











No one will present better………..

Examples 13C NMR Spectra
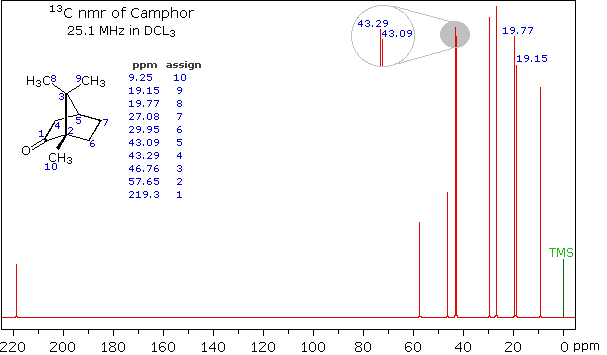
Here are a couple of examples for judging 13C chemical shifts:
Minimum description used……..mostly pictorial
Start with benzene
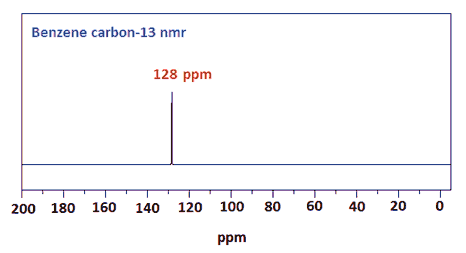
Then toluene



Remember that 13C shifts generally follow the pattern of 1H shifts, but are much larger.
Thus, in (a)
- The peak at lowest field is the caarbon with the OH attached
- The peak at highest field is the CH3 farthest from the OH
- Generally in both 13C and 1H spectra, CH2 groups fall to lower field than CH3, which assigns the remaining peaks.
For (b):
- The peak at lowest field is the C=O carbon, shifted by electronegativity and “diamagnetic anisotropy”
- Assignment of the other two peaks follows from the CH2 lower than CH3 rule.
Now try predicting the splitting pattern to be found if the decoupler is turned off.


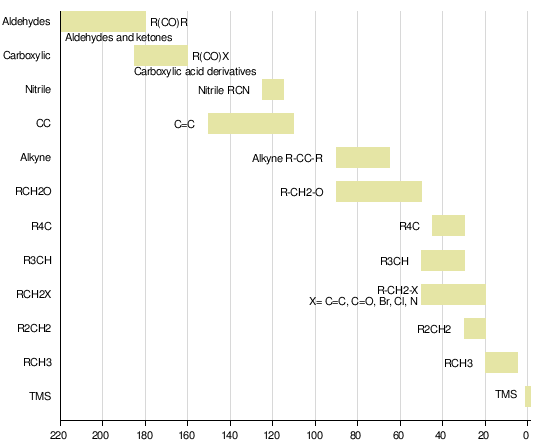
| Taking a close look at three C-13 NMR spectraThe C-13 NMR spectrum for ethanol |
Remember that each peak identifies a carbon atom in a different environment within the molecule. In this case there are two peaks because there are two different environments for the carbons.The carbon in the CH3 group is attached to 3 hydrogens and a carbon. The carbon in the CH2 group is attached to 2 hydrogens, a carbon and an oxygen.So which peak is which?You might remember from the introductory page that the external magnetic field experienced by the carbon nuclei is affected by the electronegativity of the atoms attached to them. The effect of this is that the chemical shift of the carbon increases if you attach an atom like oxygen to it. That means that the peak at about 60 (the larger chemical shift) is due to the CH2 group because it has a more electronegative atom attached.
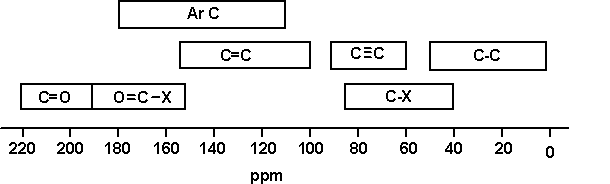
| A table of typical chemical shifts in C-13 NMR spectra |
| carbon environment | chemical shift (ppm) |
|---|---|
| C=O (in ketones) | 205 – 220 |
| C=O (in aldehydes) | 190 – 200 |
| C=O (in acids and esters) | 170 – 185 |
| C in aromatic rings | 125 – 150 |
| C=C (in alkenes) | 115 – 140 |
| RCH2OH | 50 – 65 |
| RCH2Cl | 40 – 45 |
| RCH2NH2 | 37 – 45 |
| R3CH | 25 – 35 |
| CH3CO- | 20 – 30 |
| R2CH2 | 16 – 25 |
| RCH3 | 10 – 15 |
In the table, the “R” groups won’t necessarily be simple alkyl groups. In each case there will be a carbon atom attached to the one shown in red, but there may well be other things substituted into the “R” group.
If a substituent is very close to the carbon in question, and very electronegative, that might affect the values given in the table slightly.
For example, ethanol has a peak at about 60 because of theCH2OH group. No problem!
It also has a peak due to the RCH3 group. The “R” group this time is CH2OH. The electron pulling effect of the oxygen atom increases the chemical shift slightly from the one shown in the table to a value of about 18.
A simplification of the table
At the time of writing, a draft UK syllabus (Cambridge pre-U) was expecting their students to learn the following simplification:
| carbon environment | chemical shift (ppm) |
|---|---|
| C-C | 0 – 50 |
| C-O | 50 – 100 |
| C=C | 100 – 150 |
| C=O | 150 – 200 |
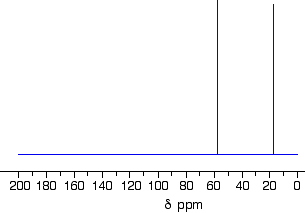
The C-13 NMR spectrum for but-3-en-2-oneThis is also known as 3-buten-2-one (amongst many other things!)

