Tout sur les médicaments הכל על תרופות كل شيئ عن الأدوية Все о наркотиках 关于药品的一切 డ్రగ్స్ గురించి అన్ని 마약에 관한 모든 것 Όλα για τα Ναρκωτικά Complete Tracking of Drugs Across the World by Dr Anthony Melvin Crasto, Worldpeacepeaker, worlddrugtracker, PH.D (ICT), MUMBAI, INDIA, Worlddrugtracker, Helping millions, 9 million hits on google on all websites, 2.5 lakh connections on all networks, “ALL FOR DRUGS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This is a compilation for educational purposes only. P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
BS B2B Bureau | Mumbai April 12, 2016 Last Updated at 10:27 IST
Novartis Healthcare will continue to market Sequadra (indacaterol/glycopyrronium inhaler), while Lupin will promote the inhaler under the brand name Loftair in India
J. Braz. Chem. Soc. vol.27 no.4 São Paulo Apr. 2016
http://dx.doi.org/10.5935/0103-5053.20150326
ARTICLES
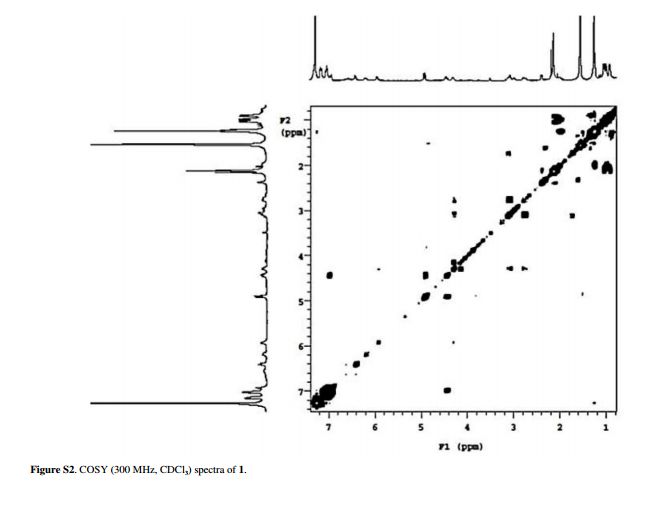
Ixorine, a New Cyclopeptide Alkaloid from the Branches of Ixora brevifolia
Rebeca P. Medinaa, Ivânia T. A. Schuquela, Armando M. Pominia, Cleuza C. Silvaa, Cecília M. A. Oliveirab, Lucília Katob, Celso V. Nakamurac, Silvana M. O. Santin*a
aDepartamento de Química, Universidade Estadual de Maringá, Av. Colombo 5790, 87020-900 Maringá-PR, Brazil
bInstituto de Química, Universidade Federal de Goiás, Campus II, Samambaia, 74001-970 Goiânia-GO, Brazil
cDepartamento de Análises Clínicas e Biomedicina, Universidade Estadual de Maringá, Av. Colombo 5790, 87020-900 Maringá-PR, Brazil
ABSTRACT
The isolation and structure determination of new cyclic peptide alkaloid ixorine, along with five known constituents frangulanine, syringaresinol, cinnamtannin B-1, daucosterol and mannitol from the branches of Ixora brevifolia are described. The cyclic peptide frangulanine is being described for the first time in the Rubiaceae family. The structures were elucidated on their spectral data basis, mainly one- (1H, 13C, DEPT) and two-dimensional (COSY, NOESY, HSQC and HMBC) nuclear magnetic resonance (NMR) and by comparison with data from the literature. The mixture of two cyclopeptide alkaloids showed weak activity against Leishmania amazonensis……..http://www.scielo.br/scielo.php?script=sci_arttext&pid=S0103-50532016000400753&lng=en&nrm=iso&tlng=en
A GABA (B) receptor agonist potentially for the treatment of muscle spasticity.
AGI-006; STX-209; OS-440
CAS No. 69308-37-8 free
847353-30-4 placarbil
Arbaclofen placarbil (ar-bac-loe-fenpla-kar-bil, also known as XP19986) is a prodrug of R–baclofen. Arbaclofen placarbil possesses more favorable pharmacokinetic profile than baclofen, with less fluctuations in plasma drug levels. It was being developed as a potential treatment for patients with GERD and spasticity due to multiple sclerosis; however, in May 2013 XenoPort announced the termination of development because of unsuccessful results in phase III clinical trials.[1]
Arbaclofen Placerbil is a prodrug of Arbaclofen, which is a selective gamma-amino-butyric acid type B receptor agonist and the R-enantiomer of baclofen. It was discovered, and has been patented by XenoPort as a new chemical entity with an improved pharmacokinetic profile compared to baclofen, which allows for sustained release properties. ArbaclofenPlacerbil was believed to have therapeutic potential in treating gastroesophogeal reflux disease (GERD) and plasticity; however due to discouraging clinical trial results, the drug was abandoned by XenoPort in 2011 for the treatment of GERD. On May 20th, 2013, XenoPort announced plans to terminate the development of Arbaclofen Placerbil for the treatment of multiple sclerosis.
