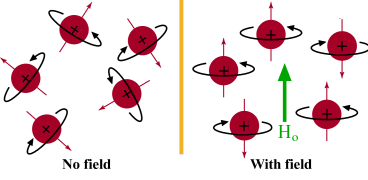
Tout sur les médicaments הכל על תרופות كل شيئ عن الأدوية Все о наркотиках 关于药品的一切 డ్రగ్స్ గురించి అన్ని 마약에 관한 모든 것 Όλα για τα Ναρκωτικά Complete Tracking of Drugs Across the World by Dr Anthony Melvin Crasto, Worldpeacepeaker, worlddrugtracker, PH.D (ICT), MUMBAI, INDIA, Worlddrugtracker, Helping millions, 9 million hits on google on all websites, 2.5 lakh connections on all networks, “ALL FOR DRUGS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This is a compilation for educational purposes only. P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
Bonan Li and Jun F. Liang, Stevens Institute of Technology, Hoboken, NJ, USA, report an approach to synthesize phospholipopeptides. They use a crosslinker with a thiol-reactive maleimide and an amine-reactive N-hydroxysuccinimide ester (pictured). Hence, the molecule is able to link the thiol group of the amino acid cystein in the peptide and the amine group of the phospholipid (phosphatidylamine).
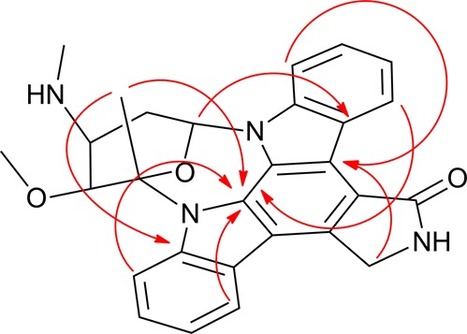
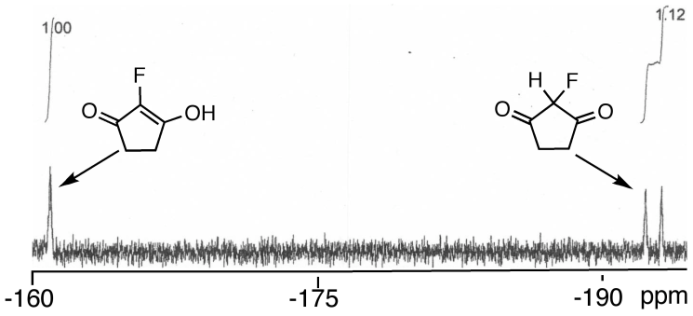
Long-range heteronuclear shift correlation methods have served as the cornerstone of modern structure elucidation protocols for several decades. The 1H–13C HMBC experiment provides a versatile and relatively sensitive means of establishing predominantly 3JCHconnectivity with the occasional 2JCH or 4JCH correlation being observed. The two-bond and four-bond outliers must be identified specifically to avoid spectral and/or structural misassignment. Despite the versatility and extensive applications of the HMBC experiment, it can still fail to elucidate structures of molecules that are highly proton-deficient, e.g., those that fall under the so-called “Crews rule”. In such cases, recourse to the ADEQUATE experiments should be considered. Thus, a study was undertaken to facilitate better investigator understanding of situations where it might be beneficial to apply 1,1- or 1,n-ADEQUATE to proton-rich or proton-deficient molecules. Equipped with a better understanding of when a given experiment might be more likely to provide the necessary correlation data, investigators can make better decisions on when it might be advisible to employ one experiment over the other. Strychnine (1) and cervinomycin A2 (2) were employed as model compounds to represent proton-rich and proton-deficient classes of molecules, respectively. DFT methods were employed to calculate the relevant nJCHheteronuclear proton–carbon and nJCC homonuclear carbon–carbon coupling constants for this study.
NMR Structure Elucidation of Small Organic Molecules and Natural Products: Choosing ADEQUATE vs HMBC
† Discovery and Preclinical Sciences, Process and Analytical Chemistry, NMR Structure Elucidation, Merck Research Laboratories, Kenilworth, New Jersey 07033, United States
‡ Discovery and Preclinical Sciences, Process and Analytical Chemistry, NMR Structure Elucidation, Merck Research Laboratories, Rahway, New Jersey 07065, United States
Using HMBC and ADEQUATE NMR Data To Define and Differentiate Long-Range Coupling Pathways: Is the Crews Rule Obsolete?
It is well known that as molecules become progressively more proton-deficient, structure elucidation becomes correspondingly more challenging. When the ratio of 1H to 13C and the sum of other heavy atoms falls below 2, an axiom that has been dubbed the “Crews rule” comes into play. The general premise of the Crews rule is that highly proton-deficient molecules may have structures that are difficult, and in some cases impossible, to elucidate using conventional suites of NMR experiments that include proton and carbon reference spectra, COSY, multiplicity-edited HSQC, and HMBC (both 1H–13C and 1H–15N). However, with access to modern cryogenic probes and microcyroprobes, experiments that have been less commonly utilized in the past and new experiments such as inverted 1JCC 1,n-ADEQUATE are feasible with modest sized samples. In this light, it may well be time to consider revising the Crews rule. The complex, highly proton-deficient alkaloid staurosporine (1) is used as a model proton-deficient compound for this investigation to highlight the combination of inverted 1JCC 1,n-ADEQUATE with 1.7 mm cryoprobe technology.
Using HMBC and ADEQUATE NMR Data To Define and Differentiate Long-Range Coupling Pathways: Is the Crews Rule Obsolete?
† Discovery and Preclinical Sciences, Process and Analytical Chemistry, Structural Elucidation Group, Merck Research Laboratories, Kenilworth, New Jersey 07033, United States
‡ Discovery and Preclinical Sciences, Process and Analytical Chemistry, Structural Elucidation Group, Merck Research Laboratories, Rahway, New Jersey 07065, United States
§ Discovery and Preclinical Sciences, Process and Analytical Chemistry, Structural Elucidation Group, Merck Research Laboratories, Summit, New Jersey 07901, United States
The Tuha’a Pae or Austral Islands (French: Îles Australes or Archipel des Australes) are the southernmost group of islands in French Polynesia, an overseas …
Tubuai is in the Austral Archipelago. These island chains are spread out over an area the size of Europe with 120 islands in all, 25 of which that are …
Juliana Aristéia de Lima holds a Ph.D. in chemistry and is currently conducting research at the State University of Campinas, located in the state of São Paulo in Southeast Brazil. She works on the development of biodegradable polymers blends (biopolymers).
Research focus: Sustainable management in the chemical industry
Juliana Aristéia de Lima holds a Ph.D. in chemistry and is currently conducting research at the State University of Campinas, located in the state of São Paulo in Southeast Brazil. She works on the development of biodegradable polymers blends (biopolymers). Polymers are ubiquitous in modern everyday life, most notably in the form of plastics. Because of that, it is essential for the future that they don’t constitute a waste problem in the way they often have in the past, but instead degrade in the way natural materials like paper or food would.
With her research, Juliana Aristéia de Lima addresses an important topic in the area of sustainable resource management. In the future, the Brazilian researcher also hopes to work on conductive ionic liquids, which could serve as solvents for preparation of polymer membranes. She is aspiring to a postdoctoral research position in Germany and wants to make new contacts with German experts in industry and academia for that purpose.
Universidade Estadual de Campinas
.
.
.
Take a tour
SOLOMON ISLANDS
HONIARA
Malaita, Solomon Islands …
.
.
Gizo, on Ghizo Island, is the capital of the Solomon Islands’ far-flung Western Province, a paradise of coral cays, atolls, lagoons and volcanic islands east of Papua New Guinea where, on a rainy day in late July, crowds flocked to the local netball court for the opening of the inaugural Akuila Talasasa Arts Festival.
Motorised canoes lined up in Gizo Harbour near the daily marketplace.
Water purification is the process of removing undesirable chemicals, biological contaminants, suspended solids and gases from contaminated water. The goal is to produce water fit for a specific purpose. Most water is disinfected for human consumption (drinking water), but water purification may also be designed for a variety of other purposes, including fulfilling the requirements of medical, pharmacological, chemical and industrial applications. The methods used include physical processes such as filtration, sedimentation, anddistillation; biological processes such as slow sand filters or biologically active carbon; chemical processes such as flocculation andchlorination and the use of electromagnetic radiation such as ultraviolet light.
Purifying water may reduce the concentration of particulate matter including suspendedparticles, parasites, bacteria, algae, viruses, fungi, as well as reducing the amount of a range of dissolved and particulate material derived from the surfaces that come from runoff due torain.
The standards for drinking water quality are typically set by governments or by international standards. These standards usually include minimum and maximum concentrations of contaminants, depending on the intended purpose of water use.
Visual inspection cannot determine if water is of appropriate quality. Simple procedures such as boiling or the use of a household activated carbon filter are not sufficient for treating all the possible contaminants that may be present in water from an unknown source. Even natural spring water – considered safe for all practical purposes in the 19th century – must now be tested before determining the kind of treatment, if any, is needed. Chemical and microbiological analysis, while expensive, are the only way to obtain the information necessary for deciding on the appropriate method of purification.