Here is the structure for the compound:
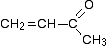
You can pick out all the peaks in this compound using the simplified table above.
The peak at just under 200 is due to a carbon-oxygen double bond. The two peaks at 137 and 129 are due to the carbons at either end of the carbon-carbon double bond. And the peak at 26 is the methyl group which, of course, is joined to the rest of the molecule by a carbon-carbon single bond.
If you want to use the more accurate table, you have to put a bit more thought into it – and, in particular, worry about the values which don’t always exactly match those in the table!
The carbon-oxygen double bond in the peak for the ketone group has a slightly lower value than the table suggests for a ketone. There is an interaction between the carbon-oxygen and carbon-carbon double bonds in the molecule which affects the value slightly. This isn’t something which we need to look at in detail for the purposes of this topic.
You must be prepared to find small discrepancies of this sort in more complicated molecules – but don’t worry about this for exam purposes at this level. Your examiners should give you shift values which exactly match the compound you are given.
The two peaks for the carbons in the carbon-carbon double bond are exactly where they would be expected to be. Notice that they aren’t in exactly the same environment, and so don’t have the same shift values. The one closer to the carbon-oxygen double bond has the larger value.
And the methyl group on the end has exactly the sort of value you would expect for one attached to C=O. The table gives a range of 20 – 30, and that’s where it is.
One final important thing to notice. There are four carbons in the molecule and four peaks because they are all in different environments. But they aren’t all the same height. In C-13 NMR, you can’t draw any simple conclusions from the heights of the various peaks.
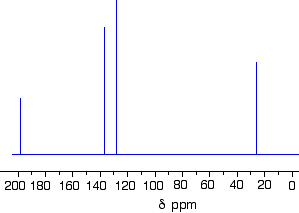
The C-13 NMR spectrum for 1-methylethyl propanoate
1-methylethyl propanoate is also known as isopropyl propanoate or isopropyl propionate.
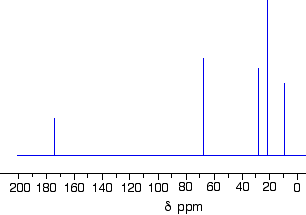
Here is the structure for 1-methylethyl propanoate:
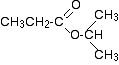
Two simple peaks
There are two very simple peaks in the spectrum which could be identified easily from the second table above.
The peak at 174 is due to a carbon in a carbon-oxygen double bond. (Looking at the more detailed table, this peak is due to the carbon in a carbon-oxygen double bond in an acid or ester.)
The peak at 67 is due to a different carbon singly bonded to an oxygen. Those two peaks are therefore due to:
If you look back at the more detailed table of chemical shifts, you will find that a carbon singly bonded to an oxygen has a range of 50 – 65. 67 is, of course, a little bit higher than that.