Autism spectrum disorder (ASD) is a behaviorally defined disorder which has increased in prevalence over the last two decades. Despite decades of research, no effective treatment is currently available. Animal models, as well as other lines of evidence, point to abnormalities in the balance of cortical excitation to inhibition in individuals with ASD, with this imbalance resulting in an overall increase in cortical excitation. To reduce cortical excitatory glutamate pathways, arbaclofen, a selective agonist of the gamma aminobutyric acid receptor type B, has been developed. This article reviews the evidence for this treatment for ASD using a systematic review methodology. Overall, a systematic search of the literature revealed 148 relevant references with the majority of these being review papers or news items that mentioned the potential promise of arbaclofen. Five original studies were identified, four of which used STX209, a form of arbaclofen developed by Seaside Therapeutics, Inc., and one which used R-baclofen. In an animal model, treatment of Fragile X, a genetic disease with ASD features, demonstrated a reversal of behavioral, neurological, and neuropathological features associated with the disease. One double-blind, placebo-controlled study treated children and adults with Fragile X. Results from this study were promising, with signs of improvement in social function, especially in the most severely socially impaired. Two studies, one open-label and one double-blind, placebo-controlled, were conducted in children, adolescents, and young adults with ASD. These studies suggested some improvements in socialization, although the effects were limited and may have been driven by individuals with ASD that were higher-functioning. These studies and others that have used arbaclofen for the treatment of gastroesophageal reflux suggest that arbaclofen is safe and well-tolerated. Clearly, further clinical studies are needed in order to refine the symptoms and characteristics of children with ASD that are best treated with arbaclofen.
Fig. 1.
The Structures of R-baclofen (1), arbaclofen placarbil (2), R-baclofen lactam (3), and the potential γ-hydroxy metabolite of R-baclofen (4).
I , Dr A.M.Crasto is writing this blog to share the knowledge/views, after reading Scientific Journals/Articles/News Articles/Wikipedia. My views/comments are based on the results /conclusions by the authors(researchers). I do mention either the link or reference of the article(s) in my blog and hope those interested can read for details. I am briefly summarising the remarks or conclusions of the authors (researchers). If one believe that their intellectual property right /copyright is infringed by any content on this blog, please contact or leave message at below email address amcrasto@gmail.com. It will be removed ASAP
Phase I studies of p.o. administered nolatrexed dihydrochloride (AG337, THYMITAQ), a nonclassical thymidylate synthase inhibitor, were performed to establish the maximum tolerated dose and a recommended dose for Phase II studies. The bioavailability and pharmacokinetic and pharmacodynamic properties of oral nolatrexed were also studied. Forty-five patients were treated with oral nolatrexed every 6 h for 5 days at doses of 288-1000 mg/m2/day. The bioavailability of the oral preparation was determined, and the effect of a standard meal on nolatrexed absorption was investigated at a dose of 800 mg/m2/day. Nolatrexed plasma concentrations were analyzed by high-performance liquid chromatography. Nolatrexed was rapidly absorbed with a median bioavailability of 89% (range 33-116%), with 88% of patients above 70%. The dose-limiting toxicities were gastrointestinal, and the recommended Phase II oral dose was 800 mg/m2/day. After a standard meal, the peak plasma nolatrexed concentration achieved was lower (median, 8.3 microg/ml versus 15.0 microg/ml; P = 0.001), and the time taken to reach the peak was longer (median, 180 min versus 45 min; P = 0.00003), but the trough concentration was higher (median, 3.6 microg/ml versus 2.1 microg/ml; P = 0.004) when compared with the fasted state. The area under the nolatrexed plasma concentration versus time curve was not affected by food. Average trough nolatrexed concentration, but not dose, was significantly related to the % decrease in both thrombocytes (r2 = 0.58; C50 = 6.0 microg/ml, where C50 is the plasma concentration associated with a 50% decrease in thrombocytes) and neutrophils (r2 = 0.63; C50 = 0.6 microg/ml). Nolatrexed can be safely administered as an oral preparation at a dose of 800 mg/m2/day for 5 days. Bioavailability was close to 100% and, because inhibition of thymidylate synthase by nolatrexed is rapidly reversible, the slower absorption after a standard meal may result in a shorter duration of noninhibitory concentrations between doses.
Catalytic hydrogenation of 2-bromo-4 -nitrotoluene (I) over Raney-Ni provided aniline (II). Reaction of (II) with chloral hydrate and hydroxylamine gave rise to the isonitrosoacetanilide (III), which was subsequently cyclized to the isatin (IV) by heating in concentrated H2SO4. Oxidative cleavage of isatin (IV) produced the anthranilic acid (V). This was converted to the benzoxazinone (VI) upon refluxing with acetic anhydride. Ring opening of benzoxazinone (VI) with MeOH, followed by acidic hydrolysis of the acetamide function, yielded the anthranilate ester (VII). The quinazoline derivative (VIII) was then obtained by treatment of anthranilate (VII) with chloroformamidine hydrochloride in refluxing diglyme. Finally, displacement of the bromide group of (VIII) with the sodium thiolate of 4-mercaptopyridine (IX) under Ullmann conditions afforded the title pyridyl sulfide.
