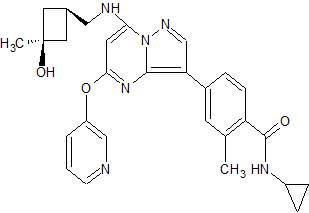
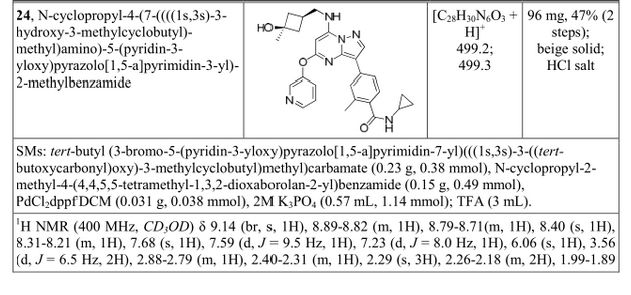
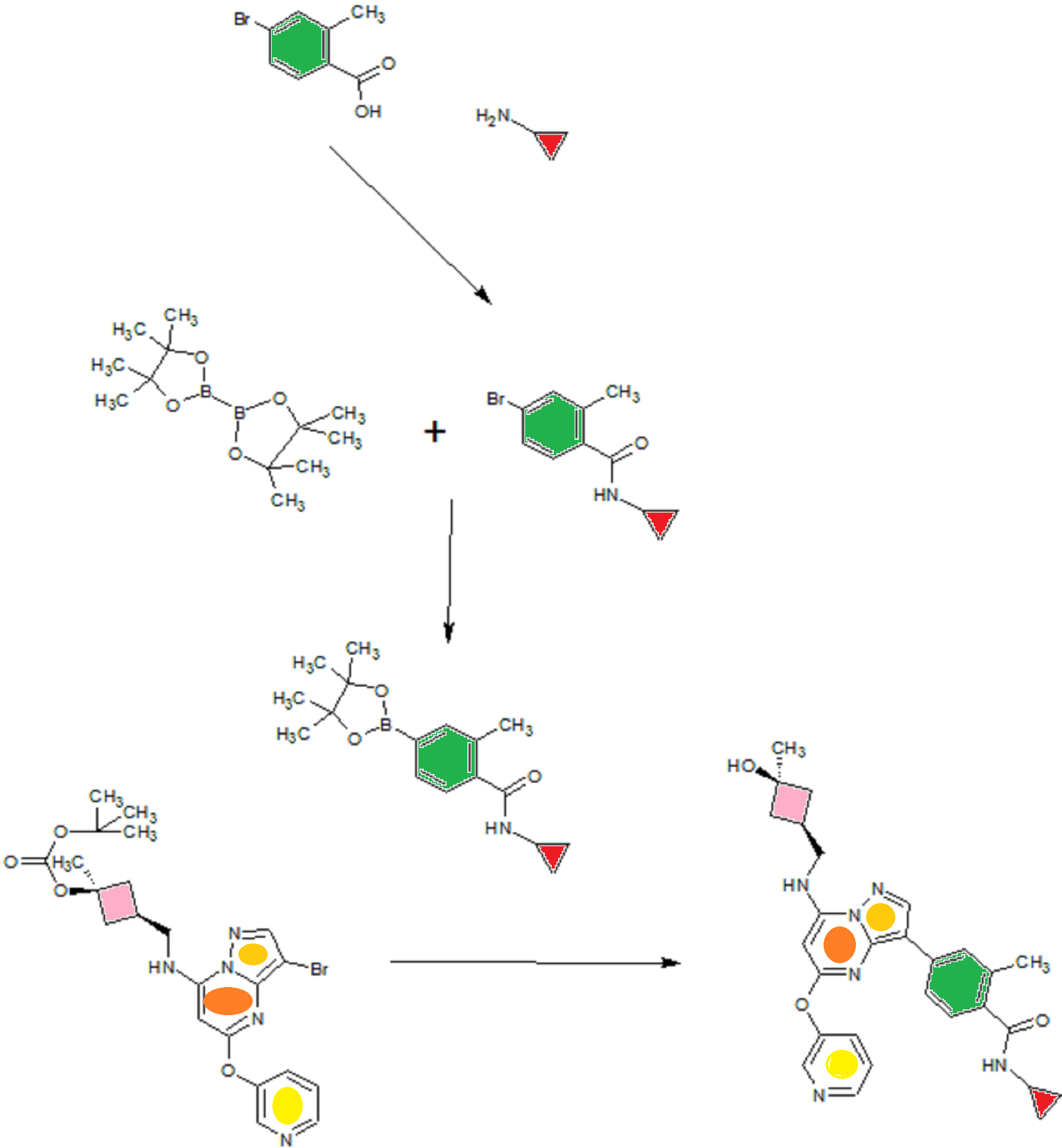
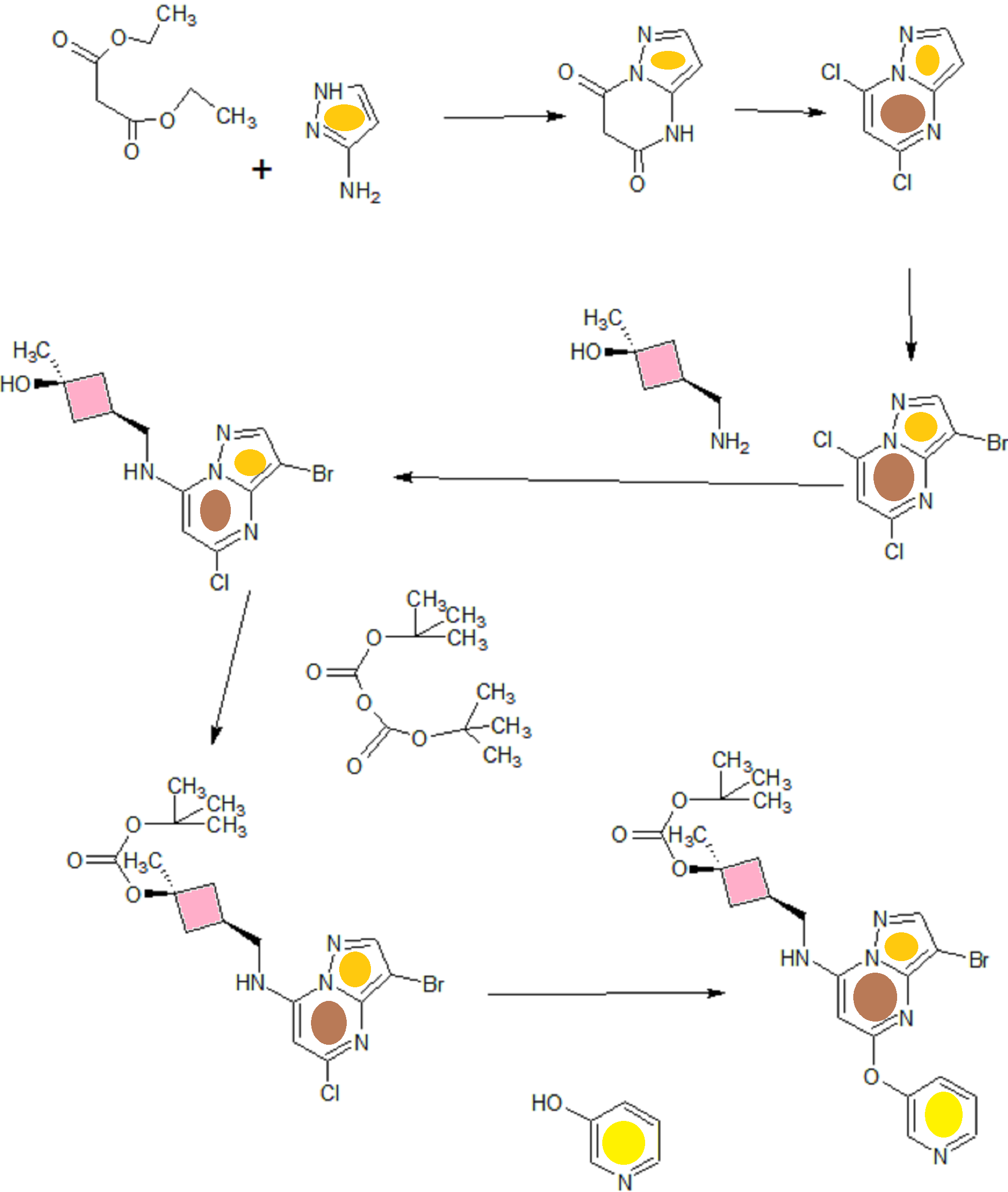
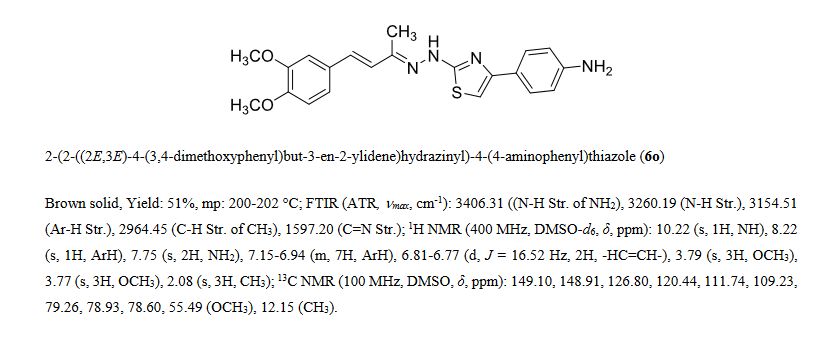
![[1860-5397-11-134-i11]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-11-134-i11.png?scale=2.0&max-width=1024&background=FFFFFF)
PIC CREDIT, The synthesis of active pharmaceutical ingredients (APIs) using continuous flow chemistry, and , Beilstein J. Org. Chem. 2015, 11, 1194–1219.,doi:10.3762/bjoc.11.134
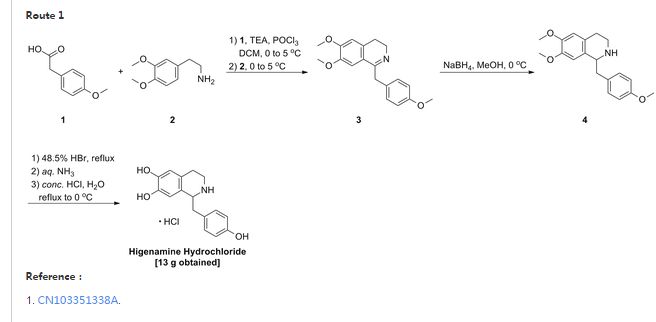
In 2012 researchers from AstraZeneca (Sweden) reported upon a scale-up campaign for their gastroesophageal reflux inhibitor programme. Specifically, flow chemical synthesis was used to efficiently and reliably provide sufficient quantities of the target compound AZD6906 (65), which had been prepared previously in batch. From these earlier batch studies concerns had been raised regarding exothermic reaction profiles as well as product instability which needed to be addressed when moving to larger scale synthesis. Flow was identified as a potential way of circumventing these specific problems and so was extensively investigated. The developed flow route [1 ] started with the reaction of methyl dichlorophosphine (66) and triethyl orthoacetate (67), which in batch could only be performed under careful addition of the reagent and external cooling using dry ice/acetone. Pleasingly, a simple flow setup in which the two streams of neat reagents were mixed in a PTFE T-piece maintained at 25 °C was found effective in order to prepare the desired adduct 68 in high yield and quality showcasing the benefits of superior heat dissipation whilst also safely handling the toxic and pyrophoric methyl dichlorophosphine reagent (Scheme 1).