As before, you must expect these small differences. No table can account for all the fine differences in environment of a carbon in a molecule. Different tables will quote slightly different ranges. At this level, you can just ignore that problem!
Before we go on to look at the other peaks, notice the heights of these two peaks we’ve been talking about. They are both due to a single carbon atom in the molecule, and yet they have different heights. Again, you can’t read any reliable information directly from peak heights in these spectra.

The three right-hand peaks
From the simplified table, all you can say is that these are due to carbons attached to other carbon atoms by single bonds. But because there are three peaks, the carbons must be in three different environments.
The more detailed table is more helpful.
Here are the structure and the spectrum again:
The easiest peak to sort out is the one at 28. If you look back at the table, that could well be a carbon attached to a carbon-oxygen double bond. The table quotes the group as CH3CO-, but replacing one of the hydrogens by a simple CH3 group won’t make much difference to the shift value.
The right-hand peak is also fairly easy. This is the left-hand methyl group in the molecule. It is attached to an admittedly complicated R group (the rest of the molecule). It is the bottom value given in the detailed table.
The tall peak at 22 must be due to the two methyl groups at the right-hand end of the molecule – because that’s all that’s left. These combine to give a single peak because they are both in exactlythe same environment.
If you are looking at the detailed table, you need to think very carefully which of the environments you should be looking at. Without thinking, it is tempting to go for the R2CH2 with peaks in the 16 – 25 region. But you would be wrong!
The carbons we are interested in are the ones in the methyl group, not in the R groups. These carbons are again in the environment: RCH3. The R is the rest of the molecule.
The table says that these should have peaks in the range 10 – 15, but our peak is a bit higher. This is because of the presence of the nearby oxygen atom. Its electronegativity is pulling electrons away from the methyl groups – and, as we’ve seen above, this tends to increase the chemical shift slightly.
Once again, don’t worry about the discrepancies. In an exam, perhaps your examiners will just want you to have learnt the simple table above – in which case, they can’t expect you to work out which peak is which in a complicated spectrum of this sort. Or they will give you tables of chemical shifts – in which case, they will give you values which match the peaks in the spectra.
Remember that you are only doing an introduction to C-13 NMR at this level. It isn’t going to be that hard in an exam!
Working out structures from C-13 NMR spectra
So far, we’ve just been trying to see the relationship between carbons in particular environments in a molecule and the spectrum produced. We’ve had all the information necessary. Now let’s make it a little more difficult – but we’ll work from much easier examples!
In each example, try to work it out for yourself before you read the explanation.
Example 1
How could you tell from just a quick look at a C-13 NMR spectrum (and without worrying about chemical shifts) whether you had propanone or propanal (assuming those were the only options)?
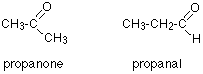
Because these are isomers, each has the same number of carbon atoms, but there is a difference between the environments of the carbons which will make a big impact on the spectra.
In propanone, the two carbons in the methyl groups are in exactly the same environment, and so will produce only a single peak. That means that the propanone spectrum will have only 2 peaks – one for the methyl groups and one for the carbon in the C=O group.
However, in propanal, all the carbons are in completely different environments, and the spectrum will have three peaks.
Example 2
Thare are four alcohols with the molecular formula C4H10O.
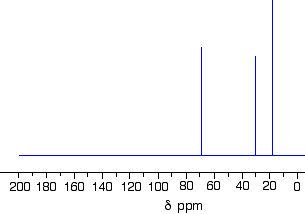
Which one produced the C-13 NMR spectrum below?
You can do this perfectly well without referring to chemical shift tables at all.
In the spectrum there are a total of three peaks – that means that there are only three different environments for the carbons, despite there being four carbon atoms.
In A and B, there are four totally different environments. Both of these would produce four peaks.
In D, there are only two different environments – all the methyl groups are exactly equivalent. D would only produce two peaks.
That leaves C. Two of the methyl groups are in exactly the same environment – attached to the rest of the molecule in exactly the same way. They would only produce one peak. With the other two carbon atoms, that would make a total of three. The alcohol is C.
Example 3
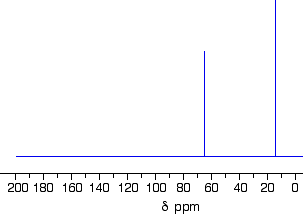
This follows on from Example 2, and also involves an isomer of C4H10O but which isn’t an alcohol. Its C-13 NMR spectrum is below. Work out what its structure is.
Because we don’t know what sort of structure we are looking at, this time it would be a good idea to look at the shift values. The approximations are perfectly good, and we will work from this table:
| carbon environment | chemical shift (ppm) |
|---|---|
| C-C | 0 – 50 |
| C-O | 50 – 100 |
| C=C | 100 – 150 |
| C=O | 150 – 200 |
There is a peak for carbon(s) in a carbon-oxygen single bond and one for carbon(s) in a carbon-carbon single bond. That would be consistent with C-C-O in the structure.
It isn’t an alcohol (you are told that in the question), and so there must be another carbon on the right-hand side of the oxygen in the structure in the last paragraph.
The molecular formula is C4H10O, and there are only two peaks. The only solution to that is to have two identical ethyl groups either side of the oxygen.
The compound is ethoxyethane (diethyl ether), CH3CH2OCH2CH3.

