
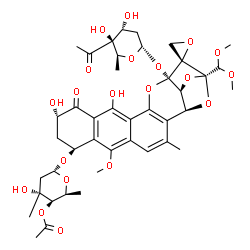
Trioxacarcin A, DC-45A
CAS No. 81552-36-5
- Molecular FormulaC42H52O20
- Average mass876.850 Da
-
17′-[(4-C-Acetyl-2,6-dideoxyhexopyranosyl)oxy]-19′-(dimethoxymethyl)-10′,13′-dihydroxy-6′-methoxy-3′-methyl-11′-oxospiro[oxirane-2,18‘-[16,20,22]trioxahexacyclo[17.2.1.02,15.05,14.07,12.017,21 ]docosa[2(15),3,5(14),6,12]pentaen]-8′-yl 4-O-acetyl-2,6-dideoxy-3-C-methylhexopyranoside
(1S,2R,3aS,4S,8S,10S,13aS)-13a-(4-C-Acetyl-2,6-dideoxy-alpha-L-xylo-hexopyranosyloxy)-2-(dimethoxymethyl)-10,12-dihydroxy-7-methoxy-5-methyl-11-oxo-4,8,9,10,11,13a-hexahydro-3aH-spiro[2,4-epoxyfuro[3,2-b]naphtho[2,3-h]-1-benzopyran-1,2′-oxiran]-8-yl 4-O-acetyl-2,6-dideoxy-3-C-methyl-alpha-L-xylo-hexopyranoside
- Kyowa Hakko Kirin INNOVATOR
Trioxacarcin B
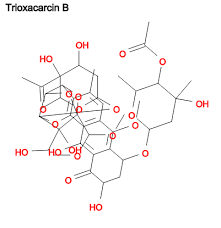
Trioxacarcin B; Antibiotic DC 45B1; DC-45-B1; Trioxacarcin A, 14,17-deepoxy-14,17-dihydroxy-; AC1MJ5N1; 81534-36-3;
| Molecular Formula: |
C42H54O21 |
| Molecular Weight: |
894.86556 g/mol |
Trioxacarcin C
(CAS NO.81781-28-4):C42H54O20
Molecular Weight: 878.8662 g/mol
Structure of Trioxacarcin C :
.png)
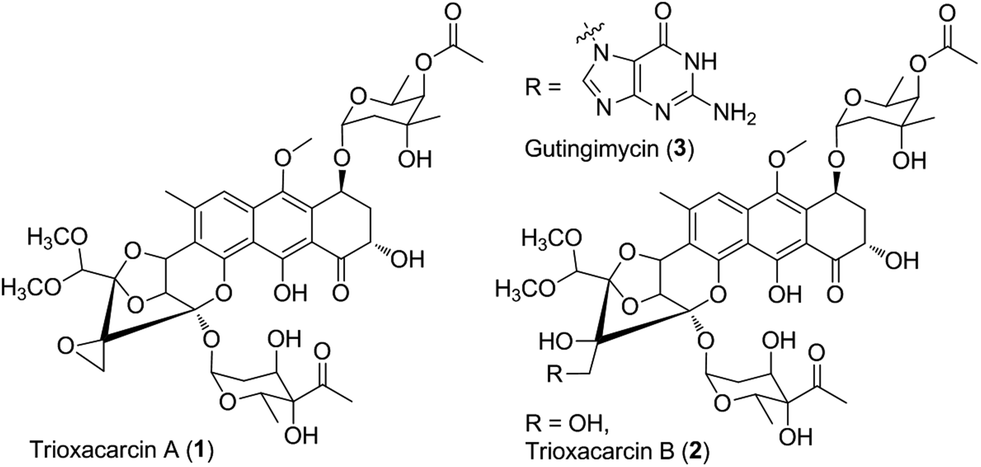
The trioxacarcins are polyoxygenated, structurally complex natural products that potently inhibit the growth of cultured human cancer cells
Natural products that bind and often covalently modify duplex DNA figure prominently in chemotherapy for human cancers. The trioxacarcins are a new class of DNA- modifying natural products with antiproliferative effects. The trioxacarcins were first described in 1981 by Tomita and coworkers (Tomita et al. , J. Antibiotics, 34( 12): 1520- 1524, 1981 ; Tamaoki et al., J. Antibiotics 34( 12): 1525- 1530, 1981 ; Fujimoto et al. , J. Antibiotics 36(9): 1216- 1221 , 1983). Trioxacarcin A, B, and C were isolated by Tomita and coworkers from the culture broth of Streptomyces bottropensis DO-45 and shown to possess anti-tumor activity in murine models as well as gram-positive antibiotic activity. Subsequent work led to the discovery of other members of this family. Trioxacarcin A is a powerful anticancer agent with subnanmolar IC70 values against lung (LXFL 529L, H-460), mammary (MCF-7), and CNS (SF-268) cancer cell lines. The trioxacarcins have also been shown to have antimicrobial activity {e.g., anti-bacterial and anti-malarial activity) (see, e.g. , Maskey et al., J. Antibiotics (2004) 57:771 -779).

trioxacarcin A
An X-ray crystal structure of trioxacarcin A bound to N-7 of a guanidylate residue in a duplex DNA oligonucleotide substrate has provided compelling evidence for a proposed pathyway of DNA modification that proceeds by duplex intercalation and alkylation (Pfoh et al, Nucleic Acids Research 36( 10):3508-3514, 2008).
All trioxacarcins appear to be derivatives of the aglycone, which is itself a bacterial isolate referred to in the patent literature as DC-45-A2. U.S. Patent 4,459,291 , issued July 10, 1984, describes the preparation of DC-45-A2 by fermentation. DC-45-A2 is the algycone of trioxacarcins A, B, and C and is prepared by the acid hydrolysis of the fermentation products trioxacarcins A and C or the direct isolation from the fermentation broth of Streptomyces bottropensis.
Based on the biological activity of the trioxacarcins, a fully synthetic route to these compounds would be useful in exploring the biological and chemical activity of known trioxacarcin compounds and intermediates thereto, as well as aid in the development of new trioxacarcin compounds with improved biological and/or chemical properties.
PAPER
Component-Based Syntheses of Trioxacarcin A, DC-45-A1, and Structural Analogs
T. Magauer, D. Smaltz, A. G. Myers, Nat. Chem. 2013, 5, 886–893. (Link)

- Nature Chemistry5,886–893(2013)
- doi:10.1038/nchem.1746
PAPER

A schematic shows a trioxacarcin C molecule, whose structure was revealed for the first time through a new process developed by the Rice lab of synthetic organic chemist K.C. Nicolaou. Trioxacarcins are found in bacteria but synthetic versions are needed to study them for their potential as medications. Trioxacarcins have anti-cancer properties. Source: Nicolaou Group/Rice University
A team led by Rice University synthetic organic chemist K.C. Nicolaou has developed a new process for the synthesis of a series of potent anti-cancer agents originally found in bacteria.