According to a 2007 World Health Organization (WHO) report, 1.1 billion people lack access to an improved drinking water supply, 88 percent of the 4 billion annual cases ofdiarrheal disease are attributed to unsafe water and inadequate sanitation and hygiene, while 1.8 million people die from diarrheal diseases each year. The WHO estimates that 94 percent of these diarrheal cases are preventable through modifications to the environment, including access to safe water.[1] Simple techniques for treating water at home, such as chlorination, filters, and solar disinfection, and storing it in safe containers could save a huge number of lives each year.[2] Reducing deaths from waterborne diseases is a major public health goal in developing countries.
Water purity is extremely important to pharmaceutical and biochemical industries. Suspended or dissolved particles, organic compounds, impurities and other contaminants prohibit the usage of tap water in laboratory applications and scientific research. Parameters such as resistivity, conductivity, size of particulate matter and concentration of microorganisms are used to categorize water quality and, therefore, specify intended uses for water. Some applications can tolerate the presence of specific impurities in the water, but others, such as High Performance Liquid Chromatography (HPLC) require removal of the majority of contaminants.
Contaminants
Water is an excellent solvent and can be sourced from almost anywhere on Earth. This property makes it prone to all kinds of contamination.
Particulates: Silt and debris which can be removed by passing water through a 10 to 20 micron filter (or less if necessary).
Microorganisms: Bacterial agents constitute a real challenge for water purification systems. Their growth rate, size and robustness require an efficient design (detection, removal from water inlet, inhibition of growth, etc.). Bacteria are measured in colony forming units per milliliter and can be killed with disinfectants. As a result, their secretions and cellular fragments must also be removed to avoid contamination.
Endotoxins, pyrogens, DNA and RNA: Cellular fragments and bacterial by-products. Harmful to tissue cultures. Can be detected with a Limus Amoebocyte Lysate (LAL) test.
Dissolved inorganic elements: Include phosphates, nitrates, calcium and magnesium, carbon dioxide, silicates, iron, chloride, fluoride, and any other natural or man-made chemicals resulting from exposure to the environment. Electrical conductivity (μSiemens/cm) is used to monitor high concentration of ions, while resistivity (MÙcm) is used to identify ions if present in small concentrations. These contaminants affect water hardness and alkalinity/acidity.
Dissolved organic elements: Pesticides, plant and animal remains or fragments. Total Organic Carbon (TOC) analyzers are used to measure CO2 emitted by organics subjected to oxidization. Organic-free water is mainly used in applications where analysis of organic substances is carried out (e.g. HPLC, chromatography and mass spectrometry).
Scientific applications require elimination of certain types of contaminants. On the other hand, pharmaceutical productions require, in most cases, near-total removal of impurities (criteria dictated by specific standards or local/international regulatory bodies).
Purification Process
There are a number of methods commonly used to purify water. Their effectiveness is linked to the type of contaminant being treated and the type of application the water will be used for.
Filtration: This process can take the form of any of the following:
Coarse filtration: Also called particle filtration, it can utilize anything from a 1 mm sand filter, to a 1 micron cartridge filter.
Micro filtration: Uses 1 to 0.1 micron devices to filter out bacteria. A typical implementation of this technique can be found in the brewing process.
Ultra filtration: Removes pyrogens, endotoxins, DNA and RNA fragments.
Reverse osmosis: Often referred to as RO, reverse osmosis is the most refined degree of liquid filtration. Instead of a filter, it uses a porous material acting as a unidirectional sieve that can separate molecular-sized particles.
Distillation: Oldest method of purification. Inexpensive but cannot be used for an on-demand process. Water must be distilled and then stored for later use, making it again prone to contamination if not stored properly.
Activated carbon adsorption: Operates like a magnet on chlorine and organic compounds.
Ultraviolet radiation: At a certain wavelength, this might cause bacteria to be sterilized and other micro organics to be broken down.
Deionization: Also known as ion exchange, it is used for producing purified water on-demand, by passing water through resin beds. Negatively charged (cationic) resin removes positive ions, while positively charged one (anionic) removes negative ions. Continuous monitoring and maintenance of the cartridges can produce the purest water.
Hot Water Sanitization
Sanitization of water purification equipment with hot water is achieved via an appropriate combination of exposure time and temperature. A primary use for this process is to deactivate viable microbes. It is worth mentioning that Endotoxin reduction is not achieved as a direct result of the hot water sanitization process.
Based on the feed water source, system operating conditions and the end-user’s operating and maintenance procedures, traditional chemical cleaning processes may still be required.
Sanitization using hot water involves incorporating heat exchangers into the traditional clean in place (CIP) system to gradually heat and cool water circulating through the reverse osmosis membrane system. Membrane manufacturers commonly stipulate a controlled heating and cooling rate to protect against irreversible damage to the membrane and ensure the system’s long-term performance.
A typical hot water sanitization sequence would consist of the following phases:
Initialization (conditions checking)
Heating
Holding
Cooling
A control system must therefore provide flexibility in the way in which accurate and repeatable control of the sterilization is achieved and will
include the following features:
Precise loop control with setpoint profile programming
Sequential control for sanitation/sterilization
Onscreen operator messaging
Duty standby pump control
Secure collection of on-line data from the purified water system for analysis and evidence
Local operator display with clear graphics and controlled access to parameters
Control room and schematics of the water purification plant to Lac de Bret, Switzerland
Pharmaceuticals can enter the water supply in a variety of ways. Debates continue over how dangerous this is. Source: GAO
Information sheet: Pharmaceuticals in drinking-water
(This information sheet is a summary of the key findings, recommendations and conclusions of the WHO technical report on Pharmaceuticals in drinking-water and the inputs of additional expert peer-reviewers)
Background and scope
Pharmaceuticals are synthetic or natural chemicals that can be found in prescription medicines, over-the-counter therapeutic drugs and veterinary drugs. Pharmaceuticals contain active ingredients that have been designed to have pharmacological effects and confer significant benefits to society. Pharmaceuticals can be introduced into water sources through sewage, which carries the excreta of individuals and patients who have used these chemicals, from uncontrolled drug disposal (e.g. discarding drugs into toilets) and from agricultural runoff comprising livestock manure. They have become chemicals of emerging concern to the public because of their potential to reach drinking-water.
Occurrence of pharmaceuticals in drinking-water
The ubiquitous use of pharmaceuticals (both prescribed and over the counter) has resulted in a relatively continuous discharge of pharmaceuticals and their metabolites into wastewater. In addition, pharmaceuticals may be released into water sources in the effluents from poorly controlled manufacturing or production facilities, primarily those associated with generic medicines.
Following advances in the sensitivity of analytical methods for the measurement of these chemicals at very low concentrations, a number of studies found trace concentrations of pharmaceuticals in wastewater, various water sources and some drinking-waters. Concentrations in surface waters, groundwater and partially treated water were typically less than 0.1 µg/l (or 100 ng/l), whereas concentrations in treated water were generally below 0.05 µg/l (or 50 ng/l). These investigations suggested that pharmaceuticals are present, albeit at trace concentrations, in many water sources receiving wastewater effluents.
The presence of specific pharmaceuticals in a water source will vary from place to place depending upon the type of pharmaceutical and the extent of discharge into water bodies. Key factors include the pharmaceuticals prescribed, used or manufactured in the area and the size of the population in the catchment. The occurrence and concentration of pharmaceuticals in receiving water sources, which are the primary pathway into drinking-water, are dependent on dilution, natural attenuation and the degree of wastewater treatment applied.
Risk assessment of pharmaceuticals in drinking-water
There are currently few systematic monitoring programmes or comprehensive studies available on human exposure to pharmaceuticals from drinking-water. Therefore, a key challenge in assessing the potential human health risk associated with exposure to very low concentrations of pharmaceuticals in drinking-water is the limited occurrence data available for the diverse group of pharmaceuticals in use today and their active metabolites.
However, several approaches for screening and prioritizing pharmaceuticals for human health risk assessment for exposure through drinking-water have been published in the peer-reviewed literature. These approaches usually apply the principle of the “minimum therapeutic dose” (also known as the “lowest clinically effective dose”) or the acceptable daily intake, in conjunction with safety factors or uncertainty factors for different groups of pharmaceuticals, to derive a margin of safety, or margin of exposure, between the worst-case exposure observed or predicted and the minimum therapeutic dose or acceptable daily intake.
Current observations suggest that it is very unlikely that exposure to very low levels of pharmaceuticals in drinking-water would result in appreciable adverse risks to human health, as concentrations of pharmaceuticals detected in drinking-water (typically in the nanogram per litre range) are several orders of magnitude (typically more, and often much more, than 1000-fold) lower than the minimum therapeutic dose.
Control measures and risk management
Concentrations of the vast majority of pharmaceuticals in the water environment can be reduced through natural processes (e.g. adsorption onto sediment, solar photodegradation and biological degradation) or during subsequent drinking-water and wastewater treatment processes.
