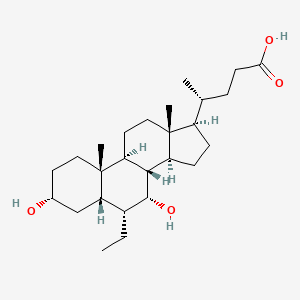
Obeticholic acid
Obeticholic acid; 6-ECDCA; INT-747; 459789-99-2; 6-Ethylchenodeoxycholic acid; 6alpha-Ethyl-chenodeoxycholic acid;
(4R)-4-[(3R,5S,6R,7R,8S,9S,10S,13R,14S,17R)-6-ethyl-3,7-dihydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl]pentanoic acid
|
| Molecular Formula: |
C26H44O4 |
| Molecular Weight: |
420.62516 g/mol |
NDA Filed
A farnesoid X receptor (FXR) agonist potentially for treatment of primary biliary cirrhosis and nonalcoholic steatohepatitis.

6-ECDCA; DSP-1747; INT-747
CAS No.459789-99-2
Obeticholic acid (abbreviated to OCA), is a semi-synthetic bile acid analogue which has the chemical structure 6α-ethyl-chenodeoxycholic acid. It has also been known as INT-747. It is undergoing development as a pharmaceutical agent for severalliver diseases and related disorders. Intercept Pharmaceuticals Inc. (NASDAQ symbol ICPT) hold the worldwide rights to develop OCA outside Japan and China, where it is licensed to Dainippon Sumitomo Pharma.[2]
REVIEW
INT-747(Obeticholic acid; 6-ECDCA) is a potent and selective FXR agonist(EC50=99 nM) endowed with anticholestatic activity. IC50 value: 99 nM(EC50) [1] Target: FXR agonist in vitro: The exposure of rat hepatocytes to 1 microM 6-ECDCA caused a 3- to 5-fold induction of small heterodimer partner (Shp) and bile salt export pump (bsep) mRNA and 70 to 80% reduction of cholesterol 7alpha-hydroxylase (cyp7a1), oxysterol 12beta-hydroxylase (cyp8b1), and Na(+)/taurocholate cotransporting peptide (ntcp) [2]. in vivo: In vivo administration of 6-ECDCA protects against cholestasis induced by E(2)17alpha [2]. high salt (HS) diet significantly increased systemic blood pressure. In addition, HS diet downregulated tissue DDAH expression while INT-747 protected the loss in DDAH expression and enhanced insulin sensitivity compared to vehicle controls [3]. Rats were gavaged with INT-747 or vehicle during 10 days after bile-duct ligation and then were assessed for changes in gut permeability, BTL, and tight-junction protein expression, immune cell recruitment, and cytokine expression in ileum, mesenteric lymph nodes, and spleen. After INT-747 treatment, natural killer cells and interferon-gamma expression markedly decreased, in association with normalized permeability selectively in ileum (up-regulated claudin-1 and occludin) and a significant reduction in BTL [4].
REFERENCES FOR ABOVE TEXT ONLY
| [1] |
Verbeke L, et al. The FXR Agonist Obeticholic Acid Prevents Gut Barrier Dysfunction and Bacterial Translocation in Cholestatic Rats. Am J Pathol. 2015 Feb;185(2):409-19. |
| [2] |
Ghebremariam YT, et al. FXR agonist INT-747 upregulates DDAH expression and enhances insulin sensitivity in high-salt fed Dahl rats. PLoS One. 2013 Apr 4;8(4):e60653. |
| [3] |
Fiorucci S, et al. Protective effects of 6-ethyl chenodeoxycholic acid, a farnesoid X receptor ligand, in estrogen-induced cholestasis. J Pharmacol Exp Ther. 2005 May;313(2):604-12. |
| [4] |
Pellicciari R, et al. 6alpha-ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J Med Chem. 2002 Aug 15;45(17):3569-72. |
Invention and development
The natural bile acid, chenodeoxycholic acid, was identified in 1999 as the most active physiological ligand for the farnesoid X receptor (FXR), which is involved in many physiological and pathological processes. A series of alkylated bile acid analogues were designed, studied and patented by Roberto Pellicciari and colleagues at the University of Perugia, with 6α-ethyl-chenodeoxycholic acid emerging as the most highly potent FXR agonist.[3] FXR-dependent processes in liver and intestine were proposed as therapeutic targets in human diseases.[4] Obeticholic acid is the first FXR agonist to be used in human drug studies.