Example 4
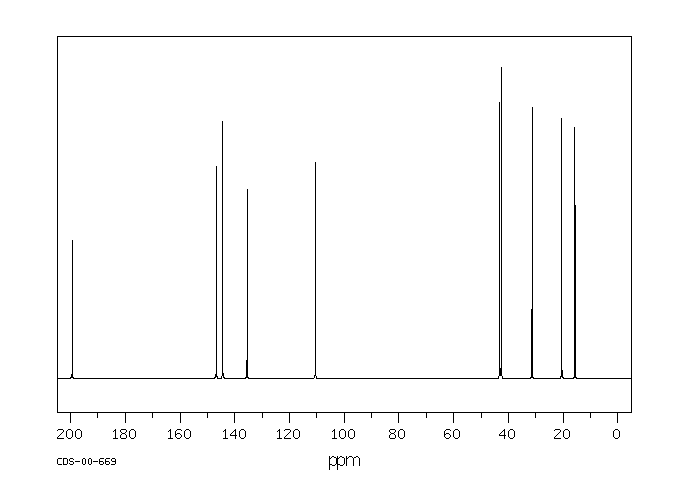
Using the simplified table of chemical shifts above, work out the structure of the compound with the following C-13 NMR spectrum. Its molecular formula is C4H6O2.
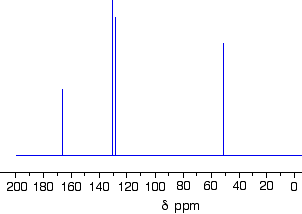
Let’s sort out what we’ve got.
- There are four peaks and four carbons. No two carbons are in exactly the same environment.
- The peak at just over 50 must be a carbon attached to an oxygen by a single bond.
- The two peaks around 130 must be the two carbons at either end of a carbon-carbon double bond.
- The peak at just less than 170 is the carbon in a carbon-oxygen double bond.
Putting this together is a matter of playing around with the structures until you have come up with something reasonable. But you can’t be sure that you have got the right structure using this simplified table.
In this particular case, the spectrum was for the compound:
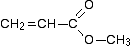
If you refer back to the more accurate table of chemical shifts towards the top of the page, you will get some better confirmation of this. The relatively low value of the carbon-oxygen double bond peak suggests an ester or acid rather than an aldehyde or ketone.
It can’t be an acid because there has to be a carbon attached to an oxygen by a single bond somewhere – apart from the one in the -COOH group. We’ve already accounted for that carbon atom from the peak at about 170. If it was an acid, you would already have used up both oxygens in the structure in the -COOH group.
Without this information, though, you could probably come up with reasonable alternative structures. If you were working from the simplified table in an exam, your examiners would have to allow any valid alternatives.