The Nicolaou lab finds ways to replicate rare, naturally occurring compounds in larger amounts so they can be studied by biologists and clinicians as potential new medications. It also seeks to fine-tune the molecular structures of these compounds through analog design and synthesis to improve their disease-fighting properties and lessen their side effects.
Such is the case with their synthesis of trioxacarcins, reported this month in the Journal of the American Chemical Society.
PAPER

PATENT
http://www.google.com/patents/EP2550285A1?cl=en
(S)-9-Hvdrox v- 10-methoxy-5-(4-methoxybenzylox v)- 1 -(methoxymethox y)-3- methyl-8-oxo-5,6.7.8-tetrahvdroanthracene-2-carbaldehvde. Potassium osmate dihydrate (29 mg, 0.079 mmol, 0.05 equiv) was added to an ice -cooled mixture of (S,£)-9-hydroxy- 10- methoxy-4-(4-methoxybenzyloxy)-8-(methoxymethoxy)-6-methyl-7-(prop- l -enyl)-3,4- dihydroanthracen-l -one (780 mg, 1.58 mmol, 1 equiv), 2,6-lutidine (369 μί, 3.17 mmol, 2.0 equiv), and sodium periodate ( 1.36 g, 6.33 mmol, 4.0 equiv) in a mixture of tetrahydrofuran (20 mL) and water ( 10 mL). After 10 min, the cooling bath was removed and the reaction flask was allowed to warm to 23 °C. After 1.5 h, the reaction mixture was partitioned between water ( 100 mL) and ethyl acetate (150 mL). The layers were separated. The organic layer was washed with aqueous sodium chloride solution (50 mL) and the washed solution was dried over sodium sulfate. The dried solution was filtered and the filtrate was concentrated. The residue was purified by flash-column chromatography (20% ethyl acetate- hexanes) to provide 498 mg of the product, (5)-9-hydroxy- 10-methoxy-5-(4- methoxybenzyloxy)- l -(methoxymethoxy)-3-methyl-8-oxo-5,6,7,8-tetrahydroanthracene-2- carbaldehyde, as an orange foam (65%). Ή NMR (500 MHz, CDC13): 15.17 (s, 1 H), 10.74 (s, 1 H), 7.66 (s, 1 H), 7.27 (d, 2H, 7 = 8.5 Hz), 6.86 (d, 2H, 7 = 8.6 Hz), 5.30-5.18 (m, 3H), 4.63 (d, 1H,7= 11.1 Hz), 4.52 (d, 1H,7 = 12.0 Hz), 3.86 (s, 3H), 3.79 (s, 3H), 3.62 (s, 3H), 3.22 (m, 1H), 2.75 (s, 3H), 2.63 (m, 1H), 2.54 (m, 1H), 2.08 (m, 1H). I3C NMR (125 MHz, CDC13): 204.9, 193.2, 163.2, 161.7, 159.2, 144.4, 141.7, 137.0, 130.1, 129.4, 120.7, 117.9, 113.8, 110.0, 102.8, 70.4, 67.2, 62.9, 58.3, 55.2, 32.3, 26.3, 22.2. FTIR, cm-1 (thin film): 2936 (m), 2907 (m), 1684 (s), 1611 (s), 1377 (s), 1246 (s). HRMS (ESI): Calcd for
(C27H2808+K)+: 519.1416; Found 519.1368. TLC (20% ethyl acetate-hexanes): R,= 0.17 (CAM).
86% yield
[00457] (S)-l,9-Dihvdroxy-10-methoxy-5-(4-methoxybenzyloxy)-3-methyl-8-oxo-5,6,7,8- tetrahydroanthracene-2-carbaldehyde. A solution of B-bromocatecholborane (418 mg, 2.10 mmol, 2.0 equiv) in dichloromethane (15 mL) was added to a solution of (S)-9-hydroxy-10- methoxy-5-(4-methoxybenzyloxy)-l-(methoxymethoxy)-3-methyl-8-oxo-5,6,7,8- tetrahydroanthracene-2-carbaldehyde (490 mg, 1.05 mmol, 1 equiv) in dichloromethane (15 mL) at -78 °C. After 50 min, the reaction mixture was diluted with saturated aqueous sodium bicarbonate solution (25 mL) and dichloromethane (100 mL). The cooling bath was removed, and the partially frozen mixture was allowed to warm to 23 °C. The biphasic mixture was diluted with 0.2 M aqueous sodium hydroxide solution (100 mL). The layers were separated. The aqueous layer was extracted with dichloromethane (100 mL). The organic layers were combined. The combined solution was washed sequentially with 0.1 M aqueous hydrochloric acid solution (100 mL), water (2 x 100 mL), then saturated aqueous sodium chloride solution (100 mL) and the washed solution was dried over sodium sulfate. The dried solution was filtered and the filtrate was concentrated to provide 380 mg of the product, (S)-\ ,9- dihydroxy-10-methoxy-5-(4-methoxybenzyloxy)-3-methyl-8-oxo-5,6,7,8- tetrahydroanthracene-2-carbaldehyde, as a yellow foam (86%). Ή NMR (500 MHz, CDCI3):
15.89 (brs, 1H), 12.81 (br s, 1H), 10.51 (s, 1H), 7.27-7.26 (m, 3H), 6.86 (d, 2H, J = 9.2 Hz), 5.14 (app s, 1H),4.62 (d, \H,J= 11.0 Hz), 4.51 (d, 1H,7= 11.0 Hz), 3.85 (s, 3H), 3.80 (s, 3H), 3.21 (m, 1H), 2.73 (s, 3H), 2.62 (m, 1H), 2.54 (m, 1H), 2.07 (m, 1H). I3C NMR (125 MHz, CDCI3): 204.4, 192.7, 166.6, 164.3, 159.3, 144.4, 142.7, 137.9, 130.4, 130.2, 129.4, 114.9, 114.2, 113.9, 113.8, 109.4, 70.4, 67.1,62.8, 55.3, 31.8, 26.5. FTIR, cm-1 (thin film): 3316 (brw), 2938 (m), 1678 (m), 1610 (s), 1514 (m), 1393 (m), 1246 (s). HRMS (ESI): Calcd for (C25H2407+Na)+ 459.1414; Found 459.1354. TLC (50% ethyl acetate-hexanes): R = 0.30 (CAM).