Clinical studies
OCA is undergoing development in phase 2 and 3 studies for specific liver and gastrointestinal disorders.[5]
Primary biliary cirrhosis
Primary biliary cirrhosis (PBC) is an auto-immune, inflammatory liver disease which produces bile duct injury, fibrosis, cholestasisand eventual cirrhosis. It is much more common in women than men and can cause jaundice, itching (pruritus) and fatigue.Ursodeoxycholic acid therapy is beneficial, but the disease often progresses and may require liver transplantation.[6] Animal studies suggested that treatment with FXR agonists should be beneficial in cholestatic diseases such as PBC.[7] OCA at doses between 10 mg and 50 mg was shown to provide significant biochemical benefit, but pruritus was more frequent with higher doses.[8][9] The results of a randomized, double-blind phase 3 study of OCA, 5 mg or 10 mg, compared to placebo (POISE) were presented in April 2014, and showed that the drug met the trial’s primary endpoint of a significant reduction in serum alkaline phosphatase, abiomarker predictive of disease progression, liver transplantation or death.[10]

Nonalcoholic steatohepatitis (NASH)
Non-alcoholic steatohepatitis is a common cause of abnormal liver function with histological features of fatty liver, inflammation andfibrosis. It may progress to cirrhosis and is becoming an increasing indication for liver transplantation. It is increasing in prevalence. OCA is proposed to treat NASH.[11] A phase 2 trial published in 2013 showed that administration of OCA at 25 mg or 50 mg daily for 6 weeks reduced markers of liver inflammation and fibrosis and increased insulin sensitivity.[12]
The Farnesoid X Receptor Ligand Obeticholic Acid in Nonalcoholic Steatohepatitis Treatment (FLINT) trial, sponsored by NIDDK, was halted early in January 2014, after about half of the 283 subjects had completed the study, when a planned interim analysis showed that a) the primary endpoint had been met and b) lipid abnormalities were detected and arose safety concerns. Treatment with OCA (25 mg/day for 72 weeks) resulted in a highly statistically significant improvement in the primary histological endpoint, defined as a decrease in the NAFLD Activity Score of at least two points, with no worsening of fibrosis. 45% (50 of 110) of the treated group had this improvement compared with 21% (23 of 109) of the placebo-treated controls.[13] However concerns about longterm safety issues such as increased cholesterol and adverse cardiovascular events may warrant the concomitant use of statins in OCA-treated patients.[14]
Portal hypertension
Animal studies suggest that OCA improves intrahepatic vascular resistance and so may be of therapeutic benefit in portal hypertension.[15] An open label phase 2a clinical study is under way.
Bile acid diarrhea
Bile acid diarrhea (also called bile acid malabsorption) can be secondary to Crohn’s disease or be a primary condition. Reduced median levels of FGF19, an ileal hormone that regulates increased hepatic bile acid synthesis, have been found in this condition.[16] FGF19 is potently stimulated by bile acids and especially by OCA.[17] A proof of concept study of OCA (25 mg/d) has shown clinical and biochemical benefit.[18]

SYNTHESIS
CN 105541953
Take 10g of austempered cholic acid 89.6% purity crude (single hetero greater than 2%), 3 times its weight of acetone and added to their 20% by weight of triethylamine was added, was heated at reflux for 2h, cooled slowly to 10 ° C, the precipitated crystals were filtered to give Obey acid organic amine salt crystals.
Acidification [0020] The organic amine salts Obey acid crystals were dissolved with purified water after 10wt% by mass percentage to the PH value of 2.0 with dilute hydrochloric acid, filtered and dried to give purified Obey acid.