Dissertation title
[BT] A New Method for Synthesis of Nolatrexed Dihydrochloride
Hangul title
Nolatrexed dihydrochloride Synthesis Process Development
Author
Xueqing Zhao, Fei Li, Weiping Zhuang, Xiaowen Xue, Yuanyang Lian, Jianhui Fan and Dongsheng Fang
Japjimyeong
ORG PROCESS RES DEV
Issue year
2010
Gwonho details
14 (2)
The surface
346-350
ABSTRACT
A new synthetic method for nolatrexed dihydrochloride (thymitaq) has been developed. The synthesis was accomplished in three steps featuring the direct conversion of the starting 4-bromo-5-methylisatin into the methyl anthranilate by potassium peroxydisulfate / sodium methoxide. In the final Ullmann reaction potassium carbonate was employed in place of sodium hydride, and the amount of copper catalysts was significantly reduced. Moreover, sodium sulfide solution was utilized to efficiently remove copper under approximately neutral conditions instead of hydrogen sulfide / methanol under strongly acidic conditions. By means of these modifications, nolatrexed dihydrochloride was ensured to be prepared in good yield and high purity.
Contents
Nolatrexed dihydrochloride (2-Amino-6-methyl-5-(4-pyridylthio) -3 H-quinazolin-4-one dihydrochloride, thymitag, 1) is the HCC cancer therapeutic agent to the TS (thymidylate synthase) folate binding site on the TS inhibitor as DNA replication inhibition, DNA damage, S-phase cell cycle arrest, and caspase-dependent apoptosis induction and clinical 2 on theresults look HCC patients, the survival benefit of showing the current phase III study is in progress in it. under scheme 1 is conducted in a number of synthesis team Nolatrexedillustrates the development process
Scheme 1. Synthetic routes A-F from 4-bromo-5-methylisatin (2) to nolatrexed dihydrochloride (1)
The scheme 1 When the complex first synthesis process but is A : 2 – 3 – 4 – 5 – 7 · HCl – 1 or in part, 6 pass through a B step ( 2 – 3 – 6 – 5 ) to obtain the desired compound with, but However, these processes are of the desired product quality control had a disadvantage unfulfilled this . after C, D, E process was developed during the E step is a step wherein compound 8 from the first to the one-pot is the most superior process consists in the process also drug of the compound for use as a quality control has difficulty in . more recentlyWennerberg is a new process F compounds were reported for 3 compound directly from the 7fully in the process I scored quality control could be the place . in the process, each reactionstep partially changed by the use of a reagent zoom impurity to minimize the formation of .However, this process also work-up, and purification there have difficulties to process the authors reported a new efficient way .
Scheme 2. Synthetic route G from 4-bromo-5-methylisatin (2) to nolatrexed dihydrochloride (1)
Scheme 2 The process reported to also have specifically not a new process only takes the best features from several processes previously reported , significant differences that the author is proud director teen two direct compound from 5 will get the , also reported in other processes already advanced mercaptopyridine introducing Ullmann reaction in the processimpurity , to reduce the formation of NaH , instead of K2CO3 were used the copper catalyst in order to minimize the amount of copper scavenge used to H2S instead of Na2S was used . the compound obtained in the process 1 of the purity is 96.6% and 3% with impurities of the 4,4′-dithiodipyridine this was confirmed copper impurity is 20 ppm was below . last Nolatrexed dihydrochloride in the process to obtain a 99.7% purity I scored the desired product , 0.3% ofunidentified impurity, and 10 ppm less than copper because it contains should think very advanced process compared to the previous number of ways . Fortunately Ullmann key contained in the reaction impurity in 4,4′-dithiodipyridine was automatically removed from the crystallization process of the last reaction.
Korea Research Institute of Chemical Technology provides incurable disease treatment and research center, Dr. jaedu
A quinolone antibiotic potentially for the treatment of bacterial infections.
Research Code CS-940
CAS No. 153808-85-6(FREE)
Cas 128427-55-4(Cadrofloxacin HCl)
HYDROCHLORIDE
Molecular Weight
447.84
Formula
C19H20F3N3O4 • HCl
OriginatorSankyo; Ube Industries
DeveloperSankyo
ClassAntibacterials; Quinolones; Small molecules
Mechanism of ActionType II DNA topoisomerase inhibitors
20 Jun 1996An animal study has been added to the Bacterial infections pharmacodynamics section
24 Mar 1995Phase-II clinical trials for Bacterial infections in Japan (PO)
Cadrofloxacin hydrochloride was studied for the treatment of bacterial infections.The compound was originally developed by UBE and Daiichi Sankyo. However, this study was discontinued. The compound currently was developed by Hengrui.
