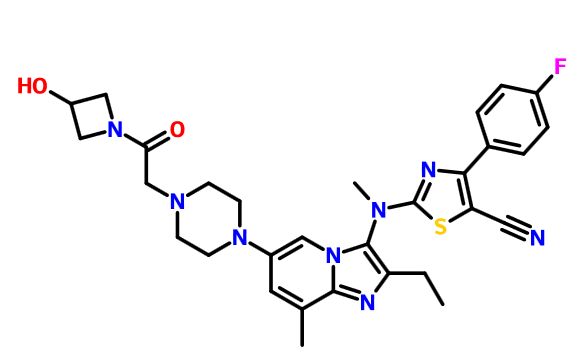

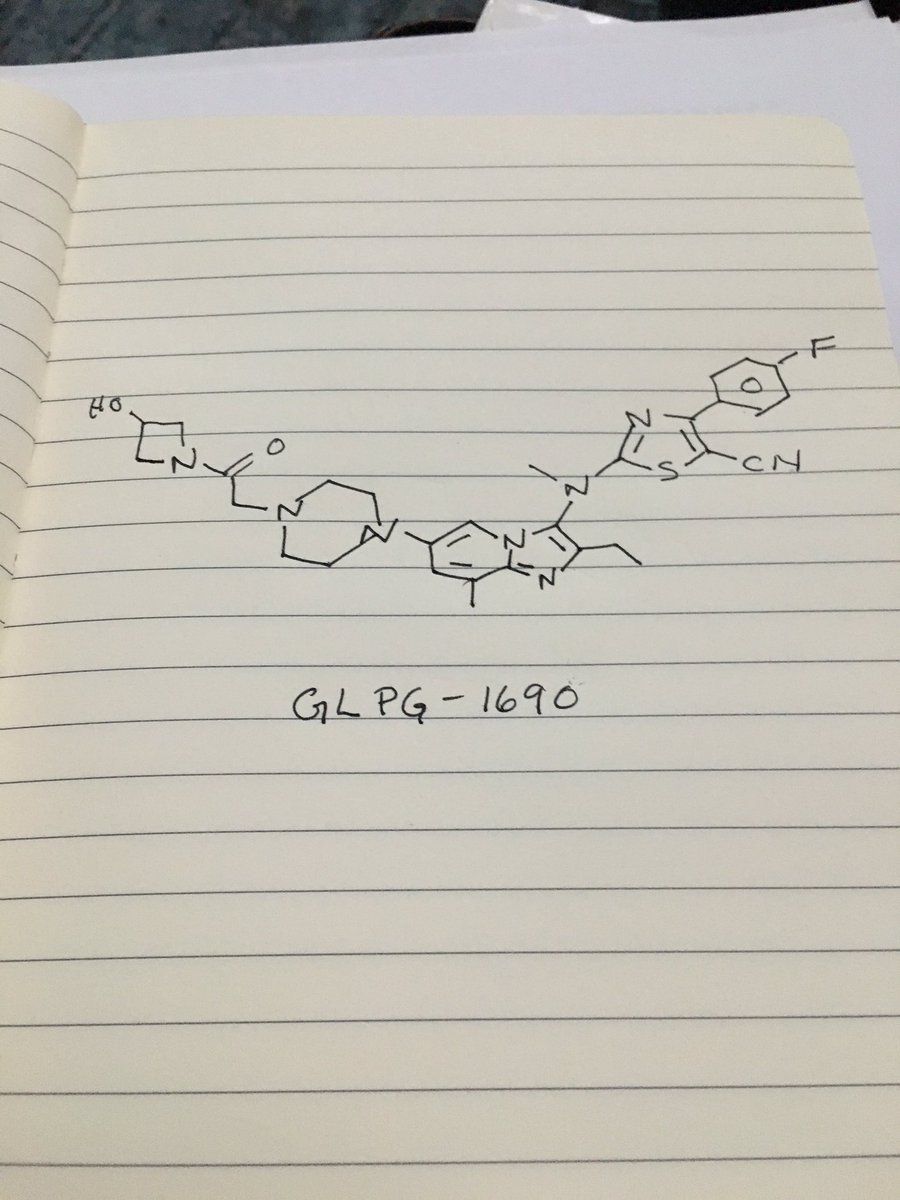
Picture credit….Bethany Halford
GLPG 1690
2-[[2-ethyl-6-[4-[2-(3-hydroxyazetidin-1-yl)-2-oxoethyl]piperazin-1-yl]-8-methylimidazo[1,2-a]pyridin-3-yl]-methylamino]-4-(4-fluorophenyl)-1,3-thiazole-5-carbonitrile
5-Thiazolecarbonitrile, 2-[[2-ethyl-6-[4-[2-(3-hydroxy-1-azetidinyl)-2-oxoethyl]-1-piperazinyl]-8-methylimidazo[1,2-a]pyridin-3-yl]methylamino]-4-(4-fluorophenyl)-
CAS 1628260-79-6
![]()
$GLPG compound for treating idiopathic pulmonary fibrosis
| Molecular Formula: | C30H33FN8O2S |
|---|---|
| Molecular Weight: | 588.698823 g/mol |
| Galapagos Nv |
http://files.glpg.com/docs/website_1/Poster_ERS_2015_final.pdf
http://www.glpg.com/docs/view/56b360a81f6b2-en
Phase I Idiopathic pulmonary fibrosis
| Description | Selective autotaxin (ENPP2; ATX) inhibitor |
| Molecular Target | Autotaxin (ENPP2) (ATX) |
- Originator Galapagos NV
- Class Anti-inflammatories; Small molecules
- Mechanism of Action ENPP2 protein inhibitors
- 23 Sep 2015 Pharmacodynamics data from a preclinical trial in Indiopathic pulmonary fibrosis released by Galapagos
- 22 Sep 2015 Pharmacokinetics data from a phase I trial in healthy volunteers released by Galapagos
- 22 Sep 2015 Updated adverse events data from a phase I trial in healthy volunteers released by Galapagos
GLPG1690
GLPG1690 is a selective autotaxin inhibitor discovered by Galapagos, with potential application in idiopathic pulmonary disease (IPF). In a Phase 1 study in healthy human volunteers, GLPG1690 demonstrated favorable safety and tolerability, as well as a strong pharmacodynamic signal implying target engagement. Galapagos is currently preparing a Phase 2 study in IPF, to be filed for approval before the end of 2015. GLPG1690 is fully proprietary to Galapagos.