[0021] The purified Obey acid ethyl acetate dissolved by heating and then cooling to 20 ° C, the precipitated crystals were filtered and dried to obtain a purity of 98.7% recrystallization Obey acid (single hetero less than 0.1%), recovery was 84.5%.
PATENT
WO 2016045480
Obey acid (as shown in formula I) is a semi-synthetic chenodeoxycholic acid derivative, for the treatment of high blood pressure, the portal vein and liver diseases, including primary biliary cirrhosis, bile acid diarrhea, non-alcoholic steatohepatitis. Obey acid through activation of FXR receptors play a role, FXR is a nuclear receptor, is expressed mainly in the liver, intestine, kidney, and it can be adjusted with acids fat and carbohydrate metabolism related gene expression in bile, also regulate immune response. FXR activation can inhibit the synthesis of bile acids, bile acids prevent excessive accumulation of toxic reactions caused.
WO2002072598 debuted Obey acid preparation method (shown below), which in strong alkaline conditions to give compound VII by alkylation with ethyl iodide compound VI directly, through reducing compound VII prepared and carboxy deprotection Obey acid. However, due to direct alkylation with ethyl iodide poor selectivity and yield is too low, the synthesis process is difficult to achieve amplification synthesis.

Obey bile acid synthesis (WO2002072598)
WO2006122977 above synthesis process has been improved (see below), the process by the silicon compound IX into protected enol compound X, compound X and acetaldehyde after dehydration condensation to give compound Vb, after compound Vb in alkaline conditions under palladium on carbon hydrogenation to give compound XI, after a carbonyl compound XI reduction system Obey acid.
Obey bile acid synthesis (WO2006122977)
The synthetic process can be achieved, although the enlarged combined, however, the compound Vb produce large amounts of byproducts under strongly alkaline conditions palladium on carbon hydrogenation process for preparing high temperature and strong alkaline compound XI during this step leading to the separation of income a lower rate (about 60%), low yield of this step may be due to compound Vb and XI in unprotected hydroxy dehydration occurs under strongly basic (30% NaOH) and high temperature (95-105 ℃) conditions side effects caused.
synthesis of bile acids Obey,
Obey bile acid synthesis route is as follows:
PATENT
CN 105175473
According to Obey acid 6 was prepared in the form of C Patent Document W02013192097A1 reaction of Example 1, as follows:
The 3 a – hydroxy -6 a – ethyl-7-keto -5 P – 24-oic acid (. 86g, 205 4mmol), water (688mL) and 50% (w / w) hydrogen sodium hydroxide solution (56. 4mL) and the mixture of sodium borohydride (7. 77g, 205. 4mmol) in a mixture of 50% (w / w) sodium hydroxide solution (1.5 mL of) and water (20 mL) in 90 ° in C to 105 ° C reaction. Was heated with stirring under reflux for at least 3 hours, the reaction was completed, the reaction solution was cooled to 80 ° C. Between 30 ° C at 50 ° C of citric acid (320. 2g, anhydrous), a mixture of n-butyl acetate (860 mL of) and water (491mL) to ensure an acidic pH of the aqueous phase was separated. Evaporation of the organic phase was distilled to give the residue was diluted with n-butyl acetate, slowly cooled to 15 ° C to 20 ° C, centrifugation. The crude product was crystallized from n-butyl acetate. After Obey acid isolated by n-butyl acetate (43mL, 4 times), dried samples were dried at 80 ° C under vacuum. To give 67. 34g (77. 9%) crystalline form C Obey acid.