SYNTHESIS
Decarboxylation of 3,5,6-trifluoro-4- hydroxyphthalic acid (I) upon heating at 140 C in an autoclave furnished 2,4,5-trifluoro-3-hydroxybenzoic acid (II). This was converted to ethyl ester (III) by refluxing in EtOH in the presence of H2SO4. Condensation of (III) with chlorodifluoromethane and NaH in hot DMF produced the corresponding difluoromethyl ether, and subsequent basic hydrolysis of the ethyl ester yielded 3- (difluoromethoxy) -2, 4,5-trifluorobenzoic acid (IV). Alternatively, acid (II) was converted to acid chloride with SOCl2 and subsequently condensed with ammonia to give amide (V). After formation of the difluoromethyl ether (VI) under similar conditions as above, acid (IV) was obtained by diazotization of the amide function of (VI) in hot sulfuric acid. The difluoromethoxy acid (IV) was also prepared by direct alkylation of hydroxy acid (II) with chlorodifluoromethane in the presence of NaOH in hot DMF. acid (IV) was activated as the corresponding acid chloride (VII) with SOCl2. Condensation of acid chloride (VII) with the magnesium salt of diethyl malonate gave rise to the benzoylmalonate (VIII). Further decarbethoxylation of (VIII) by heating in the presence of p-toluenesulfonic acid yielded keto ester (IX). This was condensed with triethyl orthoformate in the presence of Ac2O to give the ethoxyacrylate (X), which was converted to enamine (XII) by treatment with cyclopropylamine (XI). The target quinolone system (XIII) was then obtained by intramolecular cyclization of (XII) in the presence of NaH. Then, ethyl ester (XII) cleavage using boron trifluoride etherate provided the key quinolonecarboxylic acid boron chelate (XIV)
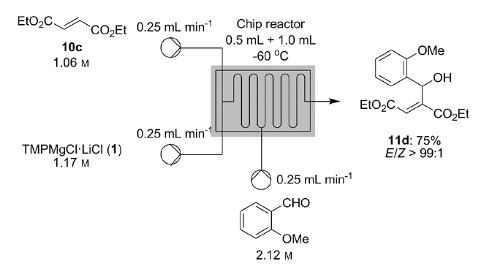
Knochel’s group in Munich have recently disclosed how a variety of functionalised heterocycles and sensitive acrylates can be rapidly magnesiated and subsequently quenched with an electrophile under continuous flow-through;conditions using a Uniqsis static mixer/reactor chip.
A key advantage is that, in contrast to typical batch procedures, these reactions required non-cryogenic conditions (typically 25C); moreover the procedure could be quickly scaled to 45 mmol without modification of the reaction conditions.
Metalations under flow-through conditions permited magnesiations that did not afford the desired product under batch conditions and acylates could be magnesiated and quenched to afford products with high stereoselectivities without concomitant polymerisation.
Continuous Flow Magnesiation of Functionalized Heterocycles and Acrylates with TMPMgCl⋅LiCl†
A flow procedure for the metalation of functionalized heterocycles (pyridines, pyrimidines, thiophenes, and thiazoles) and various acrylates using the strong, non-nucleophilic base TMPMgCl⋅LiCl is reported. The flow conditions allow the magnesiations to be performed under more convenient conditions than the comparable batch reactions, which often require cryogenic temperatures and long reaction times. Moreover, the flow reactions are directly scalable without further optimization. Metalation under flow conditions also allows magnesiations that did not produce the desired products under batch conditions, such as the magnesiation of sensitive acrylic derivatives. The magnesiated species are subsequently quenched with various electrophiles, thereby introducing a broad range of functionalities.
Successful chemical production of molecules while simultaneously reducing the environmental impact of the process relies not only on more efficient reactions but also on developments in reactor and separation technology. Recent decades have also witnessed a significant growth in industrial interest in solvent-based separations using membranes stable to organic solvents. The incorporation of membranes into a chemical process can be via a simple downstream processing method or an integrated reaction membrane method. This paper deals with homogeneous organometallic catalyzed reactions and probes the separation of a number of readily available palladium complexes from reaction mixtures with highly stable ceramic membranes. A number of different processing methods, namely, online, at-line, and off-line are compared and contrasted. A high rejection of Palladium species and consequently very low palladium contamination of reaction products with a single organic solvent nanofiltration (OSN) step has been demonstrated.
Potential of Homogeneous Pd Catalyst Separation by Ceramic Membranes. Application to Downstream and Continuous Flow Processes
SYNTHESISComments Off on Development of a Safe and Robust Process for the Large-Scale Preparation of a Vinyl Bromide from a Ketone Using a (PhO)3P/Br2-Derived Reagent
The large-scale synthesis of ethyl 4-bromocyclohex-3-enecarboxyalate, using a mild brominating reagent derived from triphenyl phosphite and bromine, is reported. The development and comparison of both continuous and batch processes are described.
A modified addition sequence was developed based on the knowledge garnered from flow-processing, resulting in a safe and efficient process for the in situ generation of the unstable active reagent and its immediate reaction with the ketone in a batch mode process.