As the subsequent Claisen condensation step was also known to generate a considerable exotherm, a similar flow setup was used in order to allow the reaction heat to dissipate. The superiority of the heat transfer process even allowed this step to be performed on kilogram quantities of both starting materials (68, 69) at a reactor temperature of 35 °C giving the desired product 72 within a residence time of only 90 seconds. Vital to the successful outcome was the efficient in situ generation of LDA from n-BuLi and diisopropylamine as well as the rapid quenching of the reaction mixture prior to collection of the crude product. Furthermore, flow processing allowed for the reaction of both substrates in a 1:1 ratio (rather than 2:1 as was required in batch) as the immediate quenching step prevented side reactions taking place under the strongly basic conditions. Having succeeded in safely preparing compound 72 on kilogram scale, the target compound 65 was then generated by global deprotection and subsequent recrystallisation where batch was reverted to as the conditions had been previously devised and worked well.

Dr Marcus Baumann
Postdoc
Marcus Baumann studied chemistry at the Philipps-University Marburg/Germany, from where he graduated in 2007. His studies involved a 6 month period as an Erasmus student at the Innovative Technology Centre at the University of Cambridge, UK (with Prof. Steven V. Ley and Dr Ian R. Baxendale), where he developed a new flow-based oxazole synthesis. He soon returned to Cambridge to pursue his doctoral studies with Prof. Steven V. Ley where he developed flow processes for Curtius rearrangements, different fluorination reactions as well as important heterocycle syntheses. Upon completion of his PhD in 2010 Marcus was awarded a Feodor Lynen Postdoctoral Fellowship (Alexander von Humboldt Foundation, Germany) allowing him to join the group of Prof. Larry E. Overman at UC Irvine, USA (2011-2013). During his time in California his research focused on the synthesis of naturally occurring terpenes as well as analogues of ETP-alkaloids. The latter project generated potent and selective histone methyltransferase inhibitors and opened routes towards new probes for epigenetic diseases which are currently under further investigation. In early 2013 Marcus returned to the UK and took up a postdoctoral position with Prof. Ian R. Baxendale at the University of Durham, where his interests concentrate on the development of flow and batch based strategies towards valuable compounds en route for regenerative medicines.

Prof. Ian R. Baxendale
(email at i.r.baxendale@durham.ac.uk)
Research Interests
My general areas of interest are: Organic synthesis (natural products, heterocyclic and medicinal chemistry), Organometallic chemistry, Catalyst design and application, Meso flow chemistry, Microfluidics, Microwave assisted synthesis, Solid supported reagents and scavengers, and facilitated reaction optimisation using Robotics and Automation.
My primary research direction is the synthesis of biologically potent molecules which encompasses the design, development and integration of new processing techniques for their preparation and solving challenges associated with the syntheses of new pharmaceutical and agrochemical compounds. In our work we utilise the latest synthesis tools and enabling technologies such as microwave reactors, solid supported reagents and scavengers, enzymes, membrane reactors and flow chemistry platforms to facilitate the bond making sequence and expedite the purification procedure. A central aspect of our investigations is our pioneering work on flow chemical synthesis and microreactor technology as a means of improving the speed, efficiency, and safety of various chemical transformations. As a part of these studies we are attempting to devise new chemical reactions that are not inherently feasible or would be problematic under standard laboratory conditions. It is our further challenge to enhance the automation associated with these reactor devices to impart a certain level of intelligence to their function so that repetitive wasteful actions currently performed by chemists can be delegated to a machine; for example, reagent screening or reaction optimisation. We use these technologies as tools to enhance our synthetic capabilities but strongly believe in not becoming slaves to any methodology or equipment.
For those interested in our research and wishing to find out more we invite you to visit our website at: http://www.dur.ac.uk/i.r.baxendale/

Early scale-up work of a promising reflux inhibitor AZD6906 is described. Two steps of an earlier route were adapted to be performed in continuous flow to avoid issues related to batch procedures, resulting in a robust method with reduced cost of goods and improved product quality. Toxic and reactive reagents and starting materials could be handled in a flow regime, thereby allowing safer and more convenient reaction optimization and production.