Patent
WO2016107575
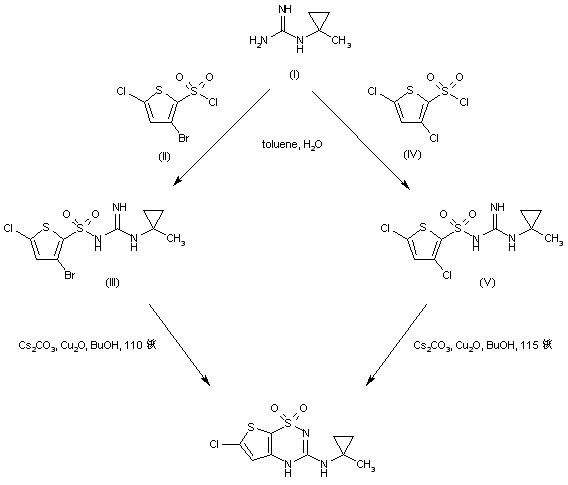
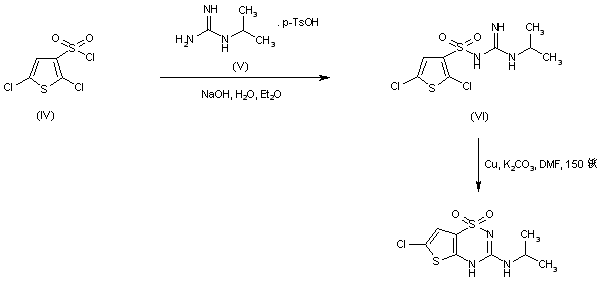
Obey acid (Obeticholic acid), developed by Intercept company farnesol X receptor (FXR) agonist, for the treatment of primary biliary cirrhosis (PBC) and nonalcoholic steatohepatitis (NASH). Obey acid is currently in Phase III clinical studies, the Phase III study shows Obey acid treatment of primary biliary cirrhosis have optimistic data, more than 20 years may become the future treatment of primary biliary cirrhosis A new method of choice, and Obey acid on improving nonalcoholic steatohepatitis important role. Obey acid, also known as 6-ethyl-chenodeoxycholic acid, and its structural formula is as follows:

(I)
Pharmaceutical polymorphs (drug polymorphism) refers to the presence of the drug has a different crystalline state of matter of two or more.Polymorphism in drugs is widespread. Different polymorphs of the same drug have significant differences in solubility, melting point, density, stability, etc., which to varying degrees, affect the stability of the drug, uniformity, bioavailability, efficacy and safety. Thus, the pharmaceutical research and development carried out comprehensive polymorph screening system to select the most suitable for the development of the crystalline form, it is one of the important research can not be ignored.
Currently, although there are reports of polymorph Obey acid but the reported crystal forms were prepared as amorphous Obey without intermediate acid product, for the purposes of purification of products, rather than as a product . For example WO2013192097 reported the first preparation Obey bile acid Form C, Form C will then be converted to amorphous Obey acid products. It has been reported as an intermediate product purified acid crystalline Obey obviously not suitable as a final product, since these polymorphic purity, and stability are poor, and there may be still other undesirable defects such WO2013192097 reported in Form C, which contains n-heptane.
Obey acid Form A preparation method:
The 216.3 mg Obey acid powder was added to 5.0 mL volume ratio of 1: ethyl acetate and n-heptane mixed solvent 9 to prepare a suspension. The suspension was put stirred for 48 hours at room temperature, filtered and the resulting cake was placed in a dry 25 ℃ vacuum oven overnight, the resulting solid was tested as Form A.
Type A crystal obtained in Example X-ray powder diffraction data are shown in Table 1 embodiment. Its XRPD pattern as shown in Figure 1, which is shown in Figure 2 DSC, TGA which is shown in Figure 3.
Also, by evaporative light scattering method Obey acid Form A purity of 99.54%.
The Form A for 90 days at 5 ℃ condition X-ray powder diffraction, FIG. 1 is substantially the same XRPD obtained in FIG.