[00458] (5)-2,2-Di-/erf-butyl-7-methoxy-8-(4-methoxybenzyloxy)-5-methyl- 1 1 -oxo- 8,9, 10, 1 1 -tetrahydroanthra[9, 1 -de \ 1 ,3,21dioxasiline-4-carbaldehyde. Όι‘-tert- butyldichlorosilane (342 μL·, 1.62 mmol, 1.8 equiv) was added to a solution of (5)-l ,9- dihydroxy- 10-methoxy-5-(4-methoxybenzyloxy)-3-methyl-8-oxo-5,6,7,8- tetrahydroanthracene-2-carbaldehyde (380 mg, 0.90 mmol, 1 equiv), hydroxybenzotriazole (60.8 mg, 0.45 mmol, 0.50 equiv) and diisopropylethylamine (786 μί, 4.50 mmol, 5.0 equiv) in dimethylformamide (30 mL). The reaction flask was heated in an oil bath at 55 °C. After 2 h, the reaction flask was allowed to cool to 23 °C. The reaction mixture was partitioned between saturated aqueous sodium bicarbonate solution (100 mL) and ethyl acetate (150 mL). The layers were separated. The organic layer was washed sequentially with water (2 x 100 mL) then saturated aqueous sodium chloride solution (100 mL) and the washed solution was dried over sodium sulfate. The dried solution was filtered and the filtrate was concentrated. The residue was purified by flash-column chromatography (10% ethyl acetate- hexanes) to provide 285 mg of the product, (S)-2,2-di-/<?ri-butyl-7-methoxy-8-(4- methoxybenzyloxy)-5-methyl- 1 1 -oxo-8,9, 10, 1 1 -tetrahydroanthra[9, 1 -de] [ 1 ,3,2]dioxasiline-4- carbaldehyde, as a yellow foam (56%). The enantiomeric compound (/?)-2,2-di-½ri-butyl-7- methoxy-8-(4-methoxybenzyloxy)-5-methyl- l 1 -oxo-8,9, 10, 1 1 -tetrahydroanthra[9, 1 – i/e][ l ,3,2]dioxasiline-4-carbaldehyde has been prepared using the same route by utilizing R- (4-methoxybenzyloxy)cyclohex-2-enone as starting material. Ή NMR (500 MHz, CDCI3): 10.84 (s, 1 H), 7.37 (s, 1 H), 7.25 (d, 2H, J = 8.8 Hz), 6.85 (d, 2H, = 8.7 Hz), 5.20 (app s, 1 H), 4.62 (d, 1 H, 7 = 10.0 Hz), 4.51 (d, 1H, J = 1 1.4 Hz), 3.88 (s, 3H), 3.78 (s, 3H), 3.03 (m, 1H), 2.73 (s, 3H), 2.57-2.53 (m, 2H), 2.07 (m, 1H), 1.16 (s, 9H), 1.14 (s, 9H). 13C NMR (125 MHz, CDCl3): 195.6, 190.9, 160.5, 159.2, 150.4, 145.7, 140.4, 134.0, 133.9, 130.3, 129.4, 1 19.5, 1 16.6, 1 15.8, 1 15.3, 1 13.8, 70.4, 67.8, 62.9, 55.2, 34.0, 26.0, 26.0, 22.5, 21.3, 21.1. FTIR, cm“1 (thin film): 2936 (m), 2862 (m), 1682 (s), 1607 (s), 1371 (s), 1244 (s) 1057 (s). HRMS (ESI): Calcd for (C33H4o07Si+H)+ 577.2616; Found 577.2584. TLC (10% ethyl acetate-hexanes): R/ = 0.19 (CAM). Alternative Routes to (4S,6S)-6-(½rt-Butyldimethylsilyloxy)-4-(4-methoxybenzyloxy) cyclohex-2-enone.
Alternative Route 1.
[00459] (25,45,55)-2,4-Bis(ferf-butyldimethylsilyloxy)-5-hvdroxycvclohexanone. Dess- Martin periodinane (6.1 1 g, 14.4 mmol, 1.1 equiv) was added to a solution of diol (5.00 g, 13.3 mmol, 1 equiv) in tetrahydrofuran (120 mL) at 23 °C (Lim, S. M.; Hill, N.; Myers, A. G. J. Am. Chem. Soc. 2009, 131, 5763-5765). After 40 min, the reaction mixture was diluted with ether (300 mL). The diluted solution was filtered through a short plug of silica gel (-5 cm) and eluted with ether (300 mL). The filtrate was concentrated. The bulk of the product was transformed as outlined in the following paragraph, without purification. Independently,
s
an analytically pure sample of the product was obtained by flash-column chromatography (20% ethyl acetate-hexanes) and was characterized by Ή NMR, l 3C NMR, IR, and HRMS. TLC: (17% ethyl acetate-hexanes) R = 0.14 (CAM); Ή NMR (500 MHz, CDCI3) δ: 4.41 (dd, 1 H, 7 = 9.8, 5.5 Hz), 4.05 (m, l H), 4.00 (m, 1H), 2.81 (ddd, 1 H, 7 = 14.0, 3.7, 0.9 Hz), 2.52 (ddd, 1 H, 7 = 14.0, 5.3, 0.9 Hz), 2.29 (br s, 1 H), 2.18 (m, 1H), 1.98 (m, 1 H), 0.91 (s, 9H), 0.89 (s, 9H), 0.13 (s, 3H), 0.1 1 (s, 3H), 0.09 (s, 3H), 0.04 (s, 3H); l 3C NMR (125 MHz, CDCI3) δ: 207.9, 73.9, 73.3, 70.5, 43.3, 39.0, 25.7, 25.6, 18.3, 17.9, -4.7, -4.8, -4.9, -5.4; FTIR (neat), cm“‘ : 3356 (br), 2954 (m), 2930 (m), 2857 (m), 1723 (m), 1472 (m). 1253 (s), 1 162 (m), 1 105 (s), 1090 (s), 1059 (s), 908 (s), 834 (s), 776 (s), 731 (s); HRMS (ESI): Calcd for (C|8H3804Si2+H)+ 375. 2381 , found 375.2381.

[00460] (4 ,6 )-4.6-Bis(fcr/-butyldimethylsilyloxy)cvclohex-2-enone. Trifluoroacetic anhydride (6.06 mL, 43.6 mmol, 3.3 equiv) was added to an ice-cooled solution of the alcohol ( 1 equiv, see paragraph above) and triethylamine ( 18.2 mL, 131 mmol, 9.9 equiv) in dichloromethane (250 mL) at 0 °C. After 20 min, the cooling bath was removed and the reaction flask was allowed to warm to 23 °C. After 18 h, the reaction flask was cooled in an ice bath at 0 °C, and the product solution was diluted with water ( 100 mL). The cooling bath was removed and the reaction flask was allowed to warm to 23 °C. The layers were separated. The aqueous layer was extracted with dichloromethane (2 x 200 mL). The organic layers were combined. The combined solution was washed with saturated aqueous sodium chloride solution ( 100 mL) and the washed solution was dried over sodium sulfate. The dried solution was filtered and the filtrate was concentrated. The residue was purified by flash- column chromatography (6% ethyl acetate-hexanes) to provide 3.02 g of the product, (4S,65)-4,6-bis(/eri-butyldimethylsilyloxy)cyclohex-2-enone, as a colorless oil (64% over two steps). TLC: (20% ethyl acetate-hexanes) R = 0.56 (CAM); Ή NMR (500 MHz, CDC13) δ: 6.76 (dd, 1 Η, / = 10.1 , 3.6 Hz), 5.88 (d, 1 H, 7 = 10.1 Hz), 4.66 (ddd, 1 H, 7 = 5.6, 4.1 , 3.6 Hz), 4.40 (dd, 1 H, 7 = 8.1 , 3.7 Hz), 2.26 (ddd, 1 H, / = 13.3, 8.0, 4.1 Hz), 2.1 1 (ddd, 1 H, J = 13.2, 5.6, 3.8 Hz), 0.91 (s, 9H), 0.89 (s, 9H), 0.12 (s, 3H), 0. 1 1 (s, 3H), 0. 10 (s, 3H), 0.10 (s, 3H); 13C NMR ( 125 MHz, CDC13) δ: 197.5, 150.3, 127.0, 71 .0, 64.8, 41.6, 25.7, 25.7, 18.3, 18.1 , -4.7, -4.8, -4.8, -5.4; FTIR (neat), cm-1 : 3038 (w), 2955 (m), 2930 (m), 1705 (m), 1472 (m), 1254 (m), 1084 (m), 835 (s), 777 (s), 675 (s); HRMS (ESI): Calcd for (C,8H3602Si2+Na)+ 379. 2095, found 379. 2080.