| Source: Galapagos NV
- Fully owned and proprietary clinical asset for pulmonary fibrosis
- GLPG1690 acts on autotaxin target
- Novel mode of action, originating from Galapagos target discovery engine
- Filing for Phase 2 clinical trial in 2015
MECHELEN, Belgium, March 16, 2015 (GLOBE NEWSWIRE) — Galapagos NV (Euronext: GLPG) announced that Janssen Pharmaceutica NV and Galapagos have mutually agreed to terminate the inflammation alliance and option agreements between the companies. Galapagos views the molecules emerging from the alliance as strong additions to its growing proprietary pipeline. Among others, all rights to candidate drug GLPG1690, a selective autotaxin inhibitor, return to Galapagos. Galapagos has successfully completed a First-in-Human Phase 1 trial for GLPG1690 and is preparing a Phase 2 clinical trial in idiopathic pulmonary fibrosis (IPF).
“We are pleased to regain the rights to GLPG1690 to pursue the most suitable clinical application of autotaxin inhibition. There is a large unmet medical need in IPF, and our pre-clinical data with GLPG1690 supports its potential as a competitive and novel approach in this disease area,” said Dr Piet Wigerinck, Chief Scientific Officer of Galapagos. “The alliance with Janssen has been underway since October 2007 and has generated three clinical molecules, two of which are now proprietary Phase 2 assets of Galapagos: GLPG1205 and GLPG1690. This program is a valuable component of our development portfolio, and regaining the rights is a next step in our transformation into a mature biotech company with a proprietary product pipeline.”
Galapagos identified autotaxin as playing a key role in inflammation, using an inflammation assay in its unique target discovery platform. Pharmacology and translational studies published by other parties in the literature since then suggest autotaxin may play a key role in metabolic disease, arthritic pain, oncology, and lung disease.
GLPG1690 is a potent and selective inhibitor of autotaxin. In a Phase 1 study in healthy human volunteers, GLPG1690 demonstrated favorable safety and tolerability, as well as a strong pharmacodynamic signal implying target engagement. Galapagos is currently preparing a Phase 2 study in IPF, to be filed for approval before the end of 2015.
About IPF
Idiopathic pulmonary fibrosis (IPF) is a chronic and ultimately fatal disease characterized by a progressive decline in lung function. Pulmonary fibrosis involves scarring of lung tissue and is the cause of shortness of breath. Fibrosis is usually associated with a poor prognosis. The term “idiopathic” is used because the cause of pulmonary fibrosis is still unknown. Estimated incidence of IPF is up to 16.3 per 100,000 persons in the US and 7.4 per 100,000 persons in Europe, with approximately 30,000-35,000 new patients diagnosed with IPF worldwide each year. The goals of treatment in IPF are essentially to reduce the symptoms, slow down disease progression, reduce acute exacerbations, and prolong survival. Approved treatments thus far have improved the overall survival of IPF patients, but unwanted side effects with these treatments are common, presenting an unmet need for effective treatments with safer side effect profiles.
| Source: Galapagos NV
MECHELEN, Belgium, Sept. 22, 2015 (GLOBE NEWSWIRE) — Galapagos NV (Euronext & NASDAQ: GLPG) presents pre-clinical and Phase 1 results for autotaxin inhibitor GLPG1690 at the European Respiratory Society Annual Meeting in Amsterdam, Netherlands. Galapagos expects to file an exploratory Phase 2 study in idiopathic pulmonary fibrosis before year end. GLPG1690 has potential application in other pulmonary diseases such as chronic obstructive pulmonary disease (COPD), as supported by the presentation on pre-clinical findings at ERS this year:
“Pharmacological profile and efficacy of GLPG1690, a novel ATX inhibitor for COPD treatment,” poster PA2129 in Poster Discussion Session: “New targets and modalities for the treatment of asthma and COPD” (September 28, 2015; Room D201-202, 10:45 AM – 12:45 PM)
Galapagos is the first to show efficacy of an autotaxin inhibitor in pre-clinical models for COPD and IPF, pointing to novel therapeutic areas for autotaxin inhibition. The poster shows how GLPG1690 acts as a potent inhibitor of mouse and human autotaxin (IC50: 100 -500 nM range). Furthermore, GLPG1690 reduces inflammation in a mouse steroid-resistant tobacco smoke model to a similar extent as a standard therapy for COPD.
Galapagos also presents the topline results with GLPG1690 in Phase 1 in healthy human volunteers: “Favorable human safety, pharmacokinetics and pharmacodynamics of the autotaxin inhibitor GLPG1690, a potential new treatment in COPD,” oral presentation OA484 in session “Advances in the future treatment of COPD” (September 27, 2015; Room 2.1, 10:45 AM – 12:45 PM)
GLPG1690 was safe and well tolerated up to a single oral dose of 1500 mg and up to 1000 mg twice daily for 14 days, with no significant adverse effects on ECGs, vital signs or laboratory parameters. The compound also showed good oral bioavailability with a half-life of 5 hours and a dose-proportional increase in exposure. GLPG1690 showed concentration-dependent reduction of a relevant biomarker (plasma LPA18:2 levels) with a maximum of approximately 90%. At steady state, continuous reduction of this biomarker levels of >60% was observed from 0 to 24 hours. The presentation will also include relevant pre-clinical model data for COPD and IPF with GLPG1690.