Despite their unique pharmacological properties, pharmaceuticals respond to treatment no differently from other organic chemicals, with removal rates depending on their physicochemical properties and the treatment technology being used. Conventional water treatment processes, such as chlorination, can remove approximately 50% of these compounds, whereas more advanced treatment processes, such as ozonation, advanced oxidation, activated carbon, nanofiltration and reverse osmosis, can achieve higher removal rates; reverse osmosis, for example, can remove more than 99% of large pharmaceutical molecules.
Funding for any water safety improvements, like any public health intervention, draws on limited resources that need to be carefully allocated with due consideration of their beneficial impact. However, implementing additional specialized and costly drinking-water treatment, specifically with the intention of reducing trace concentrations of pharmaceuticals, is not considered necessary at this time, as the human health benefit would be limited.
The most appropriate approach to minimize the presence of pharmaceuticals in drinking-water and reduce human exposure is to prevent or reduce their entry into the water environment as far as reasonably practical. This can be achieved through a combination of preventive measures, including enhanced communication to the public on rational drug use and disposal of pharmaceuticals (e.g. avoid flushing unused drugs down the toilet), education for prescribers and systematic drug take-back programmes.
However, in line with the water safety plan principle of control of contaminants at the source, it would be appropriate to investigate improvements in wastewater treatment to remove pharmaceuticals and other potential contaminants of concern from their main route of entry into the water environment.
Monitoring of pharmaceuticals in water
In the absence of regulatory mandates, routine monitoring for pharmaceuticals in water sources and drinking-water on a national basis would not be desirable except in cases where local circumstances indicate a potential for elevated concentrations (e.g. manufacturing facilities with uncontrolled effluent discharge upstream of a drinking-water source). In these circumstances, investigative monitoring of, for example, surface water, groundwater and wastewater effluent can be undertaken to assess possible occurrence levels and exposure; if necessary, screening values can be developed in conjunction with an assessment of the potential risks to human health from exposure through drinking-water.
Based on the results of this risk assessment, an evaluation of possible control options could be considered as part of a water safety plan. Practical difficulties associated with implementing monitoring programmes for pharmaceuticals include the lack of standardized sampling and analysis protocols, high costs and the limited availability of the analytical instruments required to measure the diverse range of pharmaceuticals that may be present.
Investigative surveys should be tailored to local circumstances, taking into account existing wastewater and water treatment processes and pharmaceuticals (and their metabolites) that are commonly prescribed, used or manufactured within the catchment area of concern. Such studies should be carried out with appropriate rigorous quality assurance and verification and designed to confirm whether drinking-water is a significant risk.
Knowledge gaps
Although current risk assessments indicate that the very low concentrations of pharmaceuticals found in drinking-water are very unlikely to pose any appreciable risks to human health, knowledge gaps exist. These include the assessment of risks to human health associated with long-term exposure to low concentrations of pharmaceuticals and the possible combined effects of mixtures of pharmaceuticals.
Although the margins of exposure are substantial, it would be of value to ensure that these margins are adequate for possibly sensitive subpopulations and to better characterize health risks, if any, from long-term, low-level exposures. In addition, future research should focus on developing methods or protocols for prioritizing pharmaceuticals in the context of an overall risk assessment for all drinking-water hazards.
Summary
Currently, analysis of the available data indicates that there is a substantial margin of safety between the very low concentrations of pharmaceuticals that would be consumed in drinking-water and the minimum therapeutic doses, which suggests a very low risk to human health. Based on this finding, the development of formal health-based guideline values for pharmaceuticals in the World Health Organization’s (WHO) Guidelines for drinking-water quality is currently not considered to be necessary.
Concerns over pharmaceuticals in drinking-water should not divert water suppliers and regulators from other priorities for drinking-water and health, most notably microbial risks, such as bacterial, viral and protozoan pathogens, and other chemical risks, such as naturally occurring arsenic and excessive levels of fluoride.
Laboratory-scale spray dryer.
A=Solution or suspension to be dried in, B=Atomization gas in, 1= Drying gas in, 2=Heating of drying gas, 3=Spraying of solution or suspension, 4=Drying chamber, 5=Part between drying chamber and cyclone, 6=Cyclone, 7=Drying gas is taken away, 8=Collection vessel of product, arrows mean that this is co-current lab-spraydryer
Spray drying is a method of producing a dry powder from a liquid or slurry by rapidly drying with a hot gas. This is the preferred method of drying of many thermally-sensitive materials such as foods and pharmaceuticals. A consistent particle size distribution is a reason for spray drying some industrial products such as catalysts. Air is the heated drying medium; however, if the liquid is a flammable solvent such as ethanol or the product is oxygen-sensitive then nitrogen is used.[1]
All spray dryers use some type of atomizer or spray nozzle to disperse the liquid or slurry into a controlled drop size spray. The most common of these are rotary disks and single-fluid high pressure swirl nozzles. Atomizer wheels are known to provide broader particle size distribution, but both methods allow for consistent distribution of particle size.[2] Alternatively, for some applications two-fluid or ultrasonic nozzles are used. Depending on the process needs, drop sizes from 10 to 500 µm can be achieved with the appropriate choices. The most common applications are in the 100 to 200 µm diameter range. The dry powder is often free-flowing.[3]
The most common spray dryers are called single effect as there is only one drying air on the top of the drying chamber (see n°4 on the scheme). In most cases the air is blown in co-current of the sprayed liquid. The powders obtained with such type of dryers are fine with a lot of dusts and a poor flowability. In order to reduce the dusts and increase the flowability of the powders, there is since over 20 years a new generation of spray dryers called multiple effect spray dryers. Instead of drying the liquid in one stage, the drying is done through two steps: one at the top (as per single effect) and one for an integrated static bed at the bottom of the chamber. The integration of this fluidized bed allows, by fluidizing the powder inside a humid atmosphere, to agglomerate the fine particles and to obtain granules having commonly a medium particle size within a range of 100 to 300 µm. Because of this large particle size, these powders are free-flowing.
The fine powders generated by the first stage drying can be recycled in continuous flow either at the top of the chamber (around the sprayed liquid) or at the bottom inside the integrated fluidized bed. The drying of the powder can be finalized on an external vibrating fluidized bed.
The hot drying gas can be passed as a co-current or counter-current flow to the atomiser direction. The co-current flow enables the particles to have a lower residence time within the system and the particle separator (typically a cyclone device) operates more efficiently. The counter-current flow method enables a greater residence time of the particles in the chamber and usually is paired with a fluidized bed system.
Freeze dryer: a more-expensive batch process for products that degrade in spray drying. Dry product is not free-flowing.
Drum dryer: a less-expensive continuous process for low-value products; creates flakes instead of free-flowing powder.
Pulse combustion dryer: A less-expensive continuous process that can handle higher viscosities and solids loading than a spray dryer, and that sometimes gives a freeze-dry quality powder that is free-flowing.
Spray dryer
Spray drying nozzles.
Schematic illustration of spray drying process.
A spray dryer takes a liquid stream and separates the solute or suspension as a solid and the solvent into a vapor. The solid is usually collected in a drum or cyclone. The liquid input stream is sprayed through a nozzle into a hot vapor stream and vaporised. Solids form as moisture quickly leaves the droplets. A nozzle is usually used to make the droplets as small as possible, maximising heat transfer and the rate of water vaporisation. Droplet sizes can range from 20 to 180 μm depending on the nozzle.[3] There are two main types of nozzles: high pressure single fluid nozzle (50 to 300 bars) and two-fluid nozzles: one fluid is the liquid to dry and the second is compressed gas (generally air at 1 to 7 bars).
Spray dryers can dry a product very quickly compared to other methods of drying. They also turn a solution, or slurry into a dried powder in a single step, which can be advantageous for profit maximization and process simplification.
The Spray Drying Process
The spray drying process is older than might commonly be imagined. Earliest descriptions date from 1860 with the first patented design recorded in 1872. The basic idea of spray drying is the production of highly dispersed powders from a fluid feed by evaporating the solvent. This is achieved by mixing a heated gas with an atomized (sprayed) fluid of high surface-to-mass ratio droplets, ideally of equal size, within a vessel (drying chamber), causing the solvent to evaporate uniformly and quickly through direct contact.
Spray drying can be used in a wide range of applications where the production of a free-flowing powder is required. This method of dehydration has become the most successful one in the following areas:
Pharmaceuticals
Bone and tooth amalgams
Beverages
Flavours, colourings and plant extracts
Milk and egg products
Plastics, polymers and resins
Soaps and detergents
Textiles and many more
Almost all other methods of drying, including use of ovens, freeze dryers or rotary evaporators, produce a mass of material requiring further processing (e.g. grinding and filtering) therefore, producing particles of irregular size and shape. Spray drying on the other hand, offers a very flexible control over powder particle properties such as density, size, flow characteristics and moisture content.
Design and Control
The challenges facing both designers and users are to increase production, improve powder quality and reduce costs. This requires an understanding of the process and a robust control implementation.
Spray drying consists of the following phases:
Feed preparation: This can be a homogenous, pumpable and free from impurities solution, suspension or paste.