[00461] (4S,6S)-6-(/er/-Butyldimethylsilyloxy)-4-hydroxycvclohex-2-enone. Tetra- j- butylammonium fluoride ( 1 .0 M solution in tetrahydrofuran, 8.00 mL, 8.00 mmol, 1 .0 equiv) was added to an ice-cooled solution of the enone (2.85 g, 8.00 mmol, 1 equiv) and acetic acid (485 ί, 8.00 mmol, 1 .0 equiv) in tetrahydrofuran (80 mL) at 0 °C. After 2 h, the cooling bath was removed and the reaction flask was allowed to warm to 23 °C. After 22 h, the reaction mixture was partitioned between water ( 100 mL) and ethyl acetate (300 mL). The layers were separated. The aqueous layer was extracted with ethyl acetate (2 x 300 mL). The organic layers were combined. The combined solution was washed sequentially with saturated aqueous sodium bicarbonate solution ( 100 mL) then saturated aqueous sodium chloride solution ( 100 mL) and the washed solution was dried over sodium sulfate. The dried solution was filtered and the filtrate was concentrated. The residue was purified by flash- column chromatography (25% ethyl acetate-hexanes) to provide 760 mg of the product, (4S,6S)-6-(ferNbutyldimethylsilyloxy)-4-hydroxycyclohex-2-enone, as a white solid (39%). TLC: (20% ethyl acetate-hexanes) R/ = 0.20 (CAM); Ή NMR (500 MHz, CDC13) δ: 6.87 (dd, 1 Η, 7 = 10.2, 3.2 Hz), 5.95 (dd, 1H, J = 10.3, 0.9 Hz), 4.73 (m, 1 H), 4.35 (dd, 1 H, 7 = 7.6, 3.7 Hz), 2.39 (m, 1 H), 2. 13 (ddd, 1 H, J = 13.3, 6.2, 3.4 Hz), 1.83 (d, 1 H, J = 6.2), 0.89 (s, 9H), 0.10 (s, 3H), 0. 10 (s, 3H); 13C NMR ( 125 MHz, CDCb) δ: 197.3, 150.0, 127.5, 70.9, 64.2, 41 .0, 25.7, 18.2, -4.8, -5.4; FTIR (neat), cm“1 : 2956 (w), 293 1 (w), 2858 (w), 1694 (m); HRMS (ESI): Calcd for (C |2H2203Si+H)+ 243.141 1 , found 243. 1412.

82″:.
[00462] (45.6S)-6-(fgrf-Butyldimethylsilyloxy)-4-(4-methoxybenzyloxy)cvclohex-2- enone. Triphenylmethyl tetrafluoroborate ( 16 mg, 50 μπιοΐ, 0.050 equiv) was added to a solution of 4-methoxybenzyl-2,2,2-trichloroacetimidate (445 μΙ_, 2.5 mmol, 2.5 equiv) and alcohol (242 mg, 1 .0 mmol, 1 equiv) in ether ( 10 mL) at 23 °C. After 4 h, the reaction mixture was partitioned between saturated aqueous sodium bicarbonate solution ( 15 mL) and ethyl acetate (50 mL). The layers were separated. The aqueous layer was extracted with ethyl acetate (50 mL). The organic layers were combined. The combined solution was washed with water (2 x 20 mL) and the washed solution was dried over sodium sulfate. The dried solution was filtered and the filtrate was concentrated. The residue was purified by flash column chromatography (5% ethyl acetate-hexanes initially, grading to 10% ethyl acetate-hexanes) to provide 297 mg of the product, (4S,6S)-6-(im-butyldimethylsilyloxy)-4-(4- methoxybenzyloxy)cyclohex-2-enone, as a colorless oil (82%).
Alternative Route 2.

[00463] (5)-?erf-Butyl(4-(4-methoxybenzyloxy)cvclohexa- 1.5-dienyloxy)dimethylsilane. rerr-Butyldimethylsilyl trifluoromethanesulfonate (202 iL, 0.94 mmol, 2.0 equiv) was added to an ice-cooled solution of triethylamine (262 μί, 1.88 mmol, 4.0 equiv) and enone ( 109 mg, 0.47 mmol, 1 equiv) in dichloromethane (5.0 mL). After 30 min, the reaction mixture was partitioned between saturated aqueous sodium bicarbonate solution ( 10 mL), water (30 mL), and dichloromethane (40 mL). The layers were separated. The organic layer was washed sequentially with saturated aqueous ammonium chloride solution (20 mL) then saturated aqueous sodium chloride solution (20 mL) and the washed solution was dried over sodium sulfate. The dried solution was filtered and the filtrate was concentrated. The residue was purified by flash-column chromatography with triethylamine-treated silica gel (5% ethyl acetate-hexanes), to provide 130 mg of the product, (5)-ierr-butyl(4-(4- methoxybenzyloxy)cyclohexa- l ,5-dienyloxy)dimethylsilane, as a colorless oil (80%). Ή
NMR (500 MHz, CDC13): 7.27 (d, 2H, J = 8.7 Hz), 6.88 (d, 2H, J = 8.6 Hz), 5.96 (dd, 1 H, J = 9.9, 3.5 Hz), 5.87 (d, 1 H, 7 = 9.6 Hz), 4.94 (m, l H), 4.46 (s, 2H), 4.14 (m, 1 H), 3.81 (s, 3H), 2.49 (m, 2H), 0.93 (s, 9H), 0. 16 (s, 3H), 0.15 (s, 3H). , 3C NMR ( 125 MHz, CDC13): 159.1 , 147.5, 130.9, 129.2, 128.6, 128.1 , 1 13.8, 101.4, 70.2, 69.0, 55.3, 28.5, 25.7, 18.0, ^1.5, -4.5. FTIR, cm-1 (thin film): 2957 (m), 2931 (m), 2859 (m), 1655 (w), 1613 (w), 1515 (s), 1248 (s), 1229 (s), 1037 (m), 910 (s). HRMS (ESI): Calcd for (C2oH3o03Si+H)+ 347.2037; Found 347.1912. TLC (20% ethyl acetate-hexanes): R = 0.74 (CAM).