![]()
Figure 3: 1H and 13C NMR spectra of the abcc-monomer and the ABCC-sequence-regulated copolymer.

(a) 1H and 13C NMR spectra of 7. (b) 1H and 13C NMR spectra of poly(7) obtained with CuCl/N,N,N′,N″,N″-pentamethyldiethylenetriamine (PMDETA) ([CuCl]0=200 mM; [PMDETA]0=800 mM) in bulk at 100 °C. All spectra were measured in CDCl3 at room temperature The brackets for repeating units in poly(7) are positioned differently from those in Figs. 1 and 4 so that the signals originating from the terminal groups can be assigned to the chemical structures.






























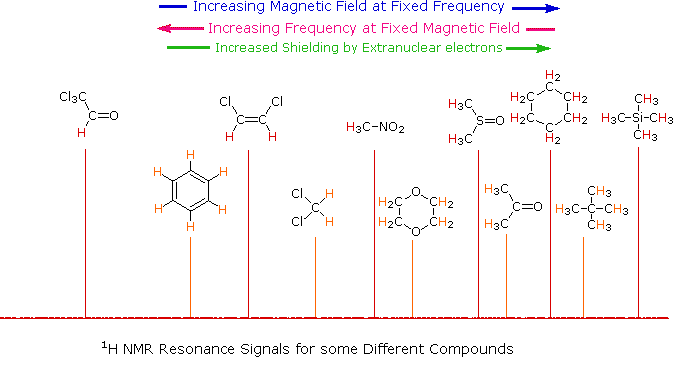
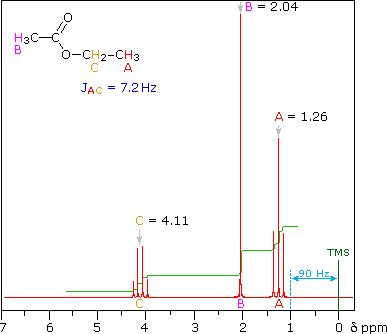
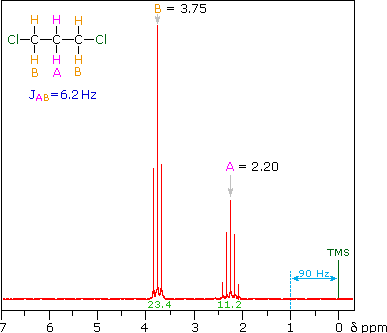
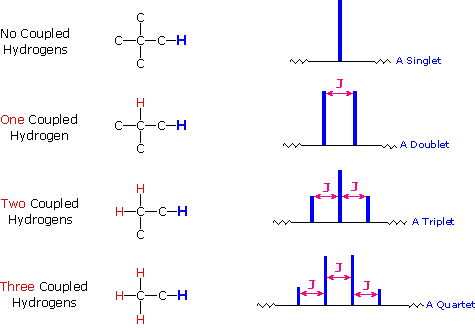
 <
< R-O-
R-O-