Atomization (transforming the feed into droplets): Most critical step in the process. The degree of atomization controls the drying rate and therefore the dryer size. The most commonly used atomization techniques are:
1. Pressure nozzle atomization: Spray created by forcing the fluid through an orifice. This is an energy efficient method which also offers the narrowest particle size distribution.
2. Two-fluid nozzle atomization: Spray created by mixing the feed with a compressed gas. Least energy efficient method. Useful for making extremely fine particles.
3. Centrifugal atomization: Spray created by passing the feed through or across a rotating disk. Most resistant to wear and can generally be run for longer periods of time.
Drying: A constant rate phase ensures moisture evaporates rapidly from the surface of the particle. This is followed by a falling rate period where the drying is controlled by diffusion of water to the surface of the particle.
Separation of powder from moist gas: To be carried out in an economical (e.g. recycling the drying medium) and pollutant-free manner. Fine particles are generally removed with cyclones, bag filters, precipitators or scrubbers.
Cooling and packaging.
A control system must therefore provide flexibility in the way in which accurate and repeatable control of the spray drying is achieved and will include the following features:
Precise loop control with setpoint profile programming
Recipe Management System for easy parameterisation
Sequential control for complex control strategies
Secure collection of on-line data from the system for analysis and evidence
Local operator display with clear graphics and controlled access to parameters
Micro-encapsulation
Spray drying often is used as an encapsulation technique by the food and other industries. A substance to be encapsulated (the load) and an amphipathic carrier (usually some sort of modified starch) are homogenized as a suspension in water (the slurry). The slurry is then fed into a spray drier, usually a tower heated to temperatures well over the boiling point of water.
As the slurry enters the tower, it is atomized. Partly because of the high surface tension of water and partly because of thehydrophobic/hydrophilic interactions between the amphipathic carrier, the water, and the load, the atomized slurry forms micelles. The small size of the drops (averaging 100 micrometers in diameter) results in a relatively large surface area which dries quickly. As the water dries, the carrier forms a hardened shell around the load.[5]
Load loss is usually a function of molecular weight. That is, lighter molecules tend to boil off in larger quantities at the processing temperatures. Loss is minimized industrially by spraying into taller towers. A larger volume of air has a lower average humidity as the process proceeds. By the osmosis principle, water will be encouraged by its difference in fugacities in the vapor and liquid phases to leave the micelles and enter the air. Therefore, the same percentage of water can be dried out of the particles at lower temperatures if larger towers are used. Alternatively, the slurry can be sprayed into a partial vacuum. Since the boiling point of a solvent is the temperature at which the vapor pressure of the solvent is equal to the ambient pressure, reducing pressure in the tower has the effect of lowering the boiling point of the solvent.
The application of the spray drying encapsulation technique is to prepare “dehydrated” powders of substances which do not have any water to dehydrate. For example, instant drink mixes are spray dries of the various chemicals which make up the beverage. The technique was once used to remove water from food products; for instance, in the preparation of dehydrated milk. Because the milk was not being encapsulated and because spray drying causes thermal degradation, milk dehydration and similar processes have been replaced by other dehydration techniques. Skim milk powders are still widely produced using spray drying technology around the world, typically at high solids concentration for maximum drying efficiency. Thermal degradation of products can be overcome by using lower operating temperatures and larger chamber sizes for increased residence times.[6]
Recent research is now suggesting that the use of spray-drying techniques may be an alternative method for crystallization of amorphous powders during the drying process since the temperature effects on the amorphous powders may be significant depending on drying residence times.[7][8]
Spray drying applications
Food: milk powder, coffee, tea, eggs, cereal, spices, flavorings, starch and starch derivatives, vitamins, enzymes, stevia, colourings, etc.
Pharmaceutical: antibiotics, medical ingredients, additives
The nano spray dryer offers new possibilities in the field of spray drying. It allows to produce particles in the range of 300 nm to 5 μm with a narrow size distribution. High yields are produced up to 90% and the minimal sample amount is 1 mL.
Pharmaceutical Spray drying is a very fast method of drying due to the very large surface area created by the atomization of the liquid feed. As a consequence, high heat transfer coefficients are generated and the fast stabilisation of the feed at moderate temperatures makes this method very attractive for heat sensitive materials.
Spray drying provides unprecedented particle control and allows previously unattainable delivery methods and molecular characteristics. These advantages allow exploration into employing previously unattainable delivery methods and molecular characteristics.
Five things you might not know about spray drying
Spray drying is suitable for heat sensitive materials
Spray drying is already used for the processing of heat sensitive materials (e.g. proteins, peptides and polymers with low Tg temperatures) on an industrial scale. Evaporation from the spray droplets starts immediately after contact with the hot process gas. Since the thermal energy is consumed by evaporation, the droplet temperature is kept at a level where no harm is caused to the product.
Spray drying turns liquid into particles within seconds The large surface of the droplets provides near instantaneous evaporation, making it possible to produce particles with a crystalline or amorphous structure. The particle morphology is determined by the operating parameters and excipients added to the feed stock.
Spray drying is relatively easy to replicate on a commercial scale GEA Niro has been producing industrial scale spray drying plants for well over half a century. Our process know-how, products and exceptional facilities put us in a unique position to advise and demonstrate how products and processes will behave on a large scale.
Spray drying is a robust process
Spray drying is a continuous process. Once the set points are established, all critical process parameters are kept constant throughout the batch. Information for the batch record can be monitored or logged, depending on the system selected.
Spray drying can be effectively validated The precise control of all critical process parameters in spray drying provides a high degree of assurance that the process consistently produces a product that meets set specifi cations.
The spray drying process
Spray drying is a very fast method of drying due to the very large surface area created by the atomization of the liquid feed and high heat transfer coefficients generated. The short drying time, and consequently fast stabilisation of feed material at moderate temperatures, means spray drying is also suitable for heat-sensitive materials.
As a technique, spray drying consists of four basic stages:
Atomization: A liquid feed stock is atomized into droplets by means of a nozzle or rotary atomizer. Nozzles use pressure or compressed gas to atomize the feed while rotary atomizers employ an atomizer wheel rotating at high speed.
Drying: Hot process gas (air or nitrogen) is brought into contact with the atomized feed guided by a gas disperser, and evaporation begins. The balance between temperature, flow rate and droplet size controls the drying process.
Particle formation: As the liquid rapidly evaporates from the droplet surface, a solid particle forms and falls to the bottom of the drying chamber.
Recovery: The powder is recovered from the exhaust gas using a cyclone or a bag filter. The whole process generally takes no more than a few seconds.
Chiou, D.; Langrish, T. A. G. (2007). “Crystallization of Amorphous Components in Spray-Dried Powders”. Drying Technology25: 1427. doi:10.1080/07373930701536718.
Ahmednagar is a medieval city. Numerous Mughal-era buildings dot the environs.Ahmednagar Fort, once considered the second most impregnable fort in India, …
2-Hydroxymalonitrile-A Useful Reagent for One-step Synthesis of α-Hydroxy Esters
YANG Jianxin1,2, YIN Yunxing2, HE Zhenmin2, MA Li2, LI Xin2, ZHANG Zhiliu2, LIN Xiaojuan2, MA Rujian2
1. Tianjin Key Laboratory for Modern Drug Delivery & High-Efficiency, School of Pharmaceutical Science and Technology, Tianjin University, Tianjin 300072, P. R. China;
2. WuXi PharmaTech Co. Ltd., Shanghai 200131, P. R. China
Corresponding Authors: MA Rujian E-mail: marj@wuxiapptec.com
Carbonyl compounds (aldehydes, ketones, carboxylic esters, carboxylic amides) react aselectrophiles at the sp2 hybridized carbon atoms and as nucleophiles if they contain an H-atom in the α-position relative to their C=O or C=N bonds. This is because this H is acidic and it can be removed by a base leaving behind an electron pair for nucleophilic attacks.
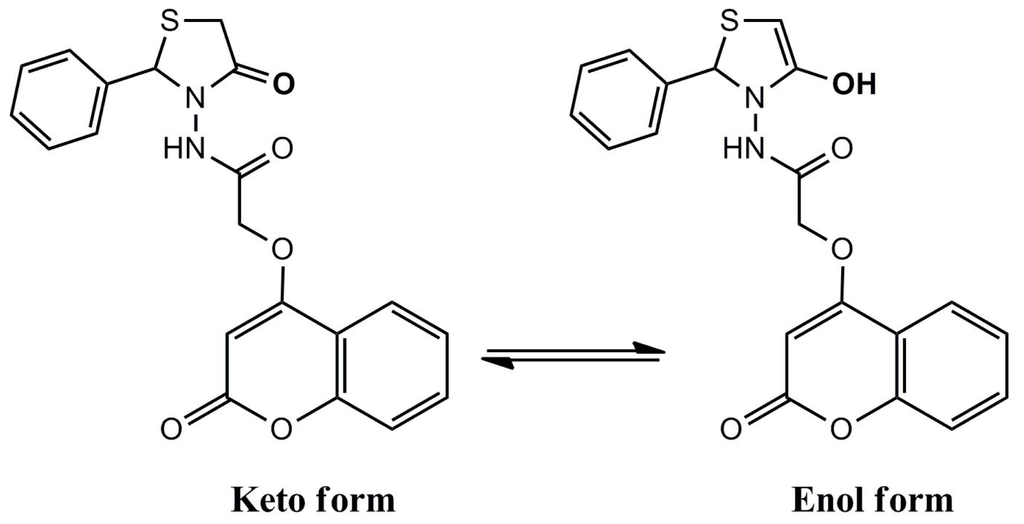
For most compounds in organic chemistry all the molecules have the same structure – even if this structure cannot satisfactory represented by a Lewis formula – but for many compounds there is a mixture of two or more structurally distinct compounds that are in rapid equilibrium. This phenomenon is called tautomerism.