OP B OPMB DM 00 ,,Α,,
c Ύ’“ -ietone ii ·η- ) ‘”OH
OTBS 82 Q
[00464] (4S,6S)-6-Hvdroxy-4-(4-methoxybenzyloxy)cvclohex-2-enone. A solution of dimethyldioxirane (0.06 M solution in acetone, 2.89 mL, 0.17 mmol, 1.2 equiv) was added to an ice-cooled solution of (S)-ieri-butyl(4-(4-methoxybenzyloxy)cyclohexa- l ,5- dienyloxy)dimethylsilane (50 mg, 0.14 mmol, 1 equiv). After 10 min, the reaction mixture was partitioned between dichloromethane ( 15 mL) and 0.5 M aqueous hydrochloric acid ( 10 mL). The layers were separated. The organic layer was washed sequentially with saturated aqueous sodium bicarbonate solution ( 10 mL) then water ( 10 mL) and the washed solution was dried over sodium sulfate. The dried solution was filtered and the filtrate was concentrated. The residue was purified by flash-column chromatography to provide 30 mg of the product, (4S,6S)-6-hydroxy-4-(4-methoxybenzyloxy)cyclohex-2-enone, as a colorless oil (82%). Ή NMR (500 MHz, CDC13): 7.28 (d, 2H, J = 8.2 Hz), 6.89 (m, 3H), 6.09 (d, 1 H, J = 10.1 Hz), 4.64 (m, 2H), 4.53 (d, 1 H, 7 = 1 1 .4 Hz), 4.24 (m, 1 H), 3.81 (s, 3H), 3.39 (d, 1 H, 7 = 1.4 Hz), 2.67 (m, 1 H), 1 .95 (ddd, 1 H, 7 = 12.8, 12.8, 3.6 Hz). I 3C NMR ( 125 MHz, CDC13): 200.4, 159.5, 146.6, 129.7, 129.4, 127.8, 1 14.0, 71.6, 69.8, 68.9, 55.3, 35.1 . FTIR, cm-1 (thin film): 3474 (br), 2934 (m), 2864 (m), 1692 (s), 1613 (m), 1512 (s), 1246 (s), 1059 (s), 1032 (s). HRMS (ESI): Calcd for (C,4Hl6O4+Na)+ 271.0941 ; Found 271.0834. TLC (50% ethyl acetate-hexanes): R/ = 0.57 (CAM).

[00465] (45,65)-6-(½rt-Butyldimethylsilyloxy)-4-(4-methoxybenzyloxy)cvclohex-2- enone. rerr-Butyldimethychlorosilane (26 mg, 0.18 mmol, 1.5 equiv) was added to an ice- cooled solution of (45,65)-6-hydroxy-4-(4-methoxybenzyloxy)cyclohex-2-enone (29 mg, 0.12 mmol, 1 equiv) and imidazole (24 mg, 0.35 mmol, 3 equiv) in dimethylformamide (0.5 mL). After 45 min, the reaction mixture was partitioned between water (15 mL), saturated aqueous sodium chloride solution (15 mL), and ethyl acetate (20 mL). The layers were separated. The organic layer was washed with water (2 x 20 mL) and the washed solution was dried over sodium sulfate. The dried solution was filtered and the filtrate was concentrated. The residue was purified by flash-column chromatography to provide 29 mg of the product, (4S,6S)-6-(rm-butyldimethylsilyloxy)-4-(4-methoxybenzyloxy)cyclohex-2- enone, as a colorless oil (87%).
Glycosylation experiments
[00466] Glycosylation experiments demonstrate that the chemical process developed allows for the preparation of synthetic, glycosylated trioxacarcins. Specifically, the C4 or CI 3 hydroxyl group may be selectively glycosylated with a glycosyl donor (for example, a glycosyl acetate) and an activating agent (for example, TMSOTf), which enables preparation of a wide array of trioxacarcin analogues.
Selective Glycosylation of the C4 Hydroxyl Group
[00467] 2,3-Dichloro-5,6-dicyanobenzoquinone ( 19.9 mg, 88 μιτιοΐ, 1.1 equiv) was added to a vigorously stirring, biphasic solution of differentially protected trioxacarcin precursor (60 mg, 80 μιτιοΐ, 1 equiv) in dichloromethane ( 1.1 mL) and pH 7 phosphate buffer (220 μί) at 23 °C. The reaction flask was covered with aluminum foil to exclude light. Over the course of 3 h, the reaction mixture was observed to change from myrtle green to lemon yellow. The product solution was partitioned between water (5 mL) and dichloromethane (50 mL). The layers were separated. The organic layer was dried over sodium sulfate. The dried solution was filtered and the filtrate was concentrated. The residue was purified by preparatory HPLC (Agilent Prep-C 18 column, 10 μιτι, 30 x 150 mm, UV detection at 270 nm, gradient elution with 40→90% acetonitrile in water, flow rate: 15 mL/min) to provide 33 mg of the product as a yellow-green powder (65%).