 , is 300 MHz. The magnetogyric ratio
, is 300 MHz. The magnetogyric ratio  of a hydrogen nucleus is 267.513
of a hydrogen nucleus is 267.513 . A quick calculation using:
. A quick calculation using:
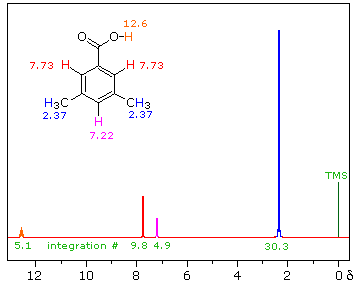
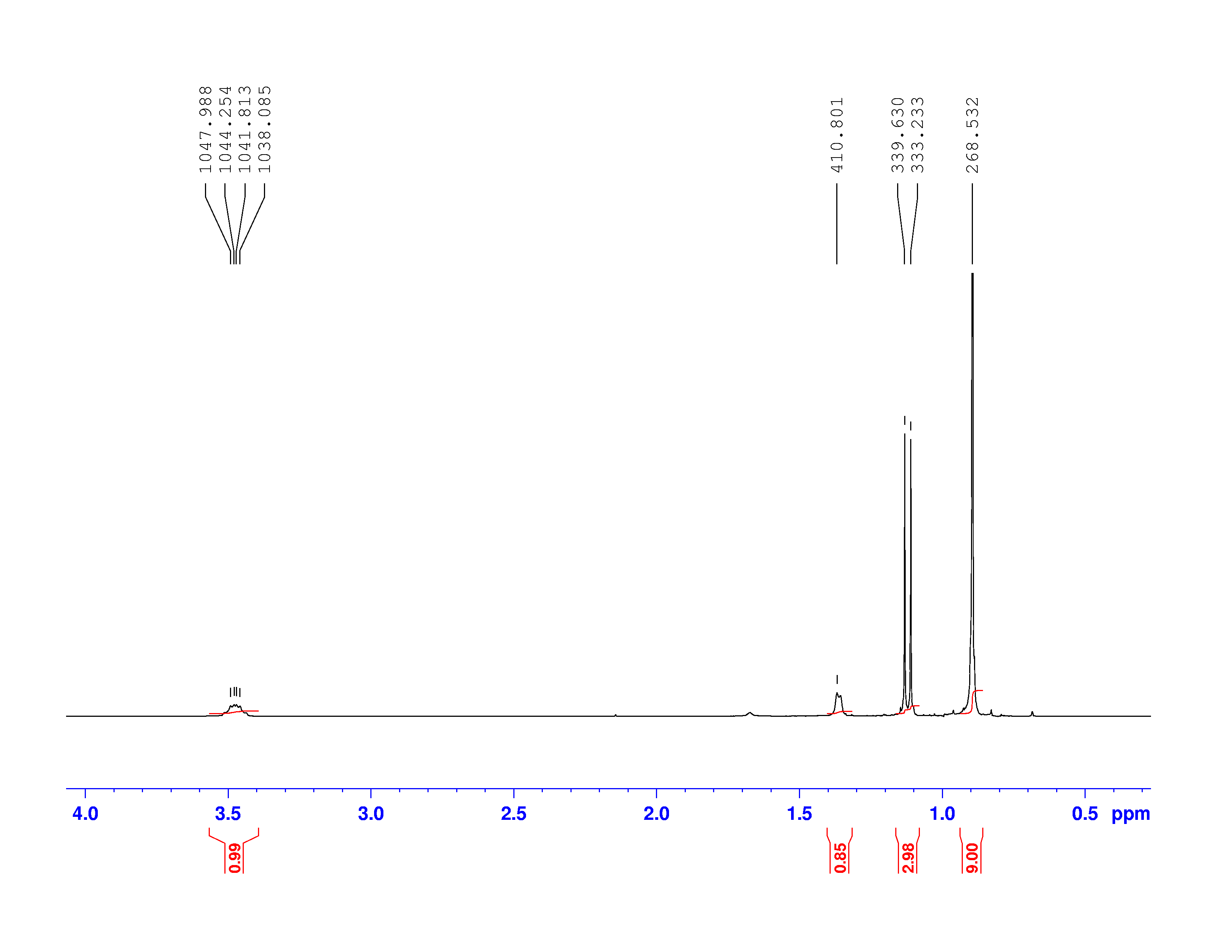
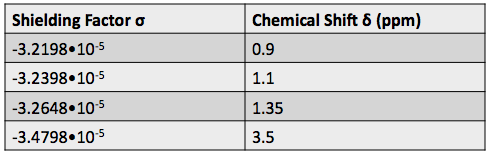
 H-NMR spectrum, we can calculate shielding factors for each type of hydrogen nucleus in 3,3-dimethyl-2-butanol, and determine what each spectrum peak actually means.
H-NMR spectrum, we can calculate shielding factors for each type of hydrogen nucleus in 3,3-dimethyl-2-butanol, and determine what each spectrum peak actually means.



 , where
, where  is the number of peaks. The definition of “nearby” is usually 1 carbon atom over in the molecule from the one the original hydrogen is attached to.
is the number of peaks. The definition of “nearby” is usually 1 carbon atom over in the molecule from the one the original hydrogen is attached to.