Tautomerism is the phenomenon that occurs in any reaction that simply involves the intramolecular transfer of a proton. An equilibrium is established between the two tautomers (structurally distinct compounds) and there is a rapid shift back and forth between the distinct compounds.
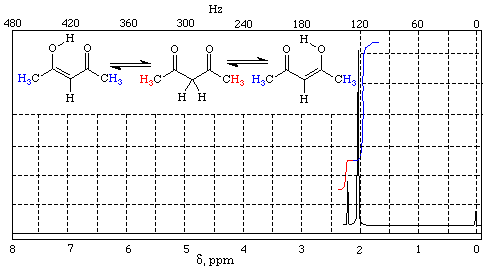
A very common form of tautomerism is that between a carbonyl compound containing an α–hydrogen and its enol form (Fig. I.1).
Fig. I.1: A keto-enol reaction
An enol is exactly what the name implies: an ene-ol. It has a C=C double bond (diene) and an OH group (alcohol) joined directly to it.
Notice that in the above reaction as in any keto-enol reaction there is no change in pH since a proton is lost from carbon and gained on oxygen. The reaction is known as enolization as it is the conversion of a carbonyl compound into its enol.
Notice also that in the above reaction the product is almost the same as the starting material since the only change is the transfer of one proton and the shift of the double bond.
In simple cases (R2 = H, alkyl, OR, etc.) the equilibrium of the keto-enol reaction lies well to the left (keto structure) (Table I.1). The reason can be seen by examining the bond energies in Table I.2.
Compound
Enol Content, %
Acetone
6 * 10-7
PhCOCH3
1.1 * 10-6
CH3CHO
6 * 10-5
Cyclohexanone
4 * 10-5
Ph2CHCHO
9.1
PhCOCH2COCH3
89.2
Table I.1: The enol content of some carbonyl compounds
If keto-enol reactions are common for aldehydes and ketones why don’t simple aldehydes and ketones exist as enols?
IR and NMR Spectra of carbonyl compounds show no signs of enols. The equilibrium lies well over towards the keto form (the equilibrium constant k for cyclohexanone is about 10-5).
Bond (Energy, kJ/mol)
Sum ( kJ/mol)
keto form
C-H (413)
C-C (350)
C=O (740)
1503
enol form
C=C (620)
C-O (367)
O-H (462)
1449
Table I.2: Bond energies in the keto and in the enol form. The keto form is thermodynamically more stable than the enol form by approximately 50 kJ/mol
The approximate sum of the bond energies in the keto form is 1503 kJ/mol while in the enol form 1449. Therefore, the keto form is thermodynamically more stable than the enol form by approximately 50 kJ/mol.
In most cases, enol forms cannot be isolated since they are less stable and are formed in minute quantities. However, there are some exceptions and in certain cases a larger amount of the enol form is present and it can be even the predominant species:
Molecules in which the enolic double bond is in conjugation with another double bond (cases are shown in Table I.1 likePh2CHCHO and PhCOCH2COCH3)
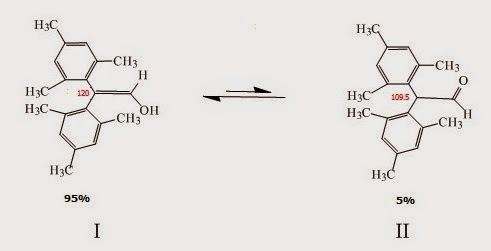
Molecules that contain two or more bulky aryl groups (Fig. I.2). Compound I in Fig. I.2 (a substituted enol) is the major species in equilibrium (~95%) while the keto form is the minor species (~5%). In cases like this steric hindrance destabilizes the keto form (the two substituted aryl groups are 109° apart) while in the enol form 120° apart.
Fig. I.2: A keto-enol reaction. The enol form (I) is the major species in this case since the keto form is destabilized by steric hindrance (the substituted aryl groups are closer in the keto form – the C-C angle is 109° and this is unfavorable due to steric hindrance)
Is there experimental evidence that keto-enol reactions are common for aldehydes and ketones?
If the NMR spectrum of a simple carbonyl compound in D2O is obtained – such as pinacolone’s (CH3)3CCOCH3 – the signal for protons next to the carbonyl group very slowly disappears. The isolated compound’s mass spectrum (after the above mentioned reaction with D2O is over) shows that those hydrogen atoms have been replaced by deuterium atoms. There is a peak at (M+1)+ or (M+2)+ or (M+3)+ instead of M+. The reaction is shown in Fig. I.3:
Fig. I.3: Evidence for a keto-enol reaction when pinacolone (CH3)3CCOCH3 reacts with D2O. When the enol form of the pinacolone reverts to the keto form it picks up a deuteron instead of a proton because the solution consists almost entirely of D2O.
What mechanism can be proposed for the above reaction?
Enolization is a slow process in neutral solution, even in D2O, and is catalyzed by acid or base in order to happen.
In the acid-catalyzed reaction the molecule is first protonated on oxygen and then loses the C-H proton in a second step (Fig. I.4). When the enol form reverts to the keto – since this is an equilibrium process – it picks up a deuteron instead of a proton since the solution is D2O.
Fig. I.4: The acid-catalyzed keto-enol reaction mechanism. If D2O is the solvent then the α-hydrogens to carbonyl group are replaced by deuterium.
In the base-catalyzed reaction the C-H proton is removed first by the base (for example hydroxide ion OH–, OD– in our case) and the proton (or D+ in our case) added to the oxygen atom in a second step (Fig. I.5).
Fig. I.5: The base-catalyzed keto-enol reaction mechanism. If D2O is the solvent then the α-hydrogens to carbonyl group are replaced by deuterium.
Notice that the enolization reactions in Fig. I.4 and Fig. I.5 are catalytic. In the acid-catalyzed mechanism the D+ (or H+ if water is the solvent) is regenerated at the end (catalyst). In the base-catalyzed mechanism OD– (or OH– if water is the solvent) is regenerated at the end (catalyst).
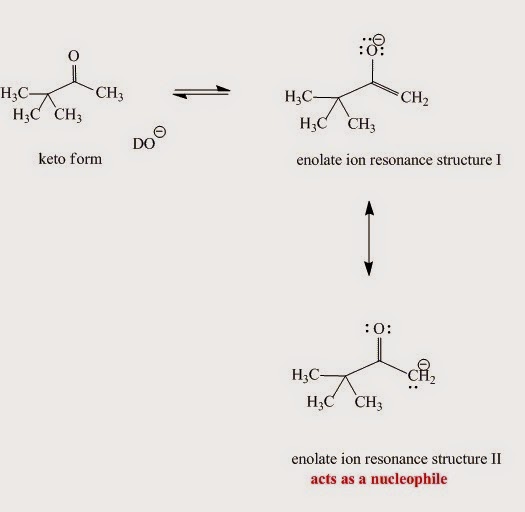
The enolate ion generated from the enol (Fig. I.6) in the base-catalyzed mechanism is nucleophilic due to:
Oxygen’s small atomic radius
Formal negative charge
An enolate ion is an ion with a negative charge on oxygen with adjacent C-C double bond.
Fig. I.6: Enolate ion resonance contributors. Although the major contributor is resonace structure I when it reacts as a nucleophile structure II is more reactive.
Enolates are reactive nucleophiles. Although the major enolate Lewis contributor shows concentration of electron density on the electronegative oxygen when it reacts as a nucleophile, it behaves like the electron density is concentrated on the α-carbon next to carbonyl group.
Enolates react with alkyl halides, aldehydes/ketones and esters and these reactions are shown in the post entitled “The chemistry of enolate ions – Enolate ion reactions”.
References
A.J. Kresge, Pure Appl. Chem., 63, 213 (1991)
B. Capon, The Chemistry of Enols, Wiley, NY, 307–322 (1990)
S.E. Biali et al., J. Am. Chem. Soc. 107, 1007 (1985).
