coupling of basic statistical training for the internal staff with a “back-room” network of external subject matter experts who supplement on project work as needed.
Conclusion
The use of statistical tools has the potential to greatly improve the process of demonstrating that scientific evidence exists to show that a process is capable of consistently delivering quality product, as required by the FDA Guidance. This more scientific approach also accelerates product development cycle-time and reduces the cost of regulatory compliance.
References
1. “Process Validation: General Principals and Practices” FDA Guidance
21st Century Validation Strategies For Drug Products
Dr. Janet Woodcock of the Federal Drug Administration (FDA) once said that a mutual goal of the pharmaceutical industry and regulatory agencies would be to have a “desired state” which is an efficient, agile flexible pharmaceutical manufacturing sector. This sector would be one that would reliably produce high quality drug products, without extensive regulatory oversight.[1] The desired state would be achieved based upon the risk based implementation of Quality by Design (QbD) outlined in the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) guidance’s: Q8 (Pharmaceutical Development), Q9 (Quality Risk Management) and Q10 (Pharmaceutical Quality System)[2].
Any pharmaceutical company that initiates and implements QbD (ICH Q8, Q9, and Q10) will effectively and efficiently fully leverage a competitive advantage in 21st century. The prudent implementation of this strategy based on good science and an accurate grasp of the available regulatory knowledge will create transformational value by providing high product quality assurance for patients, more efficient regulatory oversight and cost savings for the company. In this article, I will give an overview on how QbD relates to process validation, as well as outline the three stages to the lifecycle approach to process validation.[3]
QbD Helps Maintain The Validated State Of Your Process
Process validation of pharmaceutical dosage forms is the process of establishing documented evidence of a drug product’s quality. This evidence provides a high degree of assurance that a specific manufacturing process will consistently produce a drug product meeting pre-determined quality attributes when operated within its critical process parameters ranges (CPPs). Once a drug product manufacturing process has been validated, the validated state must be maintained through implementation of operating procedures, preventive maintenance, calibration programs, atypical investigation procedures, annual product review programs, and change control procedures. Any changes to the process must be evaluated through the process change request (PCR) system as outlined by each corporation’s quality policy, procedure, guidelines, and procedures for change control to determine if the validated state will be compromised.
Designing a process validation requires the demonstration of process controls. For example, a company must define upfront the necessary critical process parameters (CPPs) as well as the in-process and finished product critical quality attributes (CQAs) that it intends to achieve with each run of the manufacturing process. A CPP is an input process variable that is maintained within a specified range to ensure that an intermediate or final product will be within the CQA limit. A CQA is a measured property of the intermediate or final product that is considered critical for establishing the intended purity, efficacy, safety, and bioavailability of the drug product. The demonstration of consistency of the CPP and CQA is ultimately consolidated to represent a validated process for manufacture of a pharmaceutical dosage form.
A Quality Management System Is Crucial For Process Validation Stages
All systems have to be in compliance with the current regulatory guidelines prior to conducting any validation studies. A big part of this is establishing a quality management system (QMS). QMS is defined by the American Society for Quality (ASQ) as a formalized system that documents the structure, responsibilities and procedures required to achieve effective quality.[4] These systems also represent a set of interrelated processes that, working together, assure product safety, strength, identity, purity, and quality. In developing quality management systems, FDA recommends six core quality systems on which companies should base their QMS: 1) quality, 2) facilities and equipment, 3) materials, 4) production, 5) packaging and labeling and 6) laboratory control. These core systems are vital to assuring that a system is well developed and worthy of being validated. These FDA recommendations are broad enough to encompass the main aspects of a system that must be considered in creating a QMS. Performing validation studies on a poorly designed system is a waste of a company’s resources and time. Therefore, companies must be in compliance with these six core systems prior to conducting any process validation studies.
All pharmaceutical products manufactured worldwide are to be prospectively validated. A life cycle approach to process validation should be comprised of three stages over the life of the product and process.
Stage 1 – Process Design
The process design stage is the activity of defining the commercial manufacturing process that can consistently deliver a product that meets its CQAs. Process design activities should include the following:
- Identify potential CQAs of the drug product, based on target product profile (TPP);
- Determine relevant CQAs of the drug substance, excipients, and packaging in order to deliver drug product of the desired quality; and
- Define the manufacturing process to be qualified and the appropriate control strategy.
Stage 2 – Process Qualification
During this stage, the process design is evaluated to determine if the process is capable of reproducible commercial manufacturing. Process Qualification is proof of process knowledge and control. This stage has two elements. The first is the design of the facility and qualification of the equipment and utilities. The second is the process performance qualification (PPQ).
Process re-qualification is proof of knowledge and control after a planned (e.g. significant process change, API source change, etc) or unplanned (e.g. process drift, atypical event) change in the manufacturing process. The proof is demonstrated via technical justification & supporting documents and Qualification study defined by a process performance qualification protocol.
Stage 3 – Continued Process Verification
Ongoing assurance is gained during routine production that the process remains in a state of control. Once stage two process qualifications are completed, processes are monitored via the continued process verification and annual review process. Significant deviation from desired process performance uncovered through continued process verification, annual review or via a typical event, warrants consideration for process requalification. It is required that the data generated is continually documented into the process design knowledge of the product (i.e. controlled strategy, design space, and risk assessment documents).
With the FDA’s guidance document providing these general stages, each pharmaceutical company should implement a QbD approach specific for each manufacturing process of a drug product. Each process should be tailored on a case-by-case basis to the specific drug product. These validations strategies could facilitate continual improvement and innovation throughout the product life cycle. Additional product and process understanding can lead to enhanced manufacturing efficiency and regulatory flexibility, thereby providing the availability of safe, effective and high-quality drug products.
In my subsequent columns, I will address validation strategies with respect to solids and controlled release dosage forms; liquids, suspension and semisolid dosage forms; and sterile dosage forms (small and large molecules).
[1] Dr. Janet Woodcock was quoted by Dr. Moheb Nasar during a January 29, 2007 meeting in Baltimore, Md.
[2] This guidance can be found on FDA’s CDER website (http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/default.htm)
[3] The information provided in this article is based upon Guidance for Industry, Process Validation: General Principles and Practices, January 2011, Revision 1 (http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070336.pdf)
[4] American Society for Quality Website (http://asq.org/glossary/q.html)
/////////////Process Validation, Regulatory Review, Drug Approval Strategies, Fast Track, Breakthrough Therapy, Accelerated Approval
45,498 total views, 2 views today