Gustafsson, T.; Sörensen, H.; Pontén, F. Org. Process Res. Dev. 2012, 16, 925–929. doi:10.1021/op200340c
Development of a Continuous Flow Scale-Up Approach of Reflux Inhibitor AZD6906

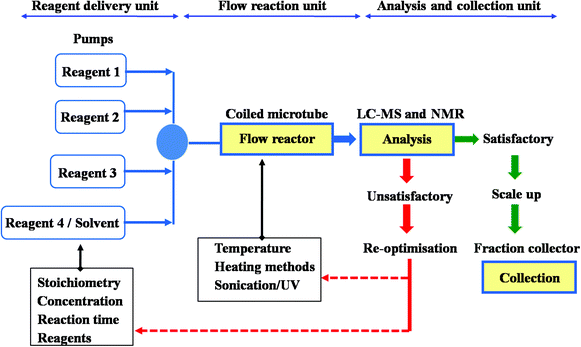
One benefit of flow reactors is improved control over reaction temperature, due to reduced reaction volume at a given time, higher surface area, and the movement of the reaction mixture. This is particularly helpful for very exothermic reactions, which often require cryogenic cooling to prevent runaway reactions – this type of cooling is very expensive and resource-intensive on a large scale. One such reaction is described in a recent paper from AstraZeneca, in which a phosphinate anion adds into a glycine derivative. The product of this reaction is an intermediate in the synthesis of a gastroesophageal reflux inhibitor drug candidate called AZD6906.
////Flow synthesis, AZD 6906
![[1860-5397-11-134-i8]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-11-134-i8.png?scale=2.0&max-width=1024&background=FFFFFF)















 Dr Friedrich Haefele, Vice President Fill & Finish Biopharma at Boehringer Ingelheim talked in his keynote speech about the revision of Annex 1 of the EU Guidelines to Good Manufacturing Practice. The first version dates already back to the year 1972. Dr Haefele stated that there had already been five revisions since this time but no fundamental review. This means the time has come to revise this fundamental document on the regulation of sterile manufacture in Europe.
Dr Friedrich Haefele, Vice President Fill & Finish Biopharma at Boehringer Ingelheim talked in his keynote speech about the revision of Annex 1 of the EU Guidelines to Good Manufacturing Practice. The first version dates already back to the year 1972. Dr Haefele stated that there had already been five revisions since this time but no fundamental review. This means the time has come to revise this fundamental document on the regulation of sterile manufacture in Europe. A further difference concerns the quality oversight. In Europe there is no requirement that the quality assurance (physically) must take place on-site during aseptic processes. But the Aseptic Guide requires a QC oversight and here, especially the media fill is mentioned. Dr Haefele invoked a harmonisation of the requirements, in order to strengthen the European philosophy, however. Quality assurance is a system and not an organisation. Mr. Haefele proposed a further change concerning the media fill in isolators. Here, interventions are carried out from the outside when carrying gloves. This means that they are “person-neutral”. The requirement that the qualification of interventions during the media fill has to be done person-specific should therefore be omitted for media fills in isolators.
A further difference concerns the quality oversight. In Europe there is no requirement that the quality assurance (physically) must take place on-site during aseptic processes. But the Aseptic Guide requires a QC oversight and here, especially the media fill is mentioned. Dr Haefele invoked a harmonisation of the requirements, in order to strengthen the European philosophy, however. Quality assurance is a system and not an organisation. Mr. Haefele proposed a further change concerning the media fill in isolators. Here, interventions are carried out from the outside when carrying gloves. This means that they are “person-neutral”. The requirement that the qualification of interventions during the media fill has to be done person-specific should therefore be omitted for media fills in isolators.