Development of a Safe and Robust Process for the Large-Scale Preparation of a Vinyl Bromide from a Ketone Using a (PhO)3P/Br2-Derived Reagent
† Chemical and Synthetic Development, Biocon Bristol-Myers Squibb Research and Development Center, Biocon Park, Jigani Link Road, Bommasandra IV, Bangalore-560099, India
‡ Chemical and Synthetic Development, Bristol-Myers Squibb, 1 Squibb Drive, New Brunswick, New Jersey 08903, United States
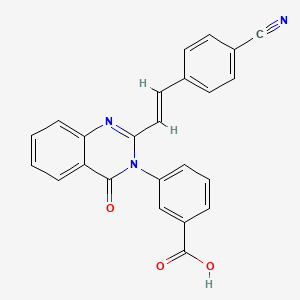
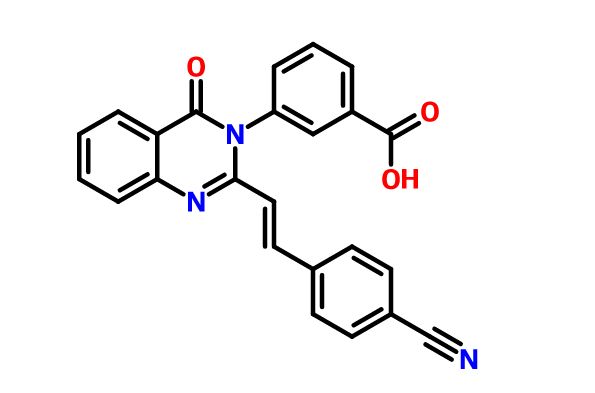
PRECLINICAL, UncategorizedComments Off on A New Antibiotic (E)-3-(3-Carboxyphenyl)-2-(4-cyanostyryl)quinazolin-4(3H)-one, from University Of Notre Dame
The emergence of resistance to antibiotics over the past few decades has created a state of crisis in the treatment of bacterial infections.Over the years, β-lactams were the antibiotics of choice for treatment of S. aureus infections. However, these agents faced obsolescence with the emergence of methicillin-resistant S. aureus (MRSA). Presently, vancomycin, daptomycin, linezolid, or ceftaroline are used for treatment of MRSA infections, although only linezolid can be dosed orally. Resistance to all four has emerged. Thus, new anti-MRSA antibiotics are sought, especially agents that are orally bioavailable. a new antibiotic (E)-3-(3-carboxyphenyl)-2-(4-cyanostyryl)quinazolin-4(3H)–one, with potent activity against S. aureus, including MRSA. We document that quinazolinones of our design are inhibitors of cell-wall biosynthesis in S. aureus and do so by binding to dd-transpeptidases involved in cross-linking of the cell wall. quinazolinones possess activity in vivo and are orally bioavailable. This antibiotic holds promise in treating difficult infections by MRSA.
PAPER
Journal of the American Chemical Society (2015), 137(5), 1738-1741.
† Department of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, Indiana 46556, United States
‡ Department of Crystallography and Structural Biology, Instituto de Química-Física “Rocasolano”, Consejo Superior de Investigaciones Científicas, Madrid, Spain
§ Freimann Life Sciences Center and Department of Biological Sciences, University of Notre Dame, Notre Dame, Indiana 46556, United States
In the face of the clinical challenge posed by resistant bacteria, the present needs for novel classes of antibiotics are genuine. In silico docking and screening, followed by chemical synthesis of a library of quinazolinones, led to the discovery of (E)-3-(3-carboxyphenyl)-2-(4-cyanostyryl)quinazolin-4(3H)–one (compound 2) as an antibiotic effective in vivo against methicillin-resistant Staphylococcus aureus (MRSA). This antibiotic impairs cell-wall biosynthesis as documented by functional assays, showing binding of 2 to penicillin-binding protein (PBP) 2a. We document that the antibiotic also inhibits PBP1 of S. aureus, indicating a broad targeting of structurally similar PBPs by this antibiotic. This class of antibiotics holds promise in fighting MRSA infections.
Staphylococcus aureus is a common bacterium found in moist areas of the body and skin. S. aureus can also grow as a biofilm, representing the leading cause of infection after implantation of medical devices. Approximately 29% (78.9 million) of the US population is colonized in the nose with S. aureus, of which 1.5% (4.1 million) is methicillin-resistant S. aureus (MRSA). In 2005, 478,000 people in the US were hospitalized with a S. aureus infection, of these 278,000 were MRSA infections, resulting in 19,000 deaths. MRSA infections have been increasing from 2% of S. aureus infections in intensive care units in 1974 to 64% in 2004, although more recent data report stabilization. Approximately 14 million outpatient visits occur every year in the US for suspected S. aureus skin and soft tissue infections. About 76% of these infections are caused by S. aureus, of which 78% are due to MRSA, for an overall rate of 59%. Spread of MRSA is not limited to nosocomial (hospital-acquired) infections, as they are also found in community-acquired infections. Over the years, β-lactams were antibiotics of choice in treatment of S. aureus infections. However, these agents faced obsolescence with the emergence of
MRSA. Presently, vancomycin, daptomycin or linezolid are agents for treatment of MRSA infections, although only linezolid can be dosed orally. Resistance to all three has emerged. Thus, new anti-MRSA therapeutic strategies are needed, especially agents that are orally bioavailable.
Clinical resistance to β-lactam antibiotics by MRSA has its basis predominantly in acquisition of the mecA gene, which encodes penicillin-binding protein 2a (PBP2a). PBP2a, a cell-wall DD- transpeptidase, is refractory to inhibition by essentially all commercially available β-lactams (ceftaroline is an exception), antibiotics that irreversibly acylate the active-site serine of typical PBPs. PBPs catalyze biosynthesis of the bacterial cell wall, which is essential for the survival of the bacterium. Accordingly, new ηοη-β-lactam antibiotics that inhibit PBP2a are needed to combat drug-resistant strains of bacteria. SUMMARY
Staphylococcus aureus is responsible for a number of human diseases, including skin and soft tissue infections. Annually, 292,000 hospitalizations in the US are due to S. aureus infections, of which 126,000 are related to methicillin-resistant Staphylococcus aureus (MRSA), resulting in 19,000 deaths. A novel structural class of antibiotics has been discovered and is described herein. A lead compound in this class shows high in vitro potency against Gram-positive bacteria comparable to those of linezolid and superior to vancomycin (both considered gold standards) and shows excellent in vivo activity in mouse models of MRSA infection.