[Table 1]
| 2theta |
d spacing |
Relative strength% |
| 4.95 |
17.87 |
55.14 |
| 5.26 |
16.79 |
51.49 |
| 6.22 |
14.20 |
100.00 |
| 7.22 |
12.24 |
39.00 |
| 7.66 |
11.54 |
49.18 |
| 8.90 |
9.93 |
42.81 |
| 9.36 |
9.44 |
38.30 |
| 9.95 |
8.89 |
43.47 |
| 10.45 |
8.46 |
30.64 |
| 10.97 |
8.06 |
20.12 |
| 12.51 |
7.08 |
99.90 |
| 14.89 |
5.95 |
32.18 |
| 15.69 |
5.65 |
62.49 |
| 15.96 |
5.55 |
37.49 |
| 16.47 |
5.38 |
49.44 |
| 17.23 |
5.15 |
30.92 |
| 17.95 |
4.94 |
28.12 |
| 18.87 |
4.70 |
36.32 |
| 19.56 |
4.54 |
31.45 |
| 20.57 |
4.32 |
21.24 |
| 21.34 |
4.16 |
15.50 |
| 22.70 |
3.92 |
7.95 |
| 23.46 |
3.79 |
6.10 |
| 24.72 |
3.60 |
6.51 |
| 25.11 |
3.55 |
7.62 |
References
- Gioiello, Antimo; Macchiarulo, Antonio; Carotti, Andrea; Filipponi, Paolo; Costantino, Gabriele; Rizzo, Giovanni; Adorini, Luciano; Pellicciari, Roberto (April 2011). “Extending SAR of bile acids as FXR ligands: Discovery of 23-N-(carbocinnamyloxy)-3α,7α-dihydroxy-6α-ethyl-24-nor-5β-cholan-23-amine”. Bioorganic & Medicinal Chemistry 19 (8): 2650–2658.doi:10.1016/j.bmc.2011.03.004.
- Wall Street Journal. “A $4 Billion Surprise for 45-Person Biotech”. Retrieved10 January 2014.
- Pellicciari R, Fiorucci S, Camaioni E, Clerici C, Costantino G, Maloney PR, Morelli A, Parks DJ, Willson TM (August 2002). “6alpha-ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity”. J. Med. Chem. 45(17): 3569–72. doi:10.1021/jm025529g. PMID 12166927.
- Rizzo G, Renga B, Mencarelli A, Pellicciari R, Fiorucci S (September 2005). “Role of FXR in regulating bile acid homeostasis and relevance for human diseases”. Curr. Drug Targets Immune Endocr. Metabol. Disord. 5 (3): 289–303. doi:10.2174/1568008054863781.PMID 16178789.
- “ClinicalTrials.gov”.
- Hirschfield GM, Gershwin ME (January 2013). “The immunobiology and pathophysiology of primary biliary cirrhosis”. Annu Rev Pathol 8: 303–30. doi:10.1146/annurev-pathol-020712-164014. PMID 23347352.
- Lindor, KD (May 2011). “Farnesoid X receptor agonists for primary biliary cirrhosis”.Current opinion in gastroenterology 27 (3): 285–8.doi:10.1097/MOG.0b013e32834452c8. PMID 21297469.
- Fiorucci S, Cipriani S, Mencarelli A, Baldelli F, Bifulco G, Zampella A (August 2011). “Farnesoid X receptor agonist for the treatment of liver and metabolic disorders: focus on 6-ethyl-CDCA”. Mini Rev Med Chem 11 (9): 753–62. doi:10.2174/138955711796355258.PMID 21707532.
- Hirschfield GM, Mason A, Luketic V, Lindor K, Gordon SC, Mayo M, Kowdley KV, Vincent C, Bodhenheimer HC, Parés A, Trauner M, Marschall HU, Adorini L, Sciacca C, Beecher-Jones T, Castelloe E, Böhm O, Shapiro D (2015). “Efficacy of obeticholic acid in patients with primary biliary cirrhosis and inadequate response to ursodeoxycholic acid”.Gastroenterology 148 (4): 751–61.e8. doi:10.1053/j.gastro.2014.12.005.PMID 25500425.
- Intercept Pharma. “Press release: Intercept Announces Positive Pivotal Phase 3 POISE Trial Results”. Retrieved March 27, 2014.