[00468] Trimethylsilyl triflate ( 10% in dichloromethane, 28.3 μί, 16 μπιοΐ, 0.3 equiv) was added to a suspension of deprotected trioxacarcin precursor (33 mg, 52 μπιοΐ, 1 equiv), 1 -0- acetyltrioxacarcinose A ( 14.1 mg, 57 μιτιοΐ, 1.1 equiv), and powdered 4- A molecular sieves (-50 mg) in dichloromethane (1 .0 mL) at -78 °C. After 5 min, the mixture was diluted with dichloromethane containing 10% triethylamine and 10% methanol (3 mL). The reaction flask was allowed to warm to 23 °C. The mixture was filtered and partitioned between
dichloromethane (40 mL) and saturated aqueous sodium chloride solution (5 mL). The layers were separated. The organic layer was dried over sodium sulfate. The dried solution was filtered and the filtrate was concentrated. The residue was purified by preparatory HPLC (Agilent Prep-C 18 column, 10 μπι, 30 x 150 mm, UV detection at 270 nm, gradient elution with 40→90% acetonitrile in water, flow rate: 15 mL/min) to provide 20 mg of the product as a yellow-green powder (47%). TLC: (5% methanol-dichloromethane) R = 0.40 (CAM); Ή NMR (500 MHz, CDC13) δ: 7.47 (s, 1H), 5.38 (d, 1H, J = 3.6 Hz), 5.35 (app s, 1 H), 5.26 ppm (d, 1 H, 7 = 4.0 Hz), 4.84 (d, 1 H, J = 4.0 Hz), 4.78 (dd, 1 H, 7 = 12.3, 5.2 Hz), 4.75 (s, 1H), 4.71 (s, 1 H), 4.52 (q, 1H, J = 6.6 Hz), 3.86 (s, 1 H), 3.83 (s, 3H), 3.62 (s, 3H), 3.47 (s, 3H), 3.15 (d, l H, y = 5.3 Hz), 3.05 (d, 1 H, 7 = 5.3 Hz), 2.60 (s, 3H), 2.58 (m, 1H), 2.35 (m, 1 H), 2.14 (s, 3H), 1.96 (dd, 1 H, 7 = 14.6, 4.1 Hz), 1.62 (d, 1 H, 7 = 14.6 Hz), 1.26 (s, 1 H), 1.23 (d, 3H, J = 6.6 Hz), 1.08 (s, 3H), 0.95 (s, 9H), 0.24 (s, 3H), 0.16 (s, 3H); ‘3C NMR ( 125 MHz, CDC13) 6: 202.8, 170.5, 163.2, 151.8, 144.4, 142.4, 135.2, 126.6, 1 16.8, 1 15.2, 1 15.1 , 108.3, 104.0, 100.3, 98.6, 98.3, 74.6, 73.4, 69.8, 69.5, 69.5, 68.9, 69.5, 69.5, 68.9, 68.4, 62.9, 62.7, 57.2, 56.8, 50.7, 38.8, 36.8, 26.0, 25.9, 21.1 , 20.6, 18.6, 17.0, -4.2, -5.3; FTIR (neat), cm“‘ : 2953 (w), 2934 (w), 2857 (w), 1749 (w), 1622 (m), 1570 (w), 1447 (w), 1391 (m), 1321 (w), 1294 (w), 1229 (m), 1 159 (m), 1 121 (s), 1084 (s), 1071 (m), 1020 (m), 995 (s), 943 (s), 868 (m), 837 (m), 779 (m); HRMS (ESI): Calcd for (C4oH540i6Si+Na)+ 841.3073, found
841.3064.
Glycosylation of a Cycloaddition Coupling Partner
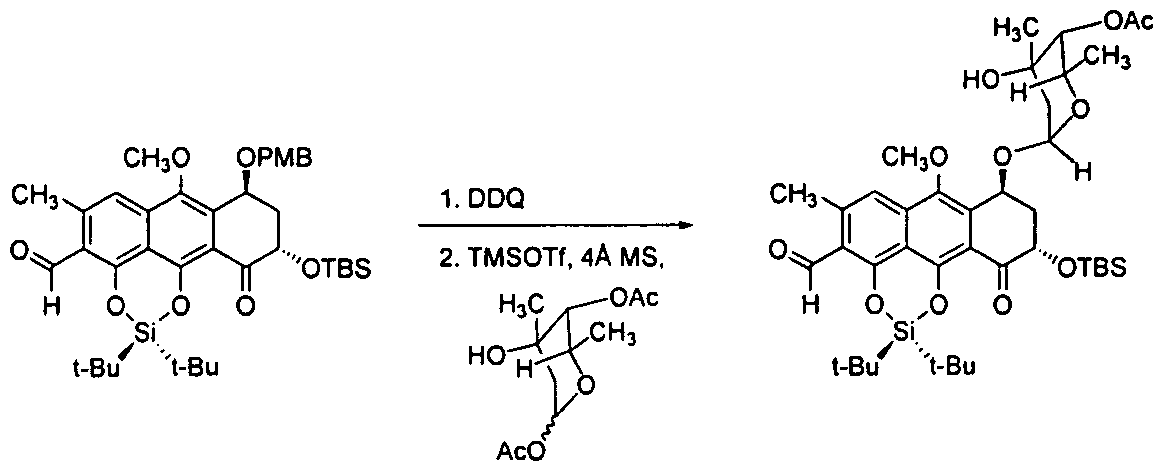
[00469] 2,3-Dichloro-5,6-dicyanobenzoquinone ( 14.3 mg, 63 μπιοΐ, 1.2 equiv) was added to a vigorously stirring, biphasic solution of differentially protected aldehyde (37 mg, 52 μιτιοΐ, 1 equiv) in dichloromethane (870 μί) and water (175 μί) at 23 °C. The reaction flask was covered with aluminum foil to exclude light. Over the course of 2 h, the reaction mixture was observed to change from myrtle green to lemon yellow. The product solution was partitioned between water (5 mL) and dichloromethane (40 mL). The layers were separated. The organic layer was dried over sodium sulfate. The dried solution was filtered and the filtrate was concentrated. The residue was purified by flash-column chromatography (5% ethyl acetate-hexanes initially, grading to 10% ethyl acetate-hexanes) to provide 28 mg of the product as a yellow powder (91 %). TLC: (20% ethyl acetate-hexanes) R/ = 0.37 (CAM); Ή NMR (500 MHz, CDC13) δ: 10.83 (s, 1H), 7.30 (s, 1 H), 5.45 (m, 1H), 4.68 (dd, 1H, / = 10.3, 4.2 Hz), 3.97 (s, 3H), 3.31 (brs, 1H), 2.72 (s, 3H), 2.51-2.45 (m, 1H), 2.41-2.37 (m, 1H), 1.15 (s, 9H), 1 , 13 (s, 9H), 0.88 (s, 9H), 0.15 (s, 3H), 0.1 1 (s, 3H); l 3C NMR (125 MHz, CDCI3) δ: 194.6, 191 , 160.5, 150.2, 146, 140.8, 135.8, 134, 1 19.6, 1 16.2, 1 15.4, 1 14.7, 72.7, 63.7, 62.4, 38.8, 29.9, 62.4, 38.8, 63.7, 62.4, 38.8, 63.7, 62.4, 38.8, 29.9, 26.2, 26.1 , 26, 22.7, 21.4; FTIR (neat), cm“1 : 3470 (br, w), 2934 (w), 2888 (w), 1684 (s), 1607 (s), 1560 (w), 1472 (m), 1445 (w), 1392 (m), 1373 (s), 1242 (s), 1 153 (s), 1 1 19 (w), 1074 (m), 1044 (s), 1013 (s), 982 (w), 934 (m), 907 (w), 870 (m), 827 (s), 795 (s), 779 (s), 733 (s), 664 (s); HRMS (ESI): Calcd for (C3iH4607Si2+H)+ 587.2855, found 587.2867.
[00470] Trimethylsilyl triflate (10% in dichloromethane, 25.9 μί, 14 μπιοΐ, 0.3 equiv) was added to a suspension of deprotected aldehyde (28 mg, 48 μηιοΐ, 1 equiv), 1-0- acetyltrioxacarcinose A (12.9 mg, 52 μπιοΐ, 1.1 equiv), and powdered 4-A molecular sieves (-50 mg) in dichloromethane ( 1.0 mL) at -78 °C. After 5 min, the mixture was diluted with dichloromethane containing 10% triethylamine and 10% methanol (3 mL). The reaction flask was allowed to warm to 23 °C. The mixture was filtered and partitioned between dichloromethane (40 mL) and saturated aqueous sodium chloride solution (5 mL). The layers were separated. The organic layer was dried over sodium sulfate. The dried solution was filtered and the filtrate was concentrated. The residue was purified by preparatory HPLC (Agilent Prep-C 18 column, 10 μπι, 30 x 150 mm, UV detection at 270 nm, gradient elution with 80→98% acetonitrile in water, flow rate: 15 mL/min) to provide 15 mg of the product as a yellow powder (41 %). TLC: (20% ethyl acetate-hexanes) R/ = 0.29 (CAM); Ή NMR (500 MHz, CDC13) δ: 10.83 (s, 1 H), 7.32 (s, 1 H), 5.43 (d, 1 H, J = 3.9 Hz), 5.32 (m, 1H), 4.74 (s, 1 H), 4.67 (dd, 1 H, J = 12.3, 5.0 Hz), 4.54 (q, 1H, J = 6.6 Hz), 3.91 (s, 1H), 3.88 (s, 3H), 2.72 (s, 3H), 2.59 (ddd, 1 H, J = 13.8, 5.0, 3.2 Hz), 2.34 (m, 1H), 2.14 (s, 3H), 1.97 (dd, 1H, J = 14.2, 4.2 Hz), 1.71 (d, 1 Η, / = 14.6 Hz), 1.22 (d, 3H, J = 6.3 Hz), 1.15 (s, 9H), 1.15 (s, 9H), 1.08 (s, 3H), 0.93 (s, 9H), 0.23 (s, 3H), 0.13 (s, 3H); 13C NMR (125 MHz, CDC13) δ: 193.9, 191.0, 170.5, 146.4, 140.9, 134.0, 132.4, 1 19.8, 1 16.8, 1 15.8, 1 15.0, 1 10.8, 99.6, 74.6, 71.5, 70.4, 68.9, 62.9, 62.7, 39.1 , 36.9, 26.2, 26.1 , 26.1 , 25.9, 24.1 , 22.7, 21.5, 21.3, 21.1 , 18.7, 16.9, -4.1 , -5.3; FTIR (neat), cm-1 : 3524 (br, w), 2934 (m), 2861 (m), 1749 (m), 1686 (s), 1607 (s), 1560 (m), 1474 (m), 1447 (m), 1424 (w), 1375 (s), 1233 (s), 1 159 (s), 1 1 17 (m), 1080 (m), 1049 (s), 1015 (s), 997 (s), 937 (m), 883 (m), 872 (m), 827 (s), 797 (m), 781 (m), 737 (w), 677 (w), 667 (m); HRMS (ESI): Calcd for (C40H60O, ,Si2+H)+773.3747, found 773.3741.
General Glycosylation Procedure of the C13 Hydroxyl Group

[00471] Crushed 4-A molecular sieves (-570 mg / 1 mmol sugar donor) was added to a stirring solution of the sugar acceptor (1 equiv.) and the sugar donor (30.0 equiv.) in dichloromethane ( 1.6 mL / 1 mmol sugar donor) and diethylether (0.228 mL / 1 mmol sugar donor) at 23 °C. The bright yellow mixture was stirred for 90 min at 23 °C and finally cooled to -78 °C. TMSOTf (10.0 equiv.) was added over the course of 10 min at -78 °C. After 4 h, a second portion of TMSOTf (5.0 equiv.) was added at -78 °C and stirring was continued for 1 h. The last portion of TMSOTf (5 equiv.) was added. After 1 h, triethylamine (20 equiv.) was added and the reaction the product mixture was filtered through a short column of silica gel deactivated with triethylamine (30% ethyl acetate-hexanes initially, grading to 50% ethyl acetate-hexanes). H NMR analysis of the residue showed minor sugar donor remainings and that the sugar acceptor had been glycosylated. The residue was purified by preparatory HPLC (Agilent Prep-C 18 column, 10 μπι, 30 x 150 mm, UV detection at 270 nm, gradient elution with 40→100% acetonitrile in water, flow rate: 15 mL/min) to provide the glycosylation product as a bright yellow oil
Three Specific Compounds Prepared by the General Glycosylation Procedure for the CI 3 Hydroxyl Group:
[00472] 10% yield; TLC: (50% ethyl acetate-hexane) R = 0.58 (UV, CAM); Ή NMR (600 MHz, CDC13) δ: 7.43 (s, 1 H), 5.84 (t, J = 3.6 Hz, 1 H), 5.29 (d, J = 4.2 Hz, 1 H), 5.19 (d, J = 4.2 Hz, 1 H), 5.01 (q, J = 6.6 Hz, 1 H), 4.75 (t, J = 3.6 Hz, 1 H), 4.73 (s, 1 H), 3.88 (s, OH), 3.77 (s, 3H), 3.63 (s, 3H), 3.47 (s, 3H), 3.03 (app q, J = 5.4 Hz, 2H), 2.84 (d, J = 6.0 Hz, 1 H), 2.77 (d, J = 6.0 Hz, 1 H), 2.72 (t, J = 6.6 Hz, 2H), 2.58 (s, 3H), 2.36 (s, 3H), 2.33 (t, J = 3.0 Hz, 2H), 2.23 (s, 3H), 2.1 1 -2.06 (m, 2H), 1.08 (d, J = 6.0 Hz, 3H). ‘

[00473] 81 % yield, TLC: (50% ethyl acetate-hexane) R = 0.30 (UV, CAM); Ή NMR (600 MHz, CDCI3) δ: 7.46 (s, 1 H), 7.28 (d, J = 9 Hz, 2H), 6.87 (d, J = 8.4 Hz, 2 H), 5.83 (dd, J = 3.6, 1.8 Hz, 1 H), 5.30 (d, J = 4.2 Hz, 1 H), 5.19 (d, J = 4.2 Hz, 1 H), 5.19 (m, 1 H), 5.00 (q, J = 6.0 Hz, 1 H), 4.96 (dd, J = 12.0, 4.8 Hz, 1 H), 4.75 (t, J = 3.6 Hz, 1 H), 4.74 (s, l H), 4.70 (d, y = 10.8 Hz, 1 H), 4.59 (d, J = 10.8 Hz, 1 H), 3.86 (s, OH), 3.83 (s, 3H), 3.80 (s, 3H), 3.63 (s, 3H), 3.47 (s, 3H), 2.81 (d, J = 6.0 Hz, 1 H), 2.73-2.68 (m, 1 H), 2.70 (d, J = 6.0 Hz, 1 H), 2.59 (s, 3H), 2.35 (s, 3H), 2.33-2.28 (m, 2H), 2.22 (s, 3H), 2.19- 2.1 3 (m, 1 H), 1 .08 (d, J = 6.0 Hz, 3H), 0.97 (s, 9H), 0.25 (s, 3H), 0.17 (s, 3H); HRMS (ESI): Calcd for (C49H62018Si+H)+ 967.3778, found 967.3795; HRMS (ESI): Calcd for (C ¾20,8Si+Na)+ 989.3598, found 989.3585.
[00474] Compound Detected by ESI Mass Spectrometry: Calculated Mass for
[C52H7| N302i Si-Hrl = 1 100.4277, Measured Mass = 1 100.4253.
PATENT
US 4511560
https://www.google.com/patents/US4511560
The physico-chemical characteristics of DC-45-A and DC-4-5-B2 according to this invention are as follows:
(1) DC-45-A
(1) Elemental analysis: H:5.74%, C:55.11%
(2) Molecular weight: 877
(3) Molecular formula: C42 H52 O20
(4) Melting point: 180° C.±3° C. (decomposed)
(5) Ultraviolet absorption spectrum: As shown in FIG. 1 (in 50% methanol)
(6) Infrared absorption spectrum: As shown in FIG. 2 (KBr tablet method)
(7) Specific rotation: [α]D 25 =-15.3° (c=1.0, ethanol)
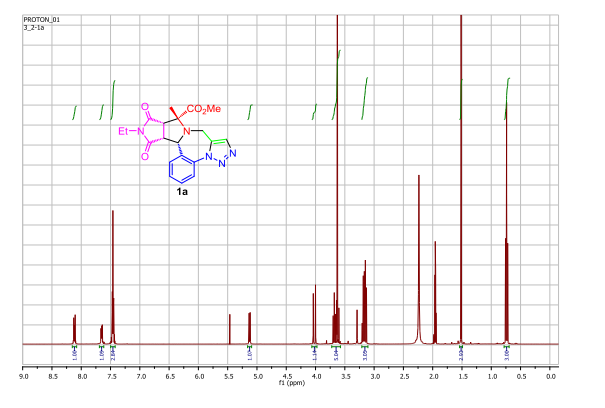
(8) PMR spectrum (in CDC]3 ; ppm): 1.07 (3H,s); 1.10 (3H, d, J=6.8); 1.24 (3H,d, J=6.5); many peaks between 1.40-2.30; 2.14 (3H,s); 2.49 (3H,s); 2.63 (3H,s); many peaks between 2.30-2.80; 2.91 (1H,d, J=5.6); 3.00 (1H,d, J=5.6); 3.49 (3H,s); 3.63 (3H,s); 3.85 (3H, s); many peaks between 3.60-4.00; 4.18 (1H,s); 4.55 (1H,q, J=6.8); many peaks between 4.70-4.90; 5.03 (1H, q, J=6.5); 5.25 (1H,d, J=4.0); 5.39 (1H, d, J=4.0); 5.87 (1H, m); 7.52 (1H,s); 14.1 (1H,s)
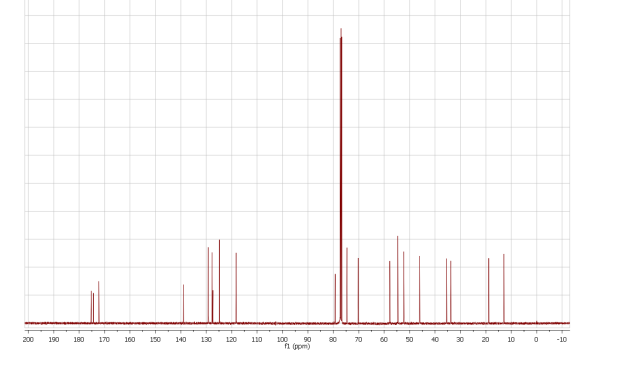
(9) CMR spectrum (in CDCl3 ; ppm): 210.9; 203.8; 170.3; 162.1; 152.5; 145.2; 142.3; 135.3; 126.7; 117.0; 114.2; 108.3; 105.3; 99.7; 97.2; 93.7; 85.1; 79.0; 74.6; 71.1; 69.6; 69.3; 68.8; 67.9; 66.3; 64.0; 62.8; 57.3; 55.9; 36.5; 32.2; 28.0; 25.7; 20.9; 20.2; 17.0; 14.7
(10) Solubility: Soluble in methanol, ethanol, water and chloroform; slightly soluble in acetone and ethyl acetate, and insoluble in ether and n-hexane
(2) DC-45-B2
(1) Elemental analysis: H: 6.03%, C: 54.34%
(2) Molecular weight: 879
(3) Molecular formula: C42 H54 O20
(4) Melting point: 181°-182° C. (decomposed)
(5) Ultraviolet absorption spectrum: As shown in FIG. 5 (in 95% ethanol)
(6) Infrared absorption spectrum: As shown in FIG. 6 (KBr tablet method)
(7) Specific rotation: [α]D 25 =-10° (c=0.2, ethanol)
(8) PMR spectrum (in CDCl3 ; ppm): 1.07 (3H,s); many peaks between 1.07-1.5; many peaks between 1.50-2.80; 2.14 (3H,s); 2.61 (3H, broad s); 2.86 (1H, d, J=5.7); 2.96 (1H, d, J=5.7); 3.46 (3H,s); 3.63 (3H, s); 3.84 (3H, s); many peaks between 3.65-4.20; many peaks between 4.40-5.00; many peaks between 5.10-5.50; 5.80 (1H, broad s); 7.49 (1H, d, J=1.0); 14.1 (1H, s)
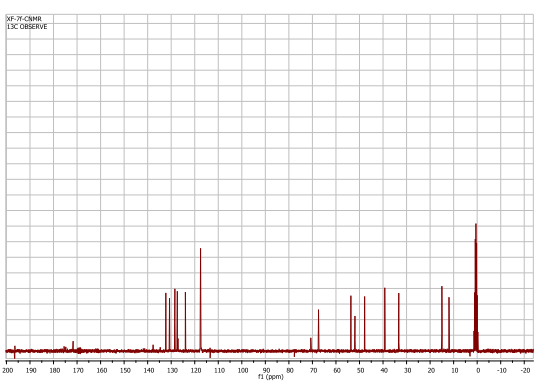
(9) CMR spectrum (in CDCl3 ; ppm): 202.8; 170.2; 163.1; 151.8; 144.8; 142.9; 135.4; 126.5; 116.8; 114.9; 107.3; 104.6; 101.5; 99.6; 98.0; 94.4; 74.4; 72.5; 71.4; 70.4; 69.1; 68.8; 68.3; 67.9; 67.5; 66.4; 62.9; 62.7; 56.8; 56.5; 48.0; 36.7; 32.3; 25.7; 20.8; 20.3; 18.2; 16.9; 15.5
(10) Solubility: Soluble in methanol, ethanol, acetone, ethyl acetate and chloroform; slightly soluble in benzene, ether and water; and insoluble in n-hexane.
//////
CC1C(C(CC(O1)OC2CC(C(=O)C3=C(C4=C5C(=C(C=C4C(=C23)OC)C)C6C7C(O5)(C8(CO8)C(O6)(O7)C(OC)OC)OC9CC(C(C(O9)C)(C(=O)C)O)O)O)O)(C)O)OC(=O)C
















































{kind=link}