The invention thus provides a novel class of ηοη-β-lactam antibiotics, the quinazolinones, which inhibit PBP2a by an unprecedented mechanism of targeting both its allosteric and active sites. This inhibition leads to the impairment of the formation of cell wall in living bacteria. The quinazolinones described herein are effective as anti-MRSA agents both in vitro and in vivo. Furthermore, they exhibit activity against other Gram-positive bacteria. The quinazolinones have anti-MRSA activity by themselves. However, these compounds synergize with β-lactam antibiotics. The use of a combination of a quinazolinone with a β-lactam antibiotic can revive the clinical use of β-lactam antibacterial therapy in treatment of MRSA infections. The invention provides a new class of quinazolinone antibiotics, optionally in combination with other antibacterial agents, for the therapeutic treatment of methicillin- resistant Staphylococcus aureus and other bacteria.
The quinazolinone compounds described herein can be prepared using standard synthetic techniques known to those of skill in the art. Examples of such techniques are described by Khajavi et al. (J. Chem. Res. (S), 1997, 286-287) and Mosley et al. (J. Med. Chem. 2010, 53, 5476-5490). A general preparatory scheme for preparing the compounds described herein, for example, compounds of Formula
wherein each of the variables are as defined for one or more of the formulas described herein, such as Formula (A).
EXAMPLES
Example 1. Compound Preparation
Chemistry. Organic reagents and solvents were purchased from Sigma- Aldrich. lH and 13C NMR spectra were recorded on a Varian INOVA-500. High-resolution mass spectra were obtained using a Bruker micrOTOF/Q2 mass spectrometer.
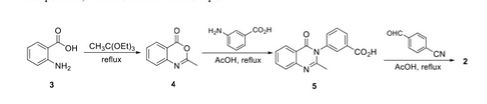
2-Methyl-4H-benzo[</| [l,3]oxazin-4-one (3). Anthranilic acid (20 g, 146 mmol) was dissolved in triethyl orthoacetate (45 mL, 245 mmol) and refluxed for 2 h. The reaction mixture was cooled on ice for 4 h to crystallize the intermediate. The resulting crystals were filtered and washed with hexanes to give 3 (17 g, 72% yield). lH NMR (500 MHz, CDC13) δ 2.47 (s, 3H), 7.50 (t, J= 7.38 Hz, 1H), 7.54 (d, J = 7.98 Hz, 1H), 7.80 (t, J= 7.18 Hz, 1H), 8.18 (d, J= 7.78 Hz, 1H). 13C NMR (126 MHz, CDCI3) δ 21.59, 1 16.84, 126.59, 128.42, 128.66, 136.77, 146.61, 159.89, 160.45. HRMS (m/z): [M + H]+, calcd for C9H8NO2, 162.0550; found , 162.0555.
2-Methyl-3-(3-carboxyphenyl)-quinazolin-4(3//)-one (4). Compound 3 (2 g, 12.4 mmol) and 3- aminophenol (1.7 g, 12.4 mmol) were suspended in glacial acetic acid (8 mL, 140 mmol), and dissolved upon heating. The reaction was refluxed for 5 h, at which point 5 mL water was added to the cooled reaction mixture. The resulting precipitate was filtered and washed with water, followed by cold ethanol and hexane to give 4 (3.19 g, 92% yield). lH (500 MHz, DMSO-d6) δ 2.87 (s, 3H), 7.52 (t, J= 7.38 Hz, 1H), 7.66-7.73 (m, 3H), 7.84 (t, J= 7.38 Hz, 1H), 8.01 (s, 1H), 8.09 (t, J= 7.58 Hz, 2H). 13C NMR (126 MHz, DMSO-de) δ 24.13, 120.48, 126.32, 126.47, 126.72, 129.52, 129.83, 130.01, 132.40, 133.07, 134.67, 138.18, 147.37, 154.13, 161.44, 166.58. HRMS (m/z): [M + H]+, calcd for C16H13N2O3 ,
281.0921 ; found, 281.0917.