- Adorini L, Pruzanski M, Shapiro D (September 2012). “Farnesoid X receptor targeting to treat nonalcoholic steatohepatitis”. Drug Discov. Today 17 (17–18): 988–97.doi:10.1016/j.drudis.2012.05.012. PMID 22652341.
- Mudaliar S, Henry RR, Sanyal AJ, Morrow L, Marschall HU, Kipnes M, Adorini L, Sciacca CI, Clopton P, Castelloe E, Dillon P, Pruzanski M, Shapiro D (September 2013). “Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease”. Gastroenterology 145 (3): 574–82.e1.doi:10.1053/j.gastro.2013.05.042. PMID 23727264.
- Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, Chalasani N, Dasarathy S, Diehl AM, Hameed B, Kowdley KV, McCullough A, Terrault N, Clark JM, Tonascia J, Brunt EM, Kleiner DE, Doo E (2015). “Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial”. Lancet 385 (9972): 956–65.doi:10.1016/S0140-6736(14)61933-4. PMID 25468160.
- http://www.thestreet.com/story/12714549/1/intercept-pharma-government-scientists-spar-over-negative-safety-of-liver-drug-emails-show.html?puc=yahoo&cm_ven=YAHOO
- Verbeke L, Farre R, Trebicka J, Komuta M, Roskams T, Klein S, Vander Elst I, Windmolders P, Vanuytsel T, Nevens F, Laleman W (November 2013). “Obeticholic acid, a farnesoid-X receptor agonist, improves portal hypertension by two distinct pathways in cirrhotic rats”. Hepatology 59 (6): :2286–98. doi:10.1002/hep.26939. PMID 24259407.
- Walters JR, Tasleem AM, Omer OS, Brydon WG, Dew T, le Roux CW (November 2009). “A new mechanism for bile acid diarrhea: defective feedback inhibition of bile acid biosynthesis”. Clin. Gastroenterol. Hepatol. 7 (11): 1189–94.doi:10.1016/j.cgh.2009.04.024. PMID 19426836.
- Zhang JH, Nolan JD, Kennie SL, Johnston IM, Dew T, Dixon PH, Williamson C, Walters JR (May 2013). “Potent stimulation of fibroblast growth factor 19 expression in the human ileum by bile acids”. Am. J. Physiol. Gastrointest. Liver Physiol. 304 (10): G940–8.doi:10.1152/ajpgi.00398.2012. PMC 3652069. PMID 23518683.
- Walters JR, Johnston IM, Nolan JD, Vassie C, Pruzanski ME, Shapiro DA (January 2015). “The response of patients with bile acid diarrhoea to the farnesoid X receptor agonist obeticholic acid”. Aliment. Pharmacol. Ther. 41 (1): 54–64.doi:10.1111/apt.12999. PMID 25329562.
External links
| Patent ID |
Date |
Patent Title |
| US8546365 |
2013-10-01 |
Bile acid derivatives as FXR ligands for the prevention or treatment of FXR-mediated diseases or conditions |
| US8377916 |
2013-02-19 |
Steroids as agonists for FXR |
| US8058267 |
2011-11-15 |
STEROIDS AS AGONISTS FOR FXR |
| US7994352 |
2011-08-09 |
Process for Preparing 3a(Beta)-7a(Beta)-Dihydroxy-6a(Beta)-Alkyl-5Beta-Cholanic Acid |
| US7932244 |
2011-04-26 |