Both the presentation and the posters will be made available on the Galapagos website after the conference.
About Galapagos
Galapagos (Euronext & NASDAQ: GLPG) is a clinical-stage biotechnology company specialized in the discovery and development of small molecule medicines with novel modes of action, with a pipeline comprising three Phase 2 programs, two Phase 1 trials, five pre-clinical studies, and 20 discovery small-molecule and antibody programs in cystic fibrosis, inflammation, and other indications. In the field of inflammation, AbbVie and Galapagos signed a collaboration agreement for the development and commercialization of filgotinib. Filgotinib is an orally-available, selective inhibitor of JAK1 for the treatment of rheumatoid arthritis and potentially other inflammatory diseases, currently in Phase 2B studies in RA and in Phase 2 in Crohn’s disease. Galapagos reported good activity and a favorable safety profile in both the DARWIN 1 and 2 trials in RA. AbbVie and Galapagos also signed a collaboration agreement in cystic fibrosis to develop and commercialize molecules that address mutations in the CFTR gene. Potentiator GLPG1837 is currently in a Phase 1 trial, and corrector GLPG2222 is at the pre-clinical candidate stage. GLPG1205, a first-in-class inhibitor of GPR84 and fully-owned by Galapagos, is currently being tested in a Phase 2 proof-of-concept trial in ulcerative colitis patients. GLPG1690, a fully proprietary, first-in-class inhibitor of autotaxin, has shown favorable safety in a Phase 1 trial and is expected to enter Phase 2 in idiopathic pulmonary fibrosis. The Galapagos Group, including fee-for-service subsidiary Fidelta, has approximately 400 employees, operating from its Mechelen, Belgium headquarters and facilities in The Netherlands, France, and Croatia. More info at www.glpg.com
CONTACT
Galapagos NV
Elizabeth Goodwin, Head of Corporate Communications & IR
Tel: +31 6 2291 6240
ir@glpg.com
MECHELEN, Belgium, Feb. 16, 2015 (GLOBE NEWSWIRE) — Galapagos NV (Euronext: GLPG) announced today that GLPG1690, a first-in-class molecule for pulmonary disease, has demonstrated target engagement, a good safety profile, and favorable drug properties in a Phase 1 study. Galapagos is developing GLPG1690 within its alliance with Janssen Pharmaceutica NV.
The aim of the Phase 1 study was to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of oral single and multiple ascending doses of GLPG1690. The randomized, double-blind, placebo-controlled, single center study was conducted in 40 healthy volunteers in Belgium. In the first part of the study, single ascending doses were evaluated. In the second part, the new compound was administered daily for 14 days.
GLPG1690 proved to be safe and well-tolerated over a wide dose range in healthy volunteers. Engagement of the thus far undisclosed novel target was confirmed using a relevant biomarker. GLPG1690 displayed a favorable pharmacokinetic and pharmacodynamic profile. The data shown in Phase 1 encourage Galapagos to explore a Phase 2 study design in pulmonary disease.
“GLPG1690 is the first molecule against this target ever to be evaluated clinically, and we are pleased with the outcome of the Phase 1 study,” said Dr Piet Wigerinck, CSO of Galapagos. “Galapagos continues to deliver novel therapeutics from its unique target and drug discovery engine.”
In 2007, Galapagos announced an alliance agreement with Janssen Pharmaceutica NV providing the option to worldwide, commercial licenses to certain Galapagos internal inflammatory disease programs. These programs are based on novel targets for inflammatory disorders that were identified and validated by Galapagos using its proprietary target discovery engine. Subsequent Galapagos research led to the discovery of GLPG1690, a first-in-class molecule that entered the clinic for inflammatory disorders. Galapagos is responsible for execution of Phase 1 and Phase 2A studies with GLPG1690.
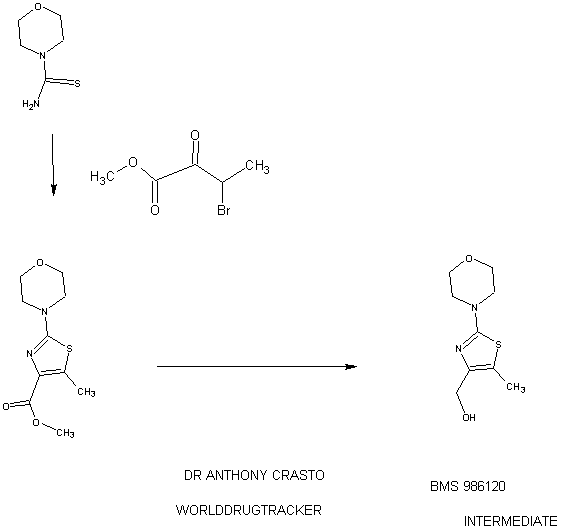
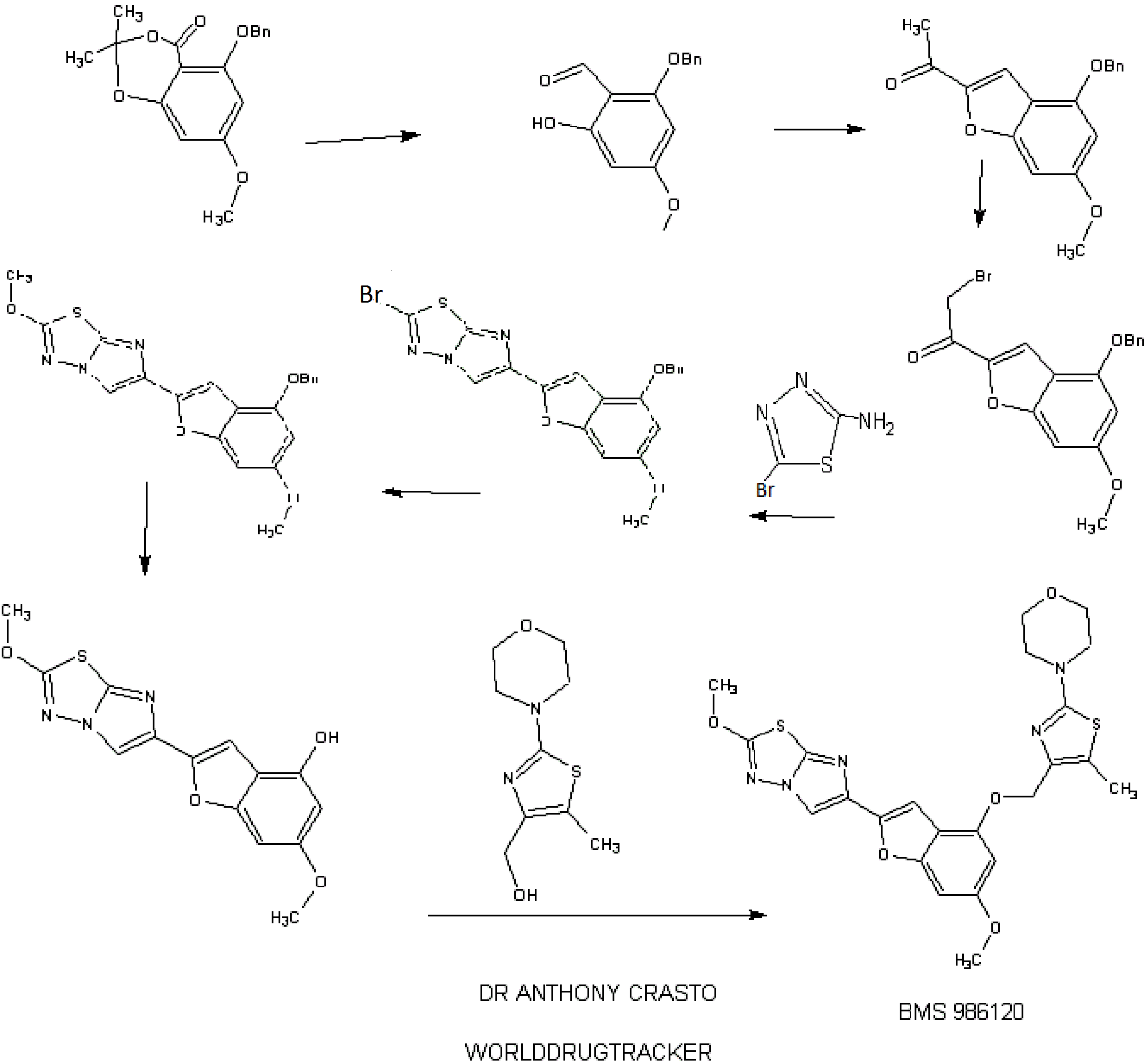
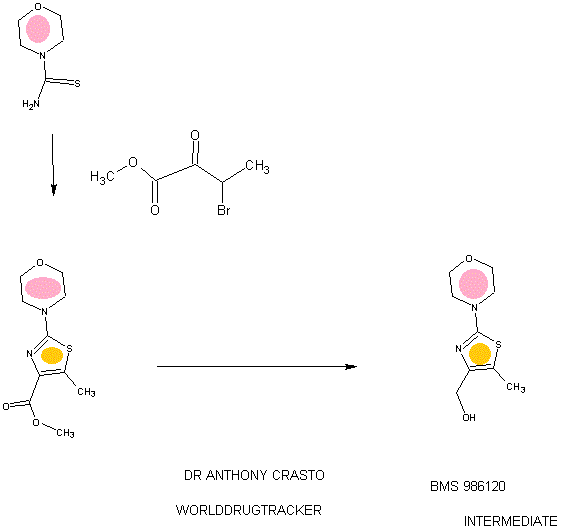
SYNTHESIS
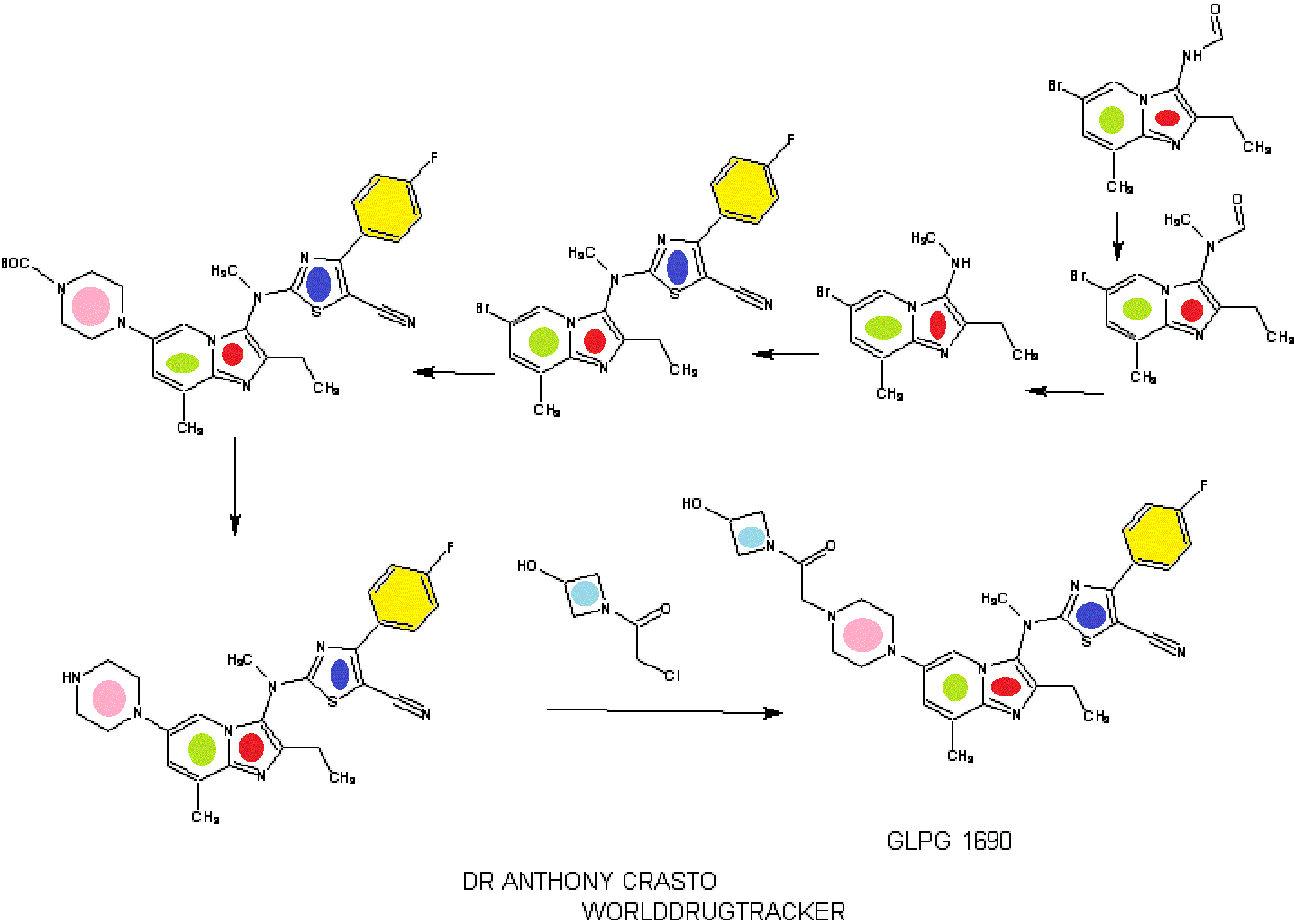
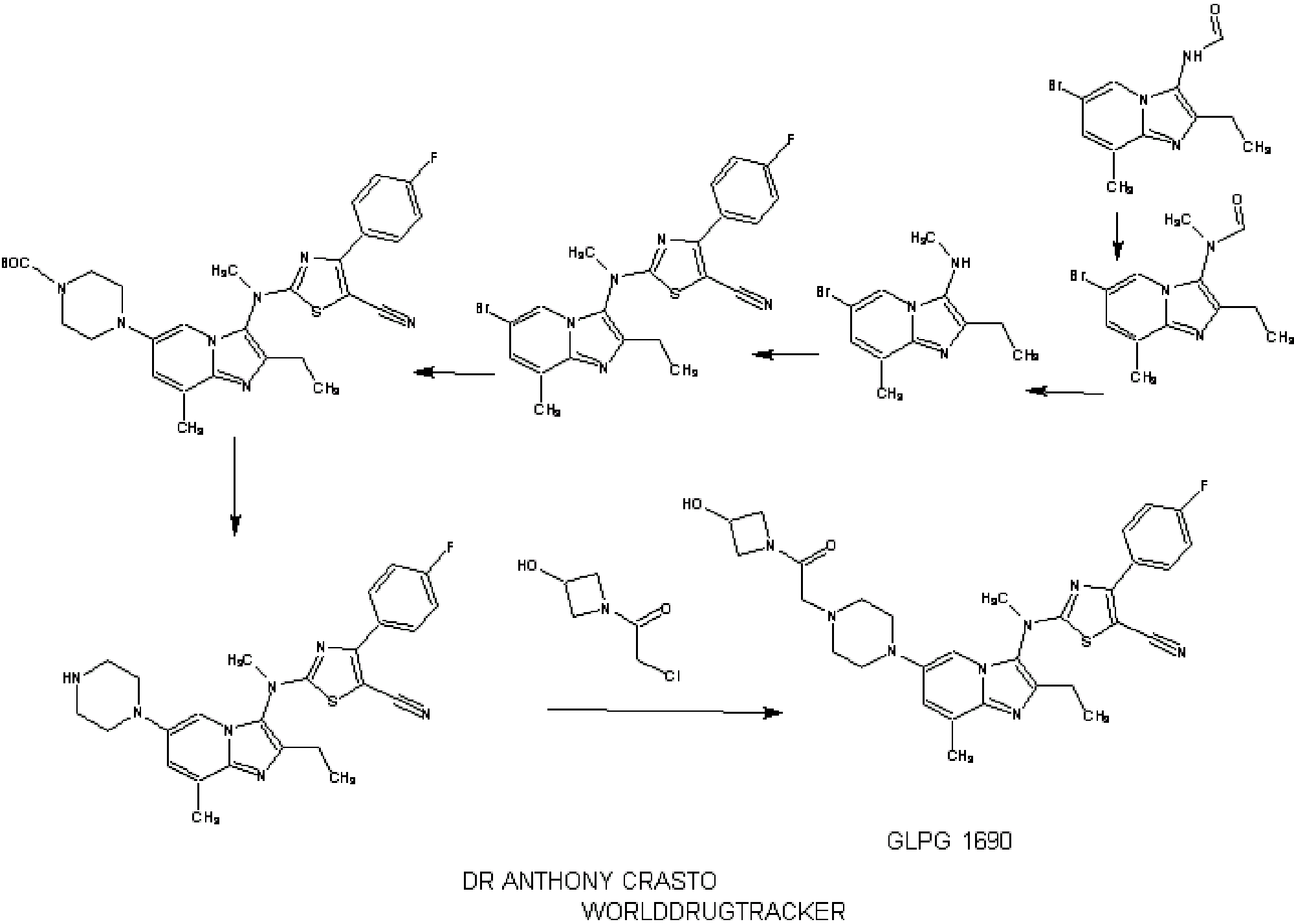
INTRODUCTION
relates to compounds that are inhibitors of autotaxin, also known as ectonucleotide pyrophosphatase/phosphodiesterase 2 (NPP2 or ENPP2), that is involved in fibrotic diseases, proliferative diseases, inflammatory diseases, autoimmune diseases, respiratory diseases, cardiovascular diseases, neurodegenerative diseases, dermatological disorders, and/or abnormal angiogenesis associated diseases. The present invention also provides methods for the production of a compound of the invention, pharmaceutical compositions comprising a compound of the invention, methods for the prophylaxis and/or treatment of diseases involving fibrotic diseases, proliferative diseases, inflammatory diseases, autoimmune diseases, respiratory diseases, cardiovascular diseases, neurodegenerative diseases, dermatological disorders, and/or abnormal angiogenesis associated diseases by administering a compound
STAGE 1

STAGE2

STAGE 3

STAGE4

STAGE 5

FINAL

PATENT
US2014303140
http://www.google.com/patents/US20140303140
1.2.4.4. Illustrative Synthesis of Intermediate Gen-3-e: N-(6-bromo-2-ethyl-8-methylimidazo[1,2-a]pyridin-3-yl)-N-methylformamide
-
-
To a suspension of formamide Gen-2-d (720 g, 2.55 mol, 1 eq.) in 5 L of acetone were added potassium carbonate (1 kg, 7.66 mol, 3 eq.) and methyl iodide (700 g, 4.93 mol, 1.9 eq.). The reaction mixture was heated to 40° C. overnight. Additional methyl iodide (25 g, 0.18 mol, 0.07 eq.) was then introduced and stirring continued for 1 h at 40° C. The reaction mixture was filtered and washed with acetone (2×300 mL) and DCM (2×300 mL). The filtrate was concentrated in vacuo and the residue was partitioned between DCM (3 L) and water (1 L). The aqueous layer was further extracted with DCM. The combined organic layers were then washed with brine, dried over Na2SO4, filtered and concentrated in vacuo. The solid was triturated with Et2O (1 L) at r.t. for 1 h, filtered off and dried to afford Intermediate Gen-3-e.
-
Rotamer A (Major): 1H NMR δ (ppm) (400 MHz, CDCl3): 8.19 (1H, s), 7.78 (1H, s), 7.15 (1H, s), 3.24 (3H, s), 2.72 (2H, q), 2.59 (3H, s), 1.31 (3H, t)
-
Rotamer B (Minor): 1H NMR δ (ppm) (400 MHz, CDCl3): 8.49 (1H, s), 7.65 (1H, s), 7.08 (1H, s), 3.36 (3H, s), 2.72 (2H, q), 2.59 (3H, s), 1.31 (3H, t)
-
LC-MS: MW (calcd): 295 (79Br), 297 (81Br); m/z MW (obsd): 296 (79Br M+1), 298 (81Br M+1)
1.2.5.2. Illustrative Synthesis of Intermediate Gen-4-d: (6-Bromo-2-ethyl-8-methyl-imidazo[1,2-a]pyridin-3-yl)-methyl-amine
-
-
Intermediate Gen-3-e (80 g, 270 mmol, 1 eq.) was dissolved in a 1.25 M HCl solution in MeOH (540 mL, 2.5 eq.) and the resulting mixture was refluxed overnight. 270 mL of 1.25 M HCl solution in MeOH were added and heating continued overnight. After 48 h, additional 70 mL of the 1.25 M HCl solution in MeOH were introduced in the reaction mixture. Heating was maintained overnight until conversion was complete. The crude mixture was then concentrated in vacuo and the residue was partitioned between EtOAc (300 mL) and water (700 mL). A saturated NaHCO3 solution was added until pH reached 8-9. The aqueous layer was extracted twice with EtOAc (2×300 mL). The combined organic layers were then washed with brine (200 mL), dried over Na2SO4, filtered and concentrated in vacuo to give Intermediate Gen-4-d (6-bromo-2-ethyl-8-methyl-imidazo[1,2-a]pyridin-3-yl)-methyl-amine) as a free base.
-
1H NMR δ (ppm) (400 MHz, CDCl3): 8.05 (1H, s), 7.04 (1H, s), 2.84-2.78 (5H, m), 2.60 (3H, s), 1.35 (3H, t)
-
LC-MS: MW (calcd): 267 (79Br), 269 (81Br); m/z MW (obsd): 268 (79Br M+1), 270 (81Br M+1)
1.2.6.4. Illustrative Synthesis of Intermediate Gen-5-t: 2-[(6-Bromo-2-ethyl-8-methyl-imidazo[1,2-a]pyridin-3-yl)-methyl-amino]-4-(4-fluoro-phenyl)-thiazole-5-carbonitrile
-
-
To a solution of amine Gen-4-d (4.4 g, 16.6 mmol, 1 eq.) in THF (44 mL) under argon was slowly added NaH (60% in oil suspension, 2.0 g, 50.0 mmol, 3 eq.). The reaction mixture was heated at 90° C. for 30 min then cooled to 40° C. before adding the chlorothiazole Gen-12-a (4.74 g, 19.9 mmol, 1.2 eq.). The reaction mixture was stirred at 90° C. overnight. After cooling to r.t. the mixture was slowly quenched by addition of water and then diluted with EtOAc. The organic layer was separated and the aqueous layer extracted with EtOAc. The combined organic layers were then washed with water and brine, dried over Na2SO4, filtered and concentrated in vacuo. The residue was triturated in Et2O, filtered and washed with Et2O and MeCN. Recrystallization was performed in MeCN (180 mL) to afford Intermediate Gen-5-t (2-[(6-Bromo-2-ethyl-8-methyl-imidazo[1,2-a]pyridin-3-yl)-methyl-amino]-4-(4-fluoro-phenyl)-thiazole-5-carbonitrile).
-
1H NMR δ (ppm) (400 MHz, CDCl3): 8.15 (2H, dd), 7.80 (1H, s), 7.22-7.14 (3H, m), 3.62 (3H, s), 2.77 (2H, q), 2.64 (3H, s), 1.35 (3H, t)
-
LC-MS: MW (calcd): 469 (79Br), 471 (81Br); m/z MW (obsd): 470 (79Br M+1), 472 (81Br M+1)
1.2.7.1.4. Illustrative Synthesis of 4-(3-{[5-Cyano-4-(4-fluoro-phenyl)-thiazol-2-yl]-methyl-amino}-2-ethyl-8-methyl-imidazo[1,2-a]pyridin-6-yl)-piperazine-1-carboxylic acid tert-butyl ester
-
-
To a solution of Intermediate Gen-5-t (24.2 g, 51.5 mmol, 1 eq.) in toluene under argon were successively added N-Boc piperazine (14.4 g, 77.3 mmol, 1.5 eq.), sodium tert-butoxide (9.9 g, 103 mmol, 2 eq.), JohnPhos (1.54 g, 5.15 mmol, 0.1 eq.) and Pd2(dba)3 (2.36 g, 2.58 mmol, 0.05 eq.). The reaction mixture was heated at 115° C. for 1 h. After cooling to r.t., the crude product was filtered on Celpure® P65 and the residue dissolved in EtOAc and washed with water. The organic layer was further washed with brine, dried over Na2SO4, filtered and concentrated in vacuo. The crude product was purified by chromatography on silica gel (elution with heptane/EtOAc:90/10 to 20/80) to afford the expected product.
-
1H NMR δ (ppm) (400 MHz, CDCl3): 8.16 (2H, dd), 7.17 (2H, app t), 6.99 (2H, bs), 3.62-3.53 (4H, m), 3.60 (3H, s), 3.04-2.93 (4H, m), 2.74 (2H, q), 2.62 (3H, s), 1.47 (9H, s), 1.33 (3H, t).
-
LC-MS: MW (calcd): 575; m/z MW (obsd): 576 (M+1)
1.2.7.8.4. Illustrative Synthesis of Compound 1: 2-[(2-Ethyl-8-methyl-6-piperazin-1-yl-imidazo[1,2-a]pyridin-3-yl)-methyl-amino]-4-(4-fluoro-phenyl)-thiazole-5-carbonitrile
-
-
4-(3-{[5-Cyano-4-(4-fluoro-phenyl)-thiazol-2-yl]-methyl-amino}-2-ethyl-8-methyl-imidazo[1,2-a]pyridin-6-yl)-piperazine-1-carboxylic acid tert-butyl ester was prepared from intermediate Gen-5-t using Boc-piperazine and method Flb.
-
To a solution of 4-(3-{[5-Cyano-4-(4-fluoro-phenyl)-thiazol-2-yl]-methyl-amino}-2-ethyl-8-methyl-imidazo[1,2-a]pyridin-6-yl)-piperazine-1-carboxylic acid tert-butyl ester (24.4 g, 42 mmol, 1 eq.) in MeOH (100 mL) was added a 2 M HCl solution in Et2O (127 mL, 254 mmol, 6 eq.). The reaction mixture was stirred at r.t. for 3.5 h then concentrated in vacuo. The residue was partitioned between EtOAc and water. The aqueous layer was extracted twice with EtOAc. A 2 M NaOH solution was added to the aqueous layer until pH reached 8-9 and further extraction with EtOAc was performed. The combined organic layers were then washed with brine, dried over Na2SO4, filtered and concentrated in vacuo. The solid was triturated with heptane (100 mL) at r.t. overnight, filtered off, washed with heptane and Et2O, and dried to afford the expected compound.
-
1H NMR δ (ppm) (400 MHz, CDCl3): 8.17 (2H, dd), 7.18 (2H, app t), 6.99 (2H, bs), 3.61 (3H, s), 3.09-2.98 (8H, m), 2.75 (2H, q), 2.61 (3H, s), 1.34 (3H, t).
-
LC-MS: MW (calcd): 475; m/z MW (obsd): 476 (M+1)
1.2.7.14. Illustrative Synthesis of Compound 2: 2-((2-ethyl-6-(4-(2-(3-hydroxyazetidin-1-yl)-2-oxoethyl)piperazin-1-yl)-8-methylimidazo[1,2-a]pyridin-3-yl)(methyl)amino)-4-(4-fluorophenyl)thiazole-5-carbonitrile
-
-
To a solution of amine compound 1 (12.6 g, 27 mmol, 1 eq.) in 100 mL of MeCN were added potassium carbonate (7.3 g, 53 mmol, 2 eq.) and Gen13-a (5.2 g, 34 mmol, 1.3 eq.). The reaction mixture was refluxed for 5.5 h then cooled to r.t. and stirred for 40 h. The crude product was filtered and washed with MeCN. The collected precipitate was then suspended in 300 mL of water, stirred for 1 h, filtered, and finally washed with water and MeCN. The solid obtained was dried in vacuo for 48 h to afford Compound 2.
-
1H NMR (400 MHz, CDCl3) δ ppm 8.20-8.12 (2H, m), 7.22-7.13 (2H, m), 6.99 (2H, s), 4.68 (1H, m), 4.43 (1H, dd), 4.26 (1H, dd), 4.14-4.05 (1H, m), 3.88 (1H, dd), 3.61 (3H, s), 3.58-3.52 (1H, m), 3.14-3.02 (6H, m), 2.74 (2H, q), 2.70-2.62 (4H, m), 2.59 (3H, s), 1.33 (3H, t)
-
LC-MS: MW (calcd): 588; m/z MW (obsd): 589 (M+1)
| US9249141 | Dec 17, 2014 | Feb 2, 2016 | Galapagos Nv | Compounds and pharmaceutical compositions thereof for the treatment of inflammatory disorders |
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015111872 | 2015-04-23 | NOVEL COMPOUNDS AND PHARMACEUTICAL COMPOSITIONS THEREOF FOR THE TREATMENT OF INFLAMMATORY DISORDERS |
| US2014303140 | 2014-10-09 | NOVEL COMPOUNDS AND PHARMACEUTICAL COMPOSITIONS THEREOF FOR THE TREATMENT OF INFLAMMATORY DISORDERS |
////////////GLPG 1690, idiopathic pulmonary fibrosis, PHASE 1, GALAPAGOS, 1628260-79-6
n12c(c(nc1c(cc(c2)N3CCN(CC3)CC(=O)N4CC(C4)O)C)CC)N(C)c5nc(c(s5)C#N)c6ccc(cc6)F
CCC1=C(N2C=C(C=C(C2=N1)C)N3CCN(CC3)CC(=O)N4CC(C4)O)N(C)C5=NC(=C(S5)C#N)C6=CC=C(C=C6)F
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE


 Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
P.S
THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT, amcrasto@gmail.com, +91 9323115463 India.
DISCLAIMER
I , Dr A.M.Crasto is writing this blog to share the knowledge/views, after reading Scientific Journals/Articles/News Articles/Wikipedia. My views/comments are based on the results /conclusions by the authors(researchers). I do mention either the link or reference of the article(s) in my blog and hope those interested can read for details. I am briefly summarising the remarks or conclusions of the authors (researchers). If one believe that their intellectual property right /copyright is infringed by any content on this blog, please contact or leave message at below email address amcrasto@gmail.com. It will be removed ASAP
 .
.









 .
.


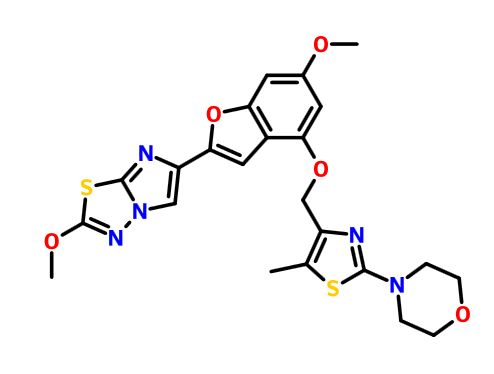
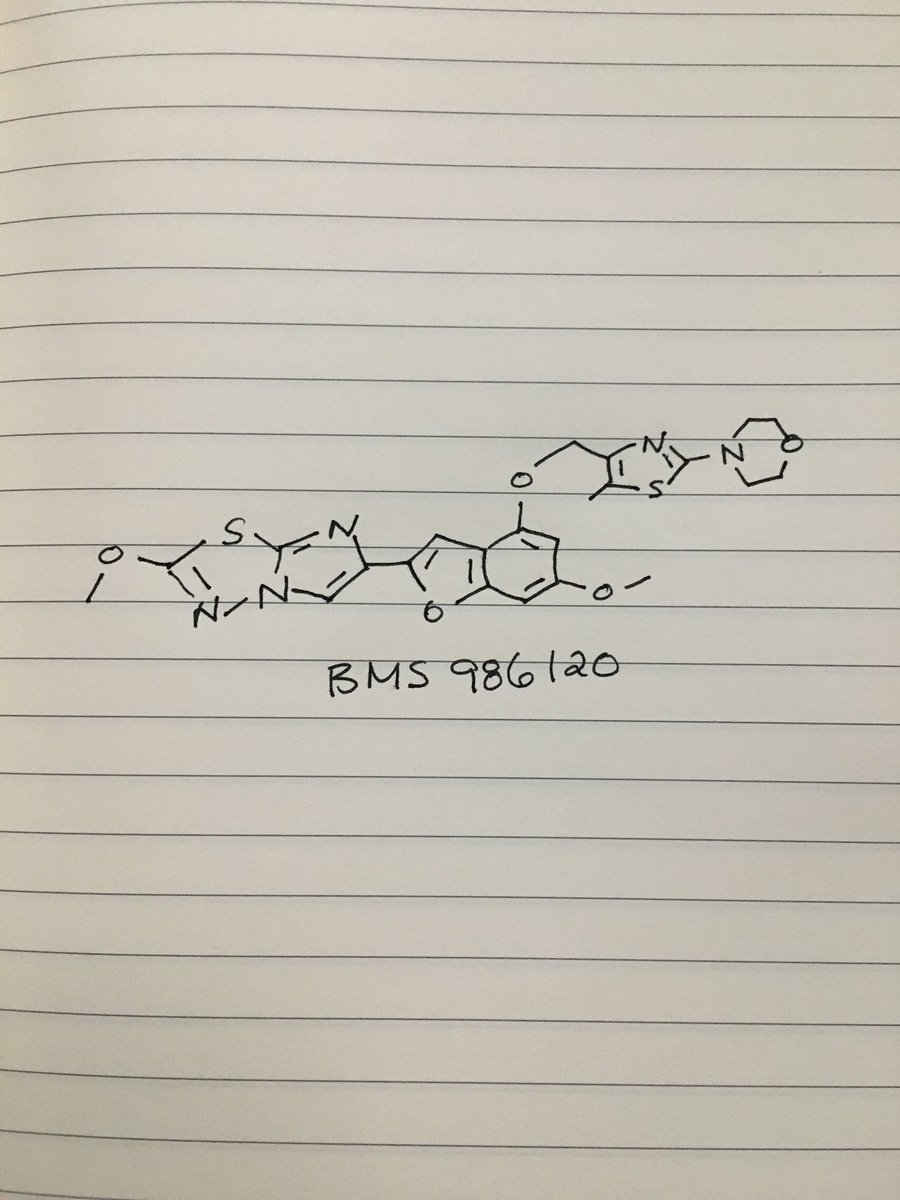 .
.