Surat, previously known as Suryapur, is a city in the Indian state of Gujarat. It is the administrative capital of the Surat district. The city is located 306 km south of …
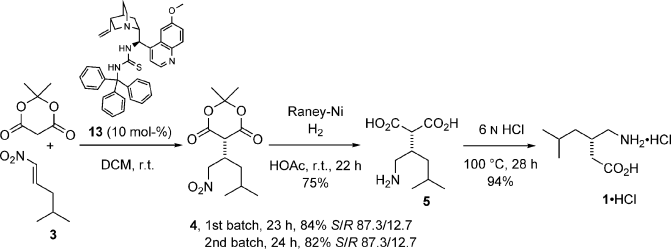
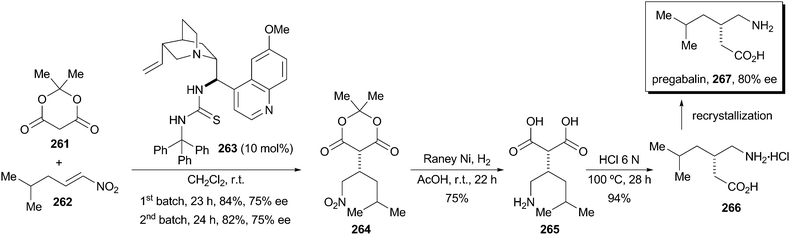
Bassas, O.; Huuskonen, J.; Rissanen, K.; Koskinen, A.M.P. ’A Simple Organocatalytic Enantioselective Synthesis of Pregabalin.’ Eur. J. Org. Chem.2009, 1340-1351.
This paper describes a new procedure for the enantioselective synthesis of the important anticonvulsant drug Pregabalin, which shows biological properties as the (S) enantiomer only. The key step of the synthetic sequence is the Michael addition reaction of Meldrum’s acid to a nitroalkene mediated by a quinidine derived thiourea. A variety of novel catalysts bearing different groups at the thiourea moiety were synthesized and tested. The most successful catalyst that incorporates a trityl substituent provided up to 75 % ee of (S)-4. The conjugate addition reaction was carried out on a multigram scale with low loadings of catalyst (10 mol-%). Moreover, the catalyst can be recycled showing the same capability in chemical yield and asymmetric induction. Then, hydrogenation of nitroalkane 4 followed by decarboxylation of diacid 5 provides Pregabalin hydrochloride in 59 % overall yield. Enantioenrichment by crystallization of the free amino acid 1 improves the (S)/(R) enantiomeric ratio to 9:1.
Author Information
1Department of Chemistry, Helsinki University of Technology, P. O. Box 6100, 02015 TKK, Espoo, Finland, Fax: +358-94512538
2NanoScience Center, Department of Chemistry, University of Jyväskylä, P. O. Box 35, 40014 Jyväskylä, Finland
Jyväskylä (Finnish pronunciation: [ˈjyvæsˌkylæ]) is a city and municipality in CentralFinland in the western part of the Finnish Lakeland. It is the largest city in …
When Women in Chemistry (WIC), which has been awarded this year’s College of Natural Sciences Student Service Award, set out to survey chemistry graduate …
Vandana Patravale met their VC and Deans along with business tycoons in a special dinner party hosted by AIBC and they were very keen to work with Institute …
… from the School of Chemistry, has won a L’Oréal Australia For Women in Science Fellowship worth $20 000 for her work on metal-organic frameworks that …
BATON ROUGE – LSU is the top university in the nation in granting Ph.D. degrees in chemistry to women and underrepresented minority students, according to a …
Roxanne Bales vividly remembers the months she spent at a graduate program in organic chemistry in New England.
Ji Qi (GRS’06,’11), a graduate student pursuing a Ph.D. in chemistry, has been awarded a 2006-2007 Novartis Fellowship in Organic Chemistry for Women and …
Design and Development of Novel Azo Prodrugs using Various Permutations by Dr.Suneela
Chemistry professor Julia Kubanek
http://chemists.princeton.edu/knowles/
The 2014 winners of the The Elsevier Foundation Awards for Early Career Women Scientists in the Developing World accept their prizes at the annual AAAS …
Ph.D. (1990, Cambridge), FASc, is a recipient of S S Bhatnagar Award, Swarnajayanti Fellowship of the DST, the Bronze Medal of the Chemical Research Society …
From left to right: Jessica Nilsson, Francesca Novara, Karen Hindson, Katja Glatz, Rachel McGlue, Anne Deveson, Elisabeth von Roedern, Susan Wilkinson, …
Tami Spector (on the left), professor of organic chemistry, in a chemistry lab with one of her current students: Amera Al-Faleh, a senior majoring in …
Kiran Mazumdar Shaw: (born 23 March 1953) She is the Chairman & Managing Director, Biocon Limited a biotechnology company based at Bangalore. She is on the Forbes list of the world’s 100 most powerful women and in business list on top 50 women released by the Financial Times’. In the year 1978, she started Biocon in the garage of her rented house in Bangalore with a seed capital of Rs. 10,000. Now the net worth of the company is more than $ 900 million. Now Biocon produces drugs for cancer, diabetes and auto-immune diseases. Product pipeline includes world’s first oral insulin, currently undergoing Phase III clinical trials.
Winners of the 2014 Elsevier Foundation Awards for Early Career Women Scientists in Developing Countries: (left to right) Dr. Eqbal Mohammed Abdu Dauqan (Biochemistry – Yemen), Dr. Simone Ann Marie Badal McCreath (Biochemistry – Jamaica), Dr. Taiwo Olayemi Elufioye (Pharmacology – Nigeria), Dr. Leni Ritmaleni (Medicinal Chemistry – Indonesia) and Dr. Nilufar Mamadalieva (Biochemistry – Uzbekistan). Photos by Alison Bert
Dr. Nilufar Mamadalieva, Senior Scientific Researcher at the Institute of the Chemistry of Plant Substances in Tashkent, Uzbekistan, was honored for her work on the phytochemical and biological investigation of active compounds derived from medicinal plants growing in Central Asia, in particular the development of efficient nutraceuticals and the discovery of new lead compounds for the pharmaceutical industry.The field of natural substances, a tradition at the Tashkent Institute, is gaining more interest in western countries for the development of efficient nutraceuticals and the discovery of new lead compounds for the pharmaceutical industry.
Dr. Mamadalieva is the recipient of a number of international fellowships, which have allowed her to travel extensively and develop a network of international collaborators.
“This award gives me confidence and confirms that I’m going for the right goal,” she said.
East and South-East Asia & the Pacific
Leni Ritmaleni, PhD
Dr. Leni Ritmaleni of the Faculty of Pharmacy at Gadjah Mada University in Yogyakarta, Indonesia, was honored for her work in the field of organic synthesis, focusing on the development of tropical medicines, in particular improved methods for the synthesis of sulfoxides and their application in the preparation of biologically active targets.She hopes her work will “encourage young women in Indonesia to love science, especially synthetic organic chemistry.”
“Women need science, science needs women and they need to work together,” she said.
Dr. Ritmaleni received her PhD from the School of Chemistry at Bristol University, UK after receiving a scholarship from the Indonesian government. She has won several awards in Indonesia and has published over 40 papers.
Dr. RItmaleni said researchers face various challenges at her institution, including a lack of access to scientific equipment and supplies and a scarcity of grants for basic science.
As a mother, she also strives to balance work and family, making “time management” an important priority.
She values the recognition provided by this award along with “the opportunity to connect with other scientists around the globe.”
Latin America & the Caribbean
Simone Ann Marie Badal McCreath, PhD
Dr. Simone Ann Marie Badal McCreath manages the biochemistry lab at the Natural Products Institute at the University of the West Indies in Jamaica, and is designing a new cell culture lab at the same Institute. She was recognized for her work in designing a new cell culture lab to investigate the cancer-fighting properties of Jamaican natural compounds.Her interest is in screening Jamaican plant isolates for their potential properties slow down block or prevent the carcinogenic process. “Our findings have so far identified several isolates that are more potent in reducing cancer cell viability as well as potentially safer than anti-cancer drugs now on the market,” she said. “This research will pave the way for future research necessary for drug development and also the propagation and culture of novel Jamaican cancer and normal cells lines.
“Since cancer is the leading cause of death in Jamaica, such findings will prove useful in cancer treatment and prevention as well as earlier diagnosis in addition to identifying molecular targets that can improve selectivity of the isolates to cancer cells only.”
Dr. Badal McCreath has received numerous awards and has published extensively.
She said the challenges she faces in her career are less gender-based and more about the long delays in getting equipment and supplies to their lab as well as a lack of funding.
“Such challenges can cost us months even years of research,” she said. “Nonetheless, women in science do face challenges, and these become more apparent the higher the ladder you climb, the top of which is male dominated.”
Winning this award, she said, means attracting funding for cancer research in Jamaica and “the motivation of young and older women in science and other areas … to never give up but to persevere through gender-based and other issues that we daily face.”
Arab region
Eqbal Mohammed Abdu Dauqan, PhD PhD
Dr. Eqbal Mohammed Abdu Dauqan is Head of the Department of Medical Laboratories Sciences at Al-Saeed University in Taizz, Yemen. She was honored for her research on the antioxidant properties of vegetable oils and specialized research in sensory evaluation and organic chemistry.She received her PhD from the National University of Malaysia. Her interests are in biochemistry and biotechnology, and she has conducted specialist research in food science, natural antioxidents and organic chemistry. She is also a dedicated teacher.