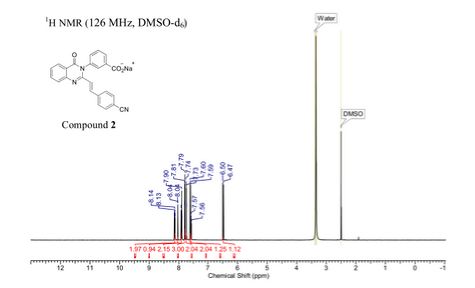
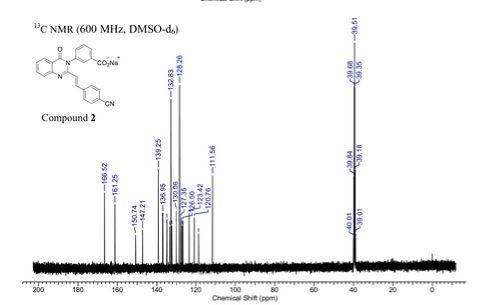
Sodium (£‘)-3-(3-carboxyphenyl)-2-(4-cyanostyryl)quinazolin-4(3H)-one (2). Compound 4 (1.0 g, 3.6 mmol) and 4-formylbenzonitrile (0.56 g, 4.3 mmol) were suspended in glacial acetic acid (5 mL, 87 mmol), a suspension that dissolved upon heating. The reaction was refluxed for 18 h and 5 mL water was added to the cooled reaction mixture. The resulting precipitate was filtered and washed with water, followed by cold ethanol and hexanes to afford the carboxylic acid (0.77g, 75% yield). HRMS (m/z): [M + H]+, calcd for C24H16N3O3, 394.1 186; found 394.1214. The carboxylic acid (0.45 g, 1.1 mmol) was dissolved in hot ethanol, to which sodium 2-ethylhexanoate (0.28 g, 1.7 mmol) was added. The reaction mixture was stirred on ice for 2 h. The precipitate was filtered and washed with cold ethanol. The product was obtained by dissolving the precipitate in about 5 mL of water and subsequent lyophilization of the solution to give 2 as the sodium salt (0.4 g, 85% yield).
ACS Editors’ Choice – This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes.
We recently reported on the discovery of a novel antibacterial (2) with a 4(3H)-quinazolinone core. This discovery was made by in silico screening of 1.2 million compounds for binding to a penicillin-binding protein and the subsequent demonstration of antibacterial activity againstStaphylococcus aureus. The first structure–activity relationship for this antibacterial scaffold is explored in this report with evaluation of 77 variants of the structural class. Eleven promising compounds were further evaluated for in vitro toxicity, pharmacokinetics, and efficacy in a mouse peritonitis model of infection, which led to the discovery of compound 27. This new quinazolinone has potent activity against methicillin-resistant (MRSA) strains, low clearance, oral bioavailability and shows efficacy in a mouse neutropenic thigh infection model.
NMR
INVENTORS
Renee Bouley
Renee Bouley selected to receive prestigious ACS Predoctoral Fellowship
Renee Bouley, a third year graduate student in the Department of Chemistry and Biochemistry, has been selected to receive a prestigious American Chemical Society (ACS) Division of Medicinal Chemistry Predoctoral Fellowship. Bouley is one of only four recipients chosen for the 2013-2014 cycle.
This award supports doctoral candidates working in the area of medicinal chemistry who have demonstrated superior achievements as graduate students and who show potential for future work as independent investigators. These fellowships have been awarded annually since 1991 and include one year stipend support and an invitation to present the fellow’s research results at a special awards session at the ACS National Meeting.
Bouley’s work, conducted under the advisement of Shahriar Mobashery, Navari Family Professor in Life Sciences, and Mayland Chang, Research Professor and Director of the Chemistry-Biochemistry-Biology Interface (CBBI) Program, centers around the discovery of a new class of antibiotics that are selective against staphylococcal species of bacteria, including hard-to-treat methicillin-resistant Staphylococcus aureus (MRSA). She has already identified a class of compounds that has in vitro activity against bacteria and demonstrated efficacy in mice. Bouley spent three months in 2012 in the laboratory of Prof. Juan Hermoso at Consejo Superior de Investigaciones Cientificas in Madrid, Spain, where she solved the crystal structure of the lead compound in complex with its target protein. Her studies have shown an unprecedented mechanism of action that opens opportunities for clinical resurrection of β-lactam antibiotics in combination with the new antibiotics. Bouley’s work during her fellowship tenure will explore structural analogs of these compounds with the goal of optimizing their potency in vivo and improving their drug-like properties.
Bouley is already the recipient of a National Institutes of Health Ruth L. Kirschstein National Research Service Award – CBBI (Chemistry-Biochemistry-Biology Interface) Program, a CBBI Research Internship Award, and an American Heart Association Predoctoral Fellowship (declined)………..https://www.linkedin.com/in/renee-bouley-43243215
Research Professor; Director, Chemistry-Biochemistry-Biology Interface (CBBI) Program
Office: 247 NSH
Phone: (574) 631-2965
Dr. Chang obtained B.S. degrees in biological sciences and chemistry from the University of Southern California, and a Ph.D. in chemistry from the University of Chicago. Subsequently, she conducted postdoctoral research at Columbia University as a National Institutes of Health postdoctoral fellow. She joined the faculty of the University of Notre Dame in 2003. Previously, Dr. Chang was Chief Operating Officer of University Research Network, Inc., Senior Scientist with Pharmacia Corporation, and Senior Chemist at Dow Chemical Company. She has characterized the ADME properties of numerous drugs, as well as prepared NDAs, INDs, Investigator’s Brochures, product development plans, and candidate drug evaluations.
Shahriar Mobashery
Shahriar Mobashery
Navari Professor at University of Notre Dame
The Mobashery research program integrates computation, biochemistry, molecular biology, and the organic synthesis of medically important molecules. Bringing together these different disciplines is required to produce both scientific and medical advances for very difficult, but critically important clinical problems.