Bile acid derivatives as FXR ligands for the prevention or treatment of FXR-mediated diseases or conditions |
| US7786102 |
2010-08-31 |
Steroids as agonists for FXR |
| US2009062526 |
2009-03-05 |
NOVEL METHOD OF SYNTHESIZING ALKYLATED BILE ACID DERIVATIVES |
| US7138390 |
2006-11-21 |
Steroids as agonists for fxr |
| US2005107475 |
2005-05-19 |
Methods of using farnesoid x receptor (frx) agonists |
| Patent ID |
Date |
Patent Title |
| US2016074419 |
2016-03-17 |
Preparation and Uses of Obeticholic Acid |
| US2015359805 |
2015-12-17 |
Bile Acid Derivatives as FXR Ligands for the Prevention or Treatment of FXR-Mediated Diseases or Conditions |
| US2015166598 |
2015-06-18 |
Steroids as Agonists for FXR |
| US2014371190 |
2014-12-18 |
Farnesoid X receptor modulators |
| US2014186438 |
2014-07-03 |
COMPOSITIONS COMPRISING EPA AND OBETICHOLIC ACID AND METHODS OF USE THEREOF |
| US2014148428 |
2014-05-29 |
Treatment of Pulmonary Disease |
| US2014057886 |
2014-02-27 |
Bile Acid Derivatives as FXR Ligands for the Prevention or Treatment of FXR-Mediated Diseases or Conditions |
| US2014024631 |
2014-01-23 |
Steroids as Agonists for FXR |
| US2013345188 |
2013-12-26 |
Preparation and Uses of Obeticholic Acid |
| US8546365 |
2013-10-01 |
Bile acid derivatives as FXR ligands for the prevention or treatment of FXR-mediated diseases or conditions |

Obeticholic acid
|
| Systematic (IUPAC) name |
(3α,5β,6α,7α)-6-Ethyl-3,7-dihydroxycholan-24-oic acidOR
(4R)-4-[(3R,5S,6R,7R,8S,9S,10S,13R,14S,17R)-6-ethyl-3,7-dihydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl]pentanoic acid
|
| Clinical data |
Routes of
administration |
Oral |
| Legal status |
| Legal status |
|
| Identifiers |
| CAS Number |
459789-99-2 |
| ATC code |
A05AA04 (WHO) |
| PubChem |
CID 447715 |
| IUPHAR/BPS |
3435 |
| ChemSpider |
394730 |
| UNII |
0462Z4S4OZ |
| KEGG |
C15636 |
| ChEMBL |
CHEMBL566315 |
| Synonyms |
6α-ethyl-chenodeoxycholic acid; INT-747 |
| Chemical data |
| Formula |
C26H44O4 |
| Molar mass |
420.62516 g/mol |
/////////6-ECDCA, DSP-1747, INT-747, 459789-99-2, Obeticholic acid
CC[C@@H]1[C@@H]2C[C@@H](CC[C@@]2([C@H]3CC[C@]4([C@H]([C@@H]3[C@@H]1O)CC[C@@H]4[C@H](C)CCC(=O)O)C)C)O
CCC1C2CC(CCC2(C3CCC4(C(C3C1O)CCC4C(C)CCC(=O)O)C)C)O




































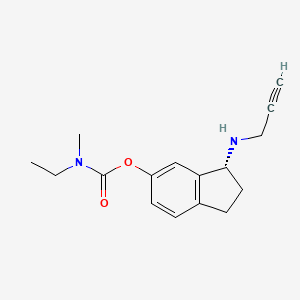
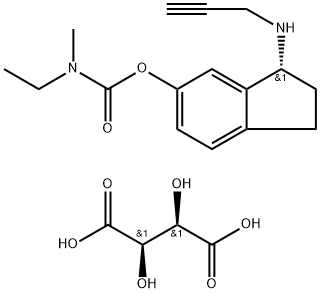




 (R)-3-(Prop-2-ynylamino)-2,3-dihydro-1H-inden-5-yl ethyl(methyl)carbamate
(R)-3-(Prop-2-ynylamino)-2,3-dihydro-1H-inden-5-yl ethyl(methyl)carbamate
 (R)-3-(Prop-2-ynylamino)-2,3-dihydro-1H-inden-5-yl ethyl(methyl)carbamate hemi((2R,3R)-2,3-dihydroxysuccinate)
(R)-3-(Prop-2-ynylamino)-2,3-dihydro-1H-inden-5-yl ethyl(methyl)carbamate hemi((2R,3R)-2,3-dihydroxysuccinate)