“Not all the people around us understand what natural antioxidents are,” she said. She and her colleagues do workshops for the public, pointing out the antioxident properties in vitamins such as C and E and how to find them in the foods they eat.
Sub-Saharan Africa
Taiwo Olayemi Elufioye, PhD
Dr. Taiwo Olayemi Elufioye is acting head of the Department of Pharmacognosy at the University of Ibadan, Nigeria. She was honored for her research on the medicinal properties of native Nigerian plants, in particular the effectiveness of different species in treating malaria, wounds, memory loss, leprosy and cancer.She said she has been able to identify a compound with good activity against a chloroquine-resistant strain of malaria parasites. Also, she and her research colleagues are creating an herb tea that that may be useful for dementia.
“My main challenge has been funding, typical for most developing world,” she said. “Also been a woman can be challenging considering the fact that prevailing conditions and policies are not necessarily woman-friendly.
“It’s just so great to know that despite these challenges, my contribution to science is being recognized. I feel so proud and definitely energized to do more.”
“The winners of the 2014 Elsevier Foundation prizes are impressive not just for their research, but also for their potential,” said TWAS Executive Director Romain Murenzi. “Certainly these awards could bring them exciting new opportunities for research. We also believe that, over time, these researchers also will fulfill their potential as teachers and mentors, as partners in international projects and as advisers to governments. Such leadership can make a long-lasting contribution to global science.”
David Ruth and Samira Omar Asam present the award to Dr. Nilufar Mamadalieva, Senior Scientific Researcher at the Institute of the Chemistry of Plant Substances in Uzbekistan.
Fang Xin, president of OWSD, said: “These five women, like all women undertaking scientific research in developing countries, will certainly have faced challenges on the road to this award. But their determination, commitment and enthusiasm have paid off. The award is recognition that they are excellent scientists and that their research has made an impact both regionally and internationally. They are an inspiration to all young women considering careers in science.”At the ceremony, Samira Omar Asem, VP for the OWSD Arab Region, said OWSD and TWAS see this award as “vital for encouraging women in developing countries to be more involved in science and technology and to make a more significant contribution to social and economic developments.”
David Ruth, Executive Director of the Elsevier Foundation, said professional visibility is crucial to developing high-profile international scientific careers, especially for women. He explained that the Elsevier Foundation provides support to early-career women scholars through its New Scholars grant programs as well as mentoring, research retreats, professional visibility, childcare, work-life integration and recognition programs.
“The awards for these impressive women scientists represent a cooperative effort supported by Elsevier, OWSD, AAAS and TWAS to build research capacity and advance scientific knowledge throughout the developing world,” he said, “and what better place than the annual AAAS conference to raise awareness among scientists, policymakers, journalists and the public about the need to retain and celebrate women scientists.”
chemistry at the Indian Institute of Science, Bangalore
As is well known the drug discovery and development process is a complex process typically starting from the target identification and validation of a target to progress to clinical studies and hopefully ending with a new drug to the market [Figure 1].
In this process, different approaches and methods are required to understand the disease’s mechanisms, to profile hit molecules that will be progressed to leads suitable for full scale lead optimization programmes and then to generate quality drug candidates to advance to clinical studies.
Focusing on small molecules’ drugs and on hit to candidate phases, a variety of techniques will be used to study the compounds’ profiles at different levels including the physicochemical profiles, purity, solid state behaviours and structures to ensure a quality hit/lead/candidate and related data to allow understanding of the mechanism of action and SAR correlation.
Nuclear Magnetic Resonance (NMR) spectroscopy will play a pivotal role generating data on the molecular interactions between ligands and biological targets, in addition to providing the structures of drug molecules, by-products, impurities, metabolites and quantification data.
NMR Screening Impact
During the drug discovery phase, NMR spectroscopy is becoming more and more relevant with application at multiple stages along the progression of a project: NMR experiments are used for hits generation, lead discovery and optimization, evaluation of in vitro/in vivo selectivity and efficacy, studies drug toxicity profiles and identification of new drug discovery targets.
Over the last years there has been a large increase in the application of NMR techniques for the rapid determination of protein-ligand structures and interactions, to powerfully screen fragment-based libraries, to identify biological relevant ligand interactions, and to monitor changes in the metabolome from bio-fluids and cells to explore compounds activity.
Focusing on the NMR-based screening techniques, the NMR experiments could be divided into two main categories: target observed and ligand observed methods.
Without doubt, high resolution protein structure is a key requirement to evaluate the biological relevance of a hit from screening and HTS (high-throughput screening) and NMR together with X-ray are playing an essential role.
The last period has witnessed the generation of fast NMR sequences and methods to allow a faster impact on the drug discovery project time but the NMR target-observed techniques still require time, material, possible labelling and difficulties in handling a number of different hits and studies on mixtures.
Nevertheless, if the resonance assignment of the labelled target is known, the exploitation of differences in chemical shifts between free and bound target in two dimensional correlation spectra (shift mapping) will provide important structural information on the site of binding. The experiments could be also be of high value on selectively labelled target decreasing the spectra complexity and so increasing the size of the target that could be studied by NMR techniques.
Considering now that the chemical shift is highly sensitive to the environment of the atom and, as a consequence, it provides information on the binding of a small molecule to a biological target, and on which part of the molecule is interacting and where, it is clear that ligand-observed techniques could generate proof and data for the binding understanding and profile.
In addition, other experiments based on molecule relaxation values are sensitive to the motion of the compound (free vs bound state) and together these experiments will allow validation of ligand binding and/or identification of ligands also in mixtures.
The ligand observed techniques benefit of:
one-dimensional experiments;
detection of the ligand’s signals (facilitating also the mixture analysis);
smaller amount of target substrate;
structural and binding information of the ligand;
detection of week binding ligands;
limited restrictions on size and type of the target, with no isotope labelling requirement or target information details.
On the undesirable side, the techniques could generate false negatives (strong binding and slow exchange equilibria) or false positives (unspecific binding) but all these aspects could be further studied to result in a substantiated answer.
During the Hits generation phase, NMR will be used for the determination of binding affinity values toward the hit validation step to generate a lead where the NMR experiments will also remove the false positives and locate the binding site (for example within the FBDD approach). In the lead optimization phase, to improve potency of the compounds, the epitope mapping will be determined, together with the conformation of the bound ligand, while in the late lead optimization stage for the candidate selection the NMR will support the bioavailability, ADME, PK and toxicological experiments.
The combination of NMR screening methods with other techniques, such as in silico computational protocol, X-ray crystallography, and biophysical experiments will decrease the number of compounds to be studied generating filters and resulting in time and cost saving and efficiency increase. The NMR will be so used to screen and profile a library (or set) of compounds with the unique ability of providing proof for binding between the ligand and the biological target and subsequently being able to detect the binding site and determining the construct of the complex.
This short note will not include technical details of the many NMR-based methods that could be found in several papers and reviews across the last decade [1-8].
The versatility of the NMR techniques is allowing the detection of target-ligand interactions through a large variety of measurements. The insights will derive from the observation of peak intensity and/or line-width changes, saturation transfer differences (STD), chemical shifts perturbations, R2 relaxation effects, R1, sel competition data, induced transferred NOEs, interligand NOEs, diffusion coefficient measurements and changes, and, in general, from monitoring any changes in the NMR spectra resulting from the ligand-target interactions.
A big impact of the NMR techniques is also evident on the “undruggable” targets when other techniques alone fail to result in relevant data and studies on protein-protein, protein-membrane macromolecular recognition are now becoming more and more frequently successfully progressed [9].
The lead compound will need then to be optimized in the bioavailability, efficacy and toxicity profile to result in a candidate to be progressed to in vivo studies, in animals, and finally on humans.
NMR will contribute heavily in all these phases will full characterization of the compound and solid state data, stability studies, formulation studies and NMR-based metabolomics experiments. All these aspects will be covered in a future contribution.
References
1. M. Pellecchia, I. Bertini, D. Cowburn, C. Dalvit, E. Giralt, W. Jahnke, T.L. James, S.W. Homans, H. Kessler and C. Luchinat, Nat. Rev. Drug Discovery, 7, 738 (2008).
2. R. Powers, Expert Opin. Drug Discov., 4(10), 1077 (2009).
3. R. Powers, J. Med. Chem., 57(14), 5860 (2014).
4. M.J. Harner, A.O. Frank and S.W.Fesik, J. Biomol. NMR, 56(2), 65 (2013).
8. W.Jahnke and D.A. Erlanson (Editors), Fragment-based Approaches in Drug Discovery, Wiley-VCH, 2006.
9. D.M. Dias, I. Van Molle, M.G.J. Baud, C. Galdeano, C.F. G. C. Geraldes and Alessio Ciulli, ACS Med. Chem. Lett., 5 (1), 23 (2014).
In Part 2 of this series, Carla Marchioro continues to offer her insights into the contribution of NMR structural techniques to the drug discovery and development process.