Tildrakizumab is a monoclonal antibody designed for the treatment of immunologically mediated inflammatory disorders.[1]
Tildrakizumab was designed to block interleukin-23, a cytokine that plays an important role in managing the immune system and autoimmune disease. Originally developed by Schering-Plough, this drug is now part of Merck‘s clinical program, following that company’s acquisition of Schering-Plough.
Sun Pharmaceutical acquired worldwide rights to tildrakizumab for use in all human indications from Merck in exchange for an upfront payment of U.S. $80 million. Upon product approval, Sun Pharmaceutical will be responsible for regulatory activities, including subsequent submissions, pharmacovigilance, post approval studies, manufacturing and commercialization of the approved product. [2]
As of March 2014, the drug was in phase III clinical trials for plaque psoriasis. The two trials will enroll a total of nearly 2000 patients, and preliminary results are expected in June, 2015. [3][4]
Sun Pharma and Merck & Co. Inc. Enter into Licensing Agreement for Tildrakizumab, MK 3222
WHITEHOUSE STATION, N.J., and MUMBAI, India, Wednesday, September 17, 2014 (BUSINESS WIRE) – Merck & Co., Inc., (NYSE:MRK), known as MSD outside the United States and Canada, and Sun Pharmaceutical Industries Ltd. (Reuters: SUN.BO, Bloomberg: SUNP IN, NSE: SUNPHARMA, BSE: 524715) through their respective subsidiaries, today announced an exclusive worldwide licensing agreement for Merck’s investigational therapeutic antibody candidate, tildrakizumab, (MK-3222), which is currently being evaluated in Phase 3 registration trials for the treatment of chronic plaque psoriasis, a skin ailment.
Under terms of the agreement, Sun Pharma will acquire worldwide rights to tildrakizumab for use in all human indications from Merck in exchange for an upfront payment of U.S. $80 million. Merck will continue all clinical development and regulatory activities, which will be funded by Sun Pharma. Upon product approval, Sun Pharma will be responsible for regulatory activities, including subsequent submissions, pharmacovigilance, post approval studies, manufacturing and commercialization of the approved product. Merck is eligible to receive undisclosed payments associated with regulatory (including product approval) and sales milestones, as well as tiered royalties ranging from mid-single digit through teen percentage rates on sales.
“Consistent with our previously announced global initiative to sharpen our commercial and R&D focus, including prioritizing our late stage pipeline candidates, we are pleased to enter into this agreement with Sun Pharma to help realize the potential of tildrakizumab for patients with chronic plaque psoriasis,” said Iain D. Dukes, Ph.D., senior vice president, Business Development and Licensing, Merck Research Laboratories.
“Sun Pharma is very pleased to enter into this collaboration with Merck, a recognized leader in the field of inflammatory/immunology therapies, for this late-stage candidate for chronic plaque psoriasis,” said Kirti Ganorkar, senior vice president, Business Development, Sun Pharma. “This collaboration is a part of our strategy towards building our pipeline of innovative dermatology products in a market with strong growth potential.”
The transaction is subject to customary closing conditions, including the requirements under the Hart Scott-Rodino Antitrust Improvements Act.
About Tildrakizumab
Tildrakizumab is an investigational humanized, anti-IL-23p19 monoclonal antibody that binds specifically to IL-23p19 and is therefore designed to selectively block the cytokine IL-23. Human genetics suggest that inhibiting IL-23 is effective for treating inflammatory conditions. In clinical studies for the treatment of chronic plaque psoriasis, tildrakizumab demonstrates efficacy in blocking inflammation by blocking IL-23. Other potential indications, which may be evaluated in future, include psoriatic arthritis and Crohn’s Disease.
Today’s Merck is a global healthcare leader working to help the world be well. Merck is known as MSD outside the United States and Canada. Through our prescription medicines, vaccines, biologic therapies, and consumer care and animal health products, we work with customers and operate in more than 140 countries to deliver innovative health solutions. We also demonstrate our commitment to increasing access to healthcare through far-reaching policies, programs and partnerships. For more information, visit www.merck.com and connect with us on Twitter, Facebook and YouTube.
About Sun Pharma
Established in 1983, listed since 1994 and headquartered in India, Sun Pharmaceutical Industries Ltd. (Reuters: SUN.BO, Bloomberg: SUNP IN, NSE: SUNPHARMA, BSE: 524715) is an international specialty pharmaceutical company with over 75% sales from global markets. It manufactures and markets a large basket of pharmaceutical formulations as branded generics as well as generics in US, India and several other markets across the world. For the year ending March 2014, overall revenues were at US$2.7 billion, of which US contributed US$1.6 billion. In India, the company is a leader in niche therapy areas of psychiatry, neurology, cardiology, nephrology, gastroenterology, orthopedics and ophthalmology. The company has strong skills in product development, process chemistry, and manufacturing of complex dosage forms. More information about the company can be found at www.sunpharma.com.


























.bmp)
.bmp)
.gif)
































{kind=link}