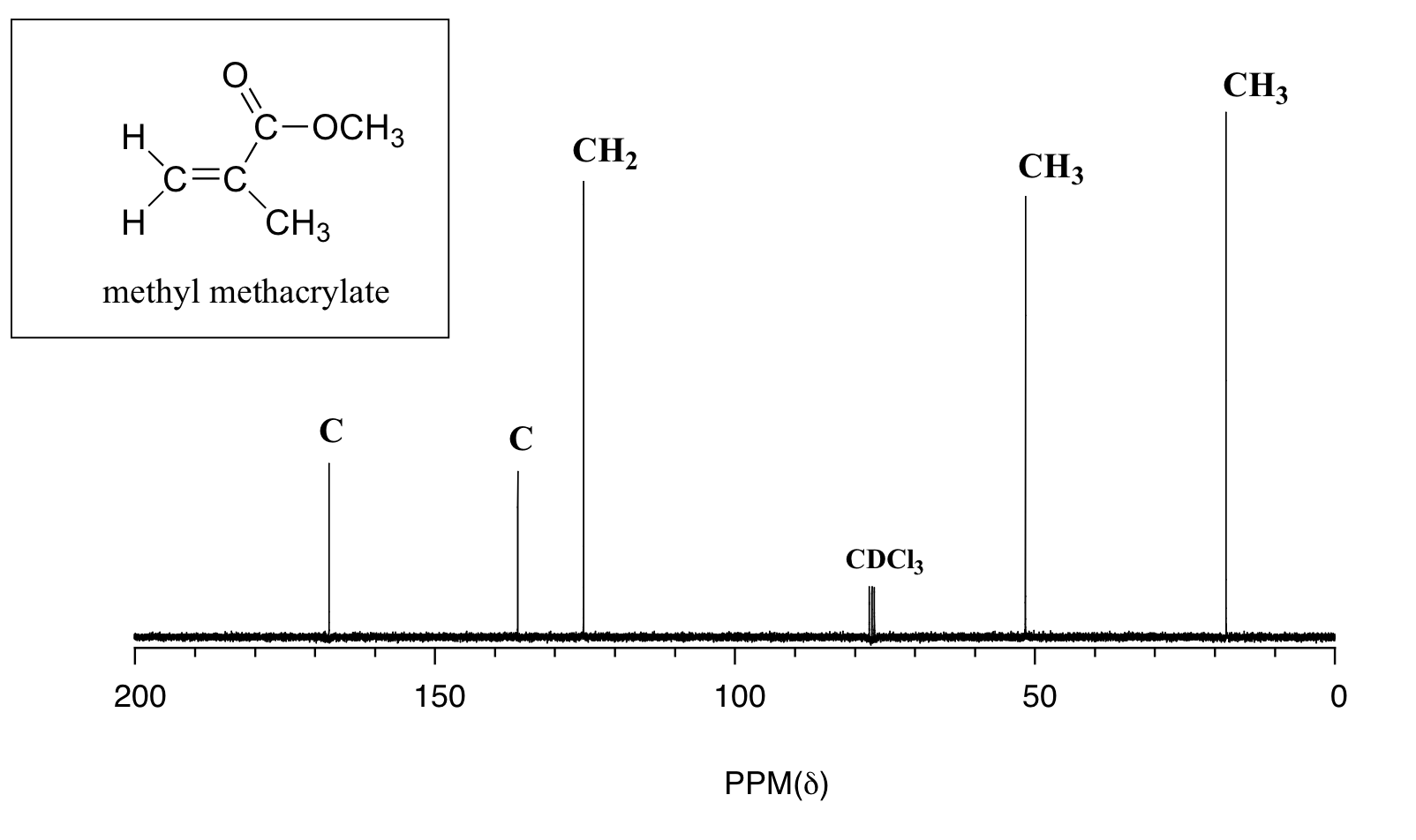
Dantrolene sodium
1-[[[5-(4-nitrophenyl)-2-furanyl]methylene]amino]-2,4-imidazolidinedione
FDA Approves Ryanodex for the Treatment of Malignant Hyperthermia
WOODCLIFF LAKE, N.J.(BUSINESS WIRE) July 23, 2014 — Eagle Pharmaceuticals, Inc. (“Eagle” or “the Company”) (Nasdaq:EGRX) today announced that the U. S. Food and Drug Administration (FDA) has approved Ryanodex (dantrolene sodium) for injectable suspension indicated for the treatment of malignant hyperthermia (MH), along with the appropriate supportive measures. MH is an inherited and potentially fatal disorder triggered by certain anesthesia agents in genetically susceptible individuals. FDA had designated Ryanodex as an Orphan Drug in August 2013. Eagle has been informed by the FDA that it will learn over the next four to six weeks if it has been granted the seven year Orphan Drug market exclusivity.
read at
Dantrium Intravenous is a sterile, non-pyrogenic, lyophilized formulation of dantrolene sodium for injection.
Dantrium Intravenous is supplied in 70 mL vials containing 20 mg dantrolene sodium, 3000 mg mannitol,
and sufficient sodium hydroxide to yield a pH of approximately 9.5 when reconstituted with 60 mL sterile water for injection USP (without a bacteriostatic agent).
Dantrium is classified as a direct-acting skeletal muscle relaxant. Chemically, Dantrium is hydrated 1-[[[5-(4-nitrophenyl)-2-furanyl]methylene]amino]-2,4-imidazolidinedione sodium salt. The structural formula for the hydrated salt is:
 |
The hydrated salt contains approximately 15% water (3-1/2 moles) and has a molecular weight of 399. The anhydrous salt (dantrolene) has a molecular weight of 336.
 |
|
| Systematic (IUPAC) name | |
|---|---|
| 1-{[5-(4-nitrophenyl)-2-furyl]methylideneamino} imidazolidine-2,4-dione |
|
| Clinical data | |
| Trade names | Dantrium |
| AHFS/Drugs.com | monograph |
| Pregnancy cat. | C (US) |
| Legal status | ? |
| Routes | Oral, intravenous |
| Pharmacokinetic data | |
| Bioavailability | 70% |
| Metabolism | Liver |
| Excretion | Biliary, renal |
| Identifiers | |
| CAS number | 7261-97-4 |
| ATC code | M03CA01 |
| PubChem | CID 2952 |
| IUPHAR ligand | 4172 |
| DrugBank | DB01219 |
| ChemSpider | 2847 |
| UNII | F64QU97QCR |
| KEGG | D02347 |
| ChEBI | CHEBI:4317 |
| ChEMBL | CHEMBL1201288 |
| Chemical data | |
| Formula | C14H10N4O5 |
| Mol. mass | 314.253 g/mol |
Dantrolene sodium is a muscle relaxant that acts by abolishing excitation-contraction coupling in muscle cells, probably by action on the ryanodine receptor. It is the only specific and effective treatment for malignant hyperthermia, a rare, life-threatening disorder triggered by general anesthesia. It is also used in the management of neuroleptic malignant syndrome, muscle spasticity (e.g. afterstrokes, in paraplegia, cerebral palsy, or patients with multiple sclerosis), 3,4-methylenedioxymethamphetamine (“ecstasy”)intoxication, serotonin syndrome, and 2,4-dinitrophenol poisoning.[1] It is marketed by JHP Pharmaceuticals LLC as Dantrium (in North America) and by Norgine BV as Dantrium, Dantamacrin, or Dantrolen (in Europe).
History
Dantrolene was first described in the scientific literature in 1967, as one of several hydantoin derivatives proposed as a new class of muscle relaxant.[2] Dantrolene underwent extensive further development, and its action on skeletal muscle was described in detail in 1973.[3]
Dantrolene was widely used in the management of spasticity before its efficacy in treating malignant hyperthermia was discovered by South African anesthesiologist Gaisford Harrison and reported in a landmark 1975 article published in the British Journal of Anaesthesia.[4] Harrison experimentally induced malignant hyperthermia with halothane anesthesia in genetically susceptible pigs, and obtained an 87.5% survival rate, where seven of his eight experiments survived after intravenous administration of dantrolene. The efficacy of dantrolene in humans was later confirmed in a large, multicenter study published in 1982,[5] and confirmed epidemiologically in 1993.[6] Before dantrolene, the only available treatment for malignant hyperthermia was procaine, which was associated with a 60% mortality rate in animal models.[4]
JULY 2014
The US Food and Drug Administration (FDA) has approved an injectable form of dantrolene sodium (Ryanodex, Eagle Pharmaceuticals) for rapid treatment of malignant hyperthermia (MH), along with the appropriate supportive measures, the company announced in a news release today.
MH is a potentially fatal inherited disorder triggered by exposure to certain drugs used for general anesthesia, including the neuromuscular blocking agent succinylcholine.
Ryanodex — which can be administered much more quickly than current formulations of dantrolene — is the first significant enhancement to MH treatment options in more than 3 decades, according to the company.
Ryanodex will be available in single-use vials containing 250 mg of dantrolene sodium in lyophilized powder form. It is formulated for rapid reconstitution and administration in less than 1 minute to patients in MH crisis. “Ryanodex should be administered by continuous rapid intravenous push beginning with a loading dose of 2.5 mg/kg, and continuing until symptoms subside,” the company says.
Ryanodex allows anesthesiologists to deliver a therapeutic dose of dantrolene sodium in a much more expedient manner than currently possible with existing IV formulations of dantrolene sodium, “potentially saving lives and reducing MH-related morbidity,” according to the company.
Other dantrolene sodium formulations require multiple 20-mg vials reconstituted in large volumes of sterile water, a process that can take 15 to 20 minutes to mix reconstitute and administer, the company notes.
MH during surgery is a “life-threatening emergency requiring immediate treatment including the administration of the ‘antidote’ drug dantrolene sodium,” Henry Rosenberg, MD, CPE, a founder and president of the Malignant Hyperthermia Association of the United States, said in the release.
“The ability for healthcare professionals in hospitals and surgery centers to more quickly prepare and administer this new formulation of the antidote dantrolene sodium is expected to bring the crisis under control more rapidly and prevent severe complications from MH,” he said.
The FDA granted Ryanodex orphan drug status in August 2013 and priority review status in March 2014.Ryanodex will be available to order through national and regional drug wholesalers in August with product shipping shortly after. More information is available at http://www.ryanodex.com/.
Contraindications
Oral dantrolene cannot be used by:
- people with a pre-existing liver disease
- people with compromised lung function
- people with severe cardiovascular impairment
- people with a known hypersensitivity to dantrolene
- pediatric patients under five years of age
- people who need good muscular balance or strength to maintain an upright position, motoric function, or proper neuromuscular balance
If the indication is a medical emergency, such as malignant hyperthermia, the only significant contraindication is hypersensitivity.
Pregnancy and breastfeeding
If needed in pregnancy, adequate human studies are lacking, therefore the drug should be given in pregnant women only if clearly indicated. It may cause hypotonia in the newborn if given closely before delivery.[1]
Dantrolene should not be given to breastfeeding mothers. If a treatment is necessary, breastfeeding should be terminated.
Adverse effects
Central nervous system side effects are quite frequently noted and encompass speech and visual disturbances, mental depression and confusion, hallucinations, headache, insomnia and exacerbation or precipitation of seizures, and increased nervousness. Infrequent cases of respiratory depression or a feeling of suffocation have been observed. Dantrolene often causes sedation severe enough to incapacitate the patient to drive or operate machinery.
Gastrointestinal effects include bad taste, anorexia, nausea, vomiting, abdominal cramps, and diarrhea.
Hepatic side effects may be seen either as asymptomatic elevation of liver enzymes and/or bilirubin or, most severe, as fatal and nonfatal hepatitis. The risk of hepatitis is associated with the duration of treatment and the daily dose. In patients treated for hyperthermia, no liver toxicity has been observed so far.
Pleural effusion with pericarditis (oral treatment only), rare cases of bone marrow damage, diffuse myalgias, backache, dermatologic reactions, transient cardiovascular reactions, and crystalluria have additionally been seen. Muscle weakness may persist for several days following treatment.
Mutagenicity and carcinogenity
Dantrolene gave positive results in animal high dose studies (with and without enzymatic activation) regarding mutagenicity and carcinogenity. No evidence for human mutagenicity and carcinogenity has been found during the long years of clinical experience.
Mechanism of action
Dantrolene depresses excitation-contraction coupling in skeletal muscle by binding to the ryanodine receptor, and decreasing free intracellular calcium concentration.[1]
Chemistry

Skeletal formula of azumolene. The bromine atom replacing the nitro group found in dantrolene may be seen at left.
Chemically it is a hydantoin derivative, but does not exhibit antiepileptic activity like other hydantoin derivates such as phenytoin.[1]
The poor water solubility of dantrolene leads to certain difficulties in its use.[1][7] A more water-soluble analog of dantrolene, azumolene, is under development for similar indications.[7] Azumolene has a bromine residue instead of the nitro group found in dantrolene, and is 30 times more water-soluble.[1]
…………………………………………….
Bioorganic and medicinal chemistry letters, 2002 , vol. 12, 22 p. 3263 – 3265
http://www.google.co.in/patents/US4543359
…………………………………………………………………………………………………
US4543359
http://www.google.co.in/patents/US4543359
Dantrolene sodium (1-[[5-(p-nitrophenyl) furfurylidene]-amino]hydantoin sodium salt) is described in U.S. Pat. No. 3,415,821. It is used as a skeletal muscle relaxant particularly in controlling the manifestations of clinical spasticity resulting from upper neuron disorders (Physicians’ Desk Reference, 36th Edition, 1982). It is also used in the prevention and treatment of malignant hyperthermia in humans (Friesen et al., Can. Anaesth. Soc. J. 26:319-321, 1979). In connection with the use of dantrolene sodium in hyperthermic crisis it was observed that there was an elimination of the arrhythmias accompanying such crisis [Salata et al., Effects of Dantrolene Sodium on the Electrophysiological Properties of Canine Cardiac Purkinje Fibers, J. Pharmacol. Exp. Ther. 220(1):157-166 (Jan.) 1982] incorporated herein by reference.
………………………………………….
Drug interactions
Dantrolene may interact with the following drugs:[8]
- Calcium channel blockers of the diltiazem/verapamil type: Intravenous treatment with dantrolene and concomitant calcium channel blocker treatment may lead to severe cardiovascular collapse, arrhythmias, myocardial depressions, and hyperkalemia.
- Nondepolarizing neuromuscular blocking agents, such as vecuronium bromide: Neuromuscular blockade is potentiated.
- CNS depressants: Sedative action is potentiated. Benzodiazepines may also cause additive muscle weakness.
- Combined oral contraceptives and hormone replacement therapy with estrogens may enhance liver toxicity of dantrolene, particularly in women over 35 years of age.
References
- Krause T, Gerbershagen MU, Fiege M, Weisshorn R, Wappler F (2004). “Dantrolene – a review of its pharmacology, therapeutic use and new developments”. Anaesthesia 59(4): 364–73. doi:10.1111/j.1365-2044.2004.03658.x. PMID 15023108.
- Snyder HR, Davis CS, Bickerton RK, Halliday RP (September 1967). “1-[(5-arylfurfurylidene)amino]hydantoins. A new class of muscle relaxants”. J Med Chem 10 (5): 807–10.doi:10.1021/jm00317a011. PMID 6048486.
- Ellis KO, Castellion AW, Honkomp LJ, Wessels FL, Carpenter JE, Halliday RP (June 1973). “Dantrolene, a direct acting skeletal muscle relaxant”. J Pharm Sci 62 (6): 948–51.doi:10.1002/jps.2600620619. PMID 4712630.
- Harrison GG (January 1975). “Control of the malignant hyperpyrexic syndrome in MHS swine by dantrolene sodium”. Br J Anaesth 47 (1): 62–5. doi:10.1093/bja/47.1.62.PMID 1148076. A reprint of the article, which became a “Citation Classic”, is available in Br J Anaesth 81 (4): 626–9. PMID 9924249 (free full text).
- Kolb ME, Horne ML, Martz R (April 1982). “Dantrolene in human malignant hyperthermia”. Anesthesiology 56 (4): 254–62. doi:10.1097/00000542-198204000-00005. PMID 7039419.
- Strazis KP, Fox AW (March 1993). “Malignant hyperthermia: review of published cases”. Anesth Analg 77 (3): 297–304. doi:10.1213/00000539-199377020-00014.
- Sudo RT, Carmo PL, Trachez MM, Zapata-Sudo G (March 2008). “Effects of azumolene on normal and malignant hyperthermia-susceptible skeletal muscle”. Basic Clin Pharmacol Toxicol 102 (3): 308–16. doi:10.1111/j.1742-7843.2007.00156.x. PMID 18047479.
- “Dantrolene Drug Interactions”. Epocrates Online. Epocrates. 2008. Retrieved on December 31, 2008.
External links
- http://www.kompendium.ch/MonographieTxt.aspx?lang=de&MonType=fi Swiss Drug Compendium on oral Dantrolene (German)
| Reference | ||
|---|---|---|
| 1 | * | Dissertation Abstracts International, 42(4), 1337 B, (1981), Malloy, K., PH.D. Thesis, 1981 . |
| 2 | Dissertation Abstracts International, 42(4), 1337-B, (1981), [Malloy, K., PH.D. Thesis, 1981]. | |
| 3 | * | Dissertation Abstracts International, 42(8), 3222 B, (1982), Salata, J., Ph.D. Thesis, 1981 . |
| 4 | Dissertation Abstracts International, 42(8), 3222-B, (1982), [Salata, J., Ph.D. Thesis, 1981]. | |
| 5 | * | Malloy, K., Ph.D. Thesis, Univ. of Rochester, 1981. |
| 6 | * | Salata, J. et al., J. Pharmacol. Exp. Ther., 220(1), 157 166, (1982). |
| 7 | Salata, J. et al., J. Pharmacol. Exp. Ther., 220(1), 157-166, (1982). | |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US4822629 * | 12 Dec 1986 | 18 Apr 1989 | Norwich Eaton Pharmaceuticals, Inc. | Azumolene dosage form |
| US4837163 * | 2 Oct 1987 | 6 Jun 1989 | Tsuyoshi Ohnishi | Simple blood test for diagnosing malignant hyperthermia |
| US4861790 * | 28 Oct 1987 | 29 Aug 1989 | Norwich Eaton Pharmaceuticals, Inc. | Use of azumolene for the treatment of malignant hyperthermia |
| US5462940 * | 3 Jun 1994 | 31 Oct 1995 | Norwich Eaton Pharmaceuticals, Inc. | 4-oxocyclic ureas useful as antiarrhythmic and antifibrillatory agents |
| US5691369 * | 7 Jun 1995 | 25 Nov 1997 | The Proctor & Gamble Company | Cardiovascular disorders |
| US5994354 * | 7 Jun 1995 | 30 Nov 1999 | The Procter & Gamble Company | Cyclic urethanes useful as antiarrhythmic and antifibrillatory agents |
| US7758890 | 1 Mar 2004 | 20 Jul 2010 | Lyotropic Therapeutics, Inc. | Treatment using dantrolene |
| US8110225 | 4 Mar 2010 | 7 Feb 2012 | Lyotropic Therapeutics, Inc. | Treatment using dantrolene |
| US8604072 | 19 Jan 2012 | 10 Dec 2013 | Lyotropic Therapeutics, Inc. | Treatment using dantrolene |
| US8685460 | 19 Jan 2012 | 1 Apr 2014 | Lyotropic Therapeutics, Inc | Treatment using dantrolene |
| EP2583670A1 | 5 Sep 2008 | 24 Apr 2013 | US Worldmeds LLC | Co-solvent compositions and methods for improved delivery of dantrolene therapeutic agents |
| WO2005013919A2 * | 1 Mar 2004 | 17 Feb 2005 | Lyotropic Therapeutics Inc | Treatment using dantrolene |




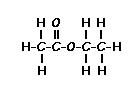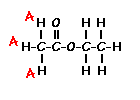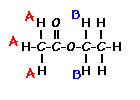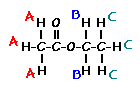
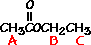


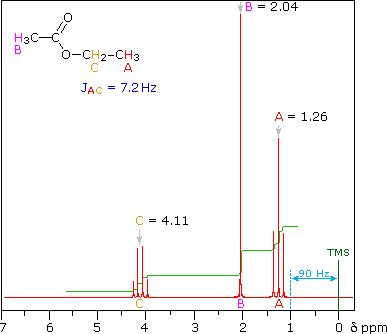
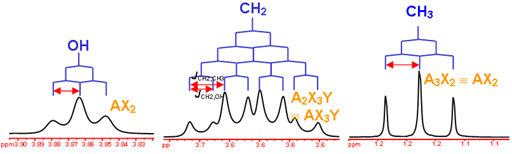



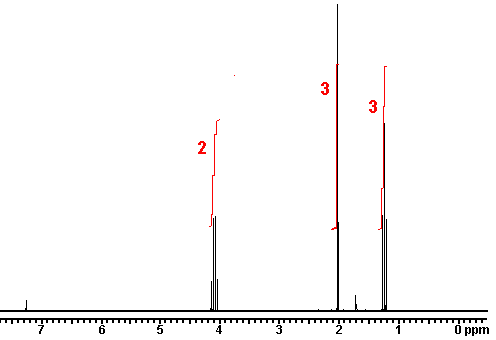
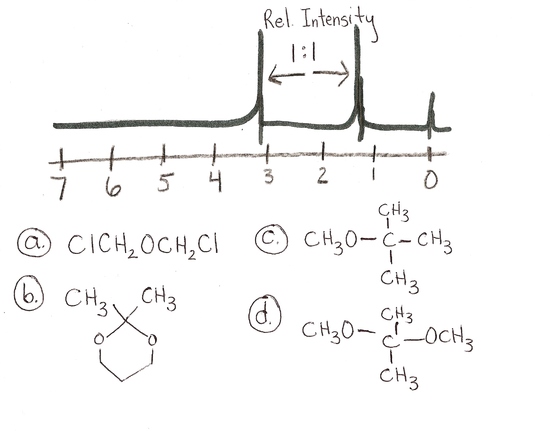 answer is a and d
answer is a and d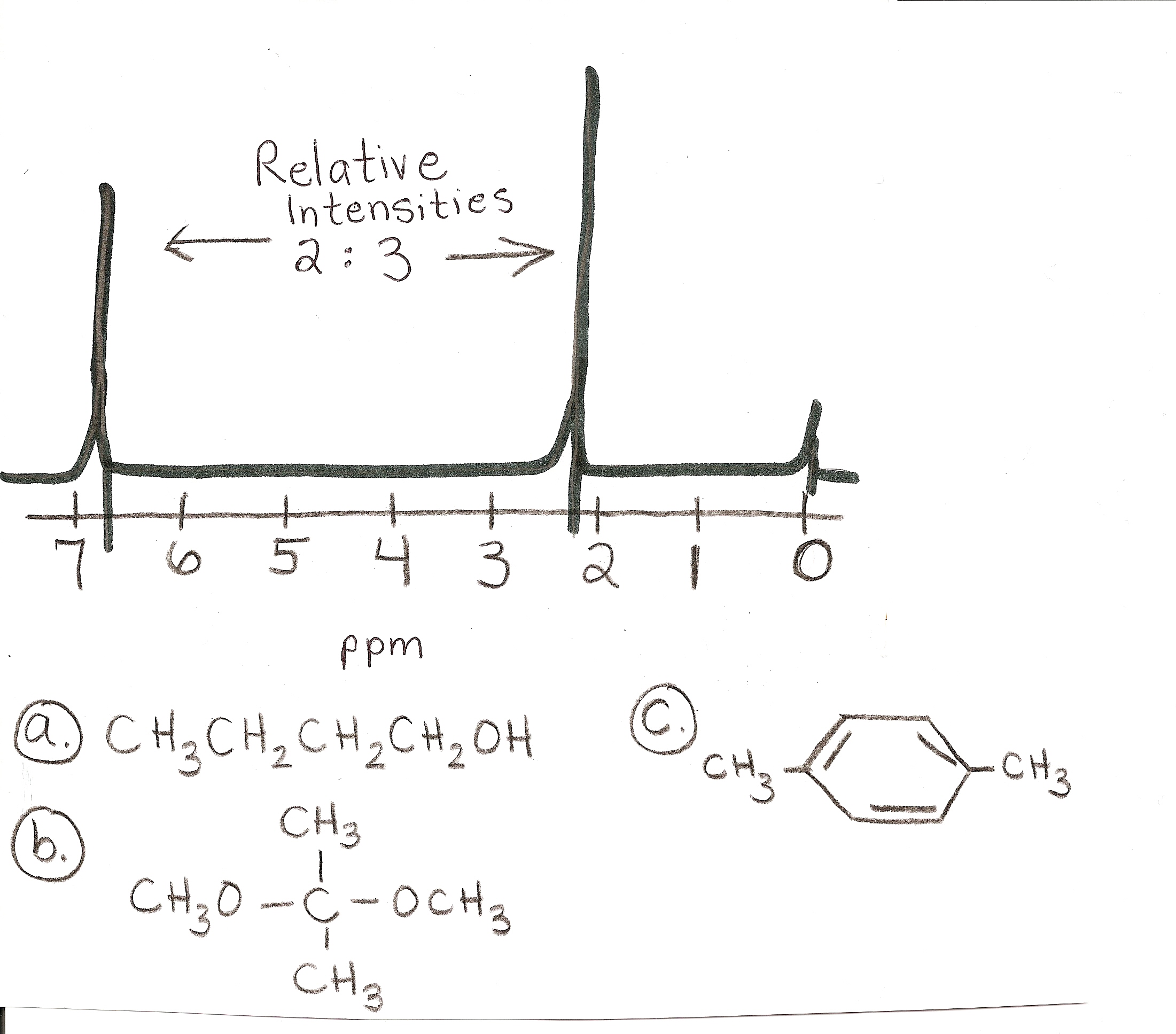
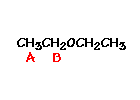
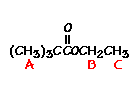




 n
n