Introduction
After some insights on the impact of NMR techniques on the initial drug discovery phase [1], NMR techniques applied in the progression of a compound from lead to candidate and to drug are clearly having, together with other techniques, a large impact with full structural determination, full understanding of chemical reactions, studies of molecules’ behaviour in solutions and solid states and stability monitoring with determination of by-products.
NMR Techniques in Lead Optimization and Drug Development
As soon as a compound has been identified as a lead to be progressed to the candidate phase, several NMR studies will be required to support the chemical effort, and to ensure a quality profile of the selected compound.
Synthesis of different compounds will be progressed to obtain the desired biological profile and structures will be characterized and studied to also support the computational effort, and to monitor and determine the purity for the biological tests.
Several techniques will be used, such a MS, IR, HPLC,…, to results together with the NMR data in a full profile of the studied compound.
Classical mono- and two-dimensional NMR techniques (1H and 13C) will be performed and, if required, experiments on additional nuclei will add further information to the full structural determination. As an example, in Figures 1 and 2, 1H-15N g-HNMQC, 19F-15N g-HNMQC, and 1H-29Si g-HMQC have been used to obtain the full structures characterizations [2, 3].
Figure 1: 1H-15N g-HNMQC and 19F-15N g-HNMQC experiments.
Figure 2: 1H-29Si g-HMQC experiment.
The selected compound(s) will be moved to candidate development with scale-up of the synthetic route, and characterization of the resulting material.
NMR will play an important role in reaction monitoring to ensure, with other techniques, a full understanding of the different steps of the chemical steps with identification of by-products and impurities.
Hyphenated HPLC- NMR has been used in the example in Figure 3 for the identification of co‑eluting low‑level impurities in key intermediate; Spectrum A has been acquired after injection of the mother liquors while Spectrum B has been acquired after injection of 100 µL of a solution of key-intermediate. Detailed analysis on the impurity in the mother liquors with a time-slice HPLC-NMR experiment (3 spectra at 10 sec. interval during peak elution) allowed the confirmation that the impurity was in fact a mixture of two co-eluting products. Structures determination has then been obtained after purification using standard NMR experiments [2].
Figure 3: Identification of co-eluting low-level impurities.
Critical experiments are also required in the case of UV transparent compounds, which will not be monitored by classical chromatographic techniques as reported in Figure 4 [2].
The final API will be fully characterized to profile the solid state profile, and to support the formulation studies. In addition to the solution phase NMR, solid-state NMR (ssNMR) will be used together with a variety of techniques to ensure a full understanding of compound behaviour.
An interesting application of solution NMR is reported in Figure 5 where experiments have been progressed for the determination of the critical micelle concentration (CMC) (value of the solute concentration at which half the total solute is present in the free monomeric form). NMR spectroscopy can be an alternative method to measure the CMC value, being the chemical shift concentration-dependent, particularly in the case of solute-solute intermolecular interactions, with typical downfield shifts of 1H NMR resonances on dilution.
In the example, the particularly large shielding for the aromatic protons allowed the assumption that the aromatic rings of the studied molecules that constitute the aggregate are placed in the inner hydrophobic part of the micelle, while the N-acetylpiperazine ring is somehow representing the hydrophilic external surface of the micelle itself. The forces that are involved in the aggregation are then those typical of π-staking. The CMC can then be evaluated plotting the chemical shift variation (Δδ, ppm) versus the reciprocal of the concentration (L/mol). No significant chemical shift variation was observed in the solutions at concentration ≤ 1 mg/mL, while a linear trend was observed in the concentration range 50 ÷ 3 mg/mL. Thus, assumption could be made that the intercepts of these lines on the x axis corresponded to the 1/CMC value. NMR measurements performed at 15 °C, 25 °C, 35 °C and 45 °C allowed the temperature dependence of the CMC to be determined and the thermodynamic parameters of the micellization process to be extrapolated [4].
In Parts 1 and 2, a few examples of the possibilities of the NMR techniques to support the drug discovery and development have been made with a focus on structures determination and characterization. The impact of NMR techniques on in vitro , ex vivo , in vivo and clinical phases will be covered in Part 3.
References
1. C. Marchioro, Spectroscopy Solutions , 3 (1), (2015).
2. S. Provera, C. Marchioro, unpublished data.
3. S. Provera, S. Davalli; G. H. Raza; S. Contini; C. Marchioro, Magn. Reson. Chem. , 39, 38 (2001).
4. S. Provera, S. Beato, Z. Cimarosti, L. Turco, A. Casazza, G. Caivano, C. Marchioro, J. Pharm. Biomed. Anal. , 54, 48 (2011).
Carla Marchioro
Scientific Director at R4R & Head of Discovery and Development; Chief Technology & Operations Advisor at AnCoreX
Carla Marchioro is Scientific Director, and Head of the Pharma & Analytical Division at Research for Rent, R4R, Italy where she is now after covering related positions in Aptuit and GlaxoSmithKline R&D where she has been leading multidisciplinary and cross national groups. In addition, she is also Chief Technology & Operations Adviser at AnCoreX Therapeutics.
She is an NMR expert with a chemistry background and has large experience in structural techniques. Over the years, she has developed an extended experience in a large part of the Research & Development process from target identification and progression to NDA filling.
In her group, in addition to classical structural and analytical approaches, state of the art techniques and technologies such as “omics”, computer-assisted drug design, fragments base screening, analytical and preparative SFC, quantitation by NMR, ssNMR methods for cells & tissues and more have been introduced and developed.
In addition to the R4R role, she is a member of a number of Scientific Boards, European and National Research funding bodies; and she has been part of the Scientific Advisory Board of the ProtEra company up to February 2010.She is author of a number of publications and presentations and she is a well-recognized member in the scientific community. She has been a member of the ENC Scientific Board, the chair of the 51” ENC (2010), member of the SMASH Conference Board, and the chair of the SMASH 2013 Conference.
Structural & Analytical expertise in the Drug Discovery, Chemical Development and Pharmaceutical Development Departments (up to transfer to Manufacturing groups). In addition, experience in the drug design and understanding of mechanism of action, metabolic pathways and safety related aspects.
Specialties: full understanding of mechanism of actions; full understanding of chemical and biological pathways; software and hardware design and needs; international experiences crossing countries and cultures.
Objective of the Verona group was to provide Structural & Analytical expertises to the Verona/Harlow Centre for Excellence in Drug Discovery (Neurosciences CEDD), Chemical Development and Pharmaceutical Development Departments (up to transfer to Manufacturing groups) at the Verona GSK site. In addition, to contribute to the international initiatives of Molecular Drug Discovery (MDR) and Analytical Chemistry.
Objective of the Verona group was to provide Structural & Analytical expertises to the Verona/Harlow/Zagreb Centre for Excellence in Drug Discovery (Neurosciences CEDD), Chemical Development and Pharmaceutical Development Departments (up to transfer to Manufacturing groups) at the Verona GSK site. In addition, to contribute to the international initiatives of Molecular Drug Discovery (MDR) and Analytical Chemistry.
Objective of the Zagreb group was to provide Structural & Analytical expertises to the Zagreb Centre for Excellence in Drug Discovery (MacrolidesCEDD), and Pharmaceutical Development Departments at the Zagreb GSK site. In addition, to contribute to the international initiatives of Molecular Drug Discovery (MDR) and Analytical Chemistry.
Contract Professor, Ferrara University (1999–2003);
Chiar for the SMASH2003 Conference, Verona, Italy
Chair for the 51th Experimental NMR Conference (ENC) (2010, Daytona Beach, US)
Chair for the SMASH2013 Conference, Santiago de Compostela, Spain
A novel, drug-like bis-amido piperidine derivative was identified as a potent dual OX1 and OX2 receptor antagonists, highly effective in a pre-clinical model of sleep.
Verona (Italian pronunciation: [veˈroːna] ( listen); Venetian: Verona, Veròna) is a city straddling the Adige river in Veneto, northern Italy, with approximately …





































 .
.
 .
.

































 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..





















 .
.
 .
.





 .
.









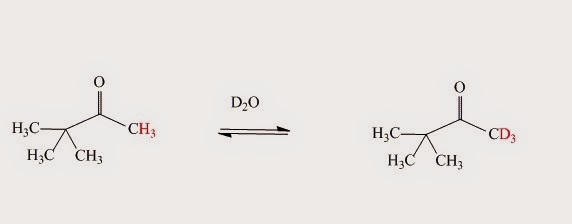




 .
.

 .
.
 ISKCON
ISKCON



.jpg)



































 .
.




















 Winners of the 2014 Elsevier Foundation Awards for Early Career Women Scientists in Developing Countries:
Winners of the 2014 Elsevier Foundation Awards for Early Career Women Scientists in Developing Countries: