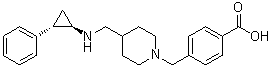
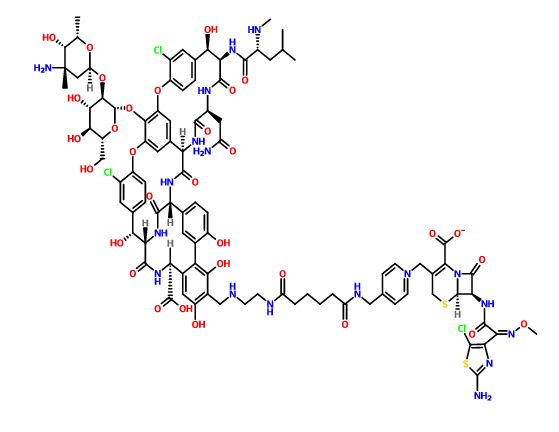
GSK-2881078
(R)-1-[1-(Methylsulfonyl)propan-2-yl]-4-(trifluoromethyl)-1H-indole-5-carbonitrile
(R)-1 -(1-(methylsulfonyl)propan-2-yl)-4-(trifluoromethyl)indoline-5-carbonitrile
Phase I
A selective androgen receptor modulator potentially for the treatment of cachexia.
| Inventors | Philip Stewart Turnbull, Rodolfo Cadilla |
| Applicant | Glaxosmithkline Intellectual Property (No.2) Limited |
![]()
| CAS Number | 1539314-06-1 |
| Chemical Name | GSK-2881078 |
| Synonyms | GSK-2881078 |
| Molecular Formula | C14H13NF3N2O2S |
| Formula Weight | 330.33 |
- Originator GlaxoSmithKline
- Mechanism of Action Selective androgen receptor modulators
- Phase I Cachexia
Most Recent Events
- 03 Sep 2015 GlaxoSmithKline initiates enrolment in a phase I trial for Cachexia (In volunteers) in USA (NCT02567773)
- 01 Mar 2015 GlaxoSmithKline completes a phase I trial in Cachexia (In volunteers) in USA (NCT02045940)
- 31 Jan 2014 Phase-I clinical trials in Cachexia (In volunteers) in USA (PO)
GSK2881078 is a selective androgen receptor modulator (SARM) that is being evaluated for effects on muscle growth and strength in subjects with muscle wasting to improve their physical function. Part A of this study will evaluate the safety, efficacy and pharmacokinetics of GSK2881078 in healthy, older men and post-menopausal women who will take daily dosing for 28 days and be followed for a total of 70 days. Part B of this study will characterize the effect of Cytochrome P450 3A4 (CYP3A4) inhibition on the GSK2881078 pharmacokinetics. Part B will only be conducted if safe and efficacious dose is identified in Part A. Part A will include healthy older males and post-menopausal females; and randomize approximately 60 subjects (about 15 per cohort [4 cohorts]) to complete approximately 48 (about 12 per cohort). Part B will enroll one cohort of approximately 15 healthy male subjects to complete approximately 12. The study duration will be approximately 115 days for Part A and 122 days for Part B.
Steroidal nuclear receptor (NR) ligands are known to play important roles in the health of both men and women. Testosterone (T) and dihydrotestosterone (DHT) are endogenous steroidal ligands for the androgen receptor (AR) that appear to play a role in every tissue type found in the mammalian body. During the development of the fetus, androgens play a role in sexual differentiation and development of male sexual organs. Further sexual development is mediated by androgens during puberty. Androgens play diverse roles in the adult, including stimulation and maintenance of male sexual accessory organs and maintenance of the musculoskeletal system. Cognitive function, sexuality, aggression, and mood are some of the behavioral aspects mediated by androgens. Androgens have a physiologic effect on the skin, bone, and skeletal muscle, as well as blood, lipids, and blood cells (Chang, C. and Whipple, G. Androgens and Androgen Receptors. Kluwer Academic Publishers: Boston, MA, 2002)
Many clinical studies with testosterone have demonstrated significant gains in muscle mass and function along with decreases in visceral fat. See, for example,
Bhasin (2003) S. J. Gerontol. A Biol. Sci. Med. Sci. 58:1002-8, and Ferrando, A. A. et al. (2002) Am. J. Phys. Endo. Met. 282: E601-E607. Androgen replacement therapy (ART) in men improves body composition parameters such as muscle mass, strength, and bone mineral density (see, for example, Asthana, S. et al. (2004) J. Ger, Series A: Biol. Sci. Med. Sci. 59: 461 -465). There is also evidence of improvement in less tangible parameters such as libido and mood. Andrologists and other specialists are increasingly using androgens for the treatment of the symptoms of androgen deficiency. ART, using T and its congeners, is available in transdermal, injectable, and oral dosage forms. All current treatment options have contraindications (e.g., prostate cancer) and side-effects, such as increased hematocrit, liver toxicity, and sleep apnoea. Side-effects from androgen therapy in women include: acne, hirsutism, and lowering of high-density lipoprotein (HDL) cholesterol levels, a notable side-effect also seen in men.
Agents that could selectively afford the benefits of androgens and greatly reduce the side-effect profile would be of great therapeutic value. Interestingly, certain NR ligands are known to exert their action in a tissue selective manner (see, for example, Smith et al. (2004) Endoc. Rev. 2545-71 ). This selectivity stems from the particular ability of these ligands to function as agonists in some tissues, while having no effect or even an antagonist effect in other tissues. The term “selective receptor modulator” (SRM) has been given to these molecules. A synthetic compound that binds to an intracellular receptor and mimics the effects of the native hormone is referred to as an agonist. A compound that inhibits the effect of the native hormone is called an antagonist. The term “modulators” refers to compounds that have a spectrum of activities ranging from full agonism to partial agonism to full antagonism.
SARMs (selective androgen receptor modulators) represent an emerging class of small molecule pharmacotherapeutics that have the potential to afford the important benefits of androgen therapy without the undesired side-effects. Many SARMs with demonstrated tissue-selective effects are currently in the early stages of development See, for example, Mohler, M. L. et al. (2009) J. Med. Chem. 52(12): 3597-617. One notable SARM molecule, Ostarine™, has recently completed phase I and II clinical studies. See, for example, Zilbermint, M. F. and Dobs, A. S. (2009) Future Oncology 5(8):121 1-20. Ostarine™ appears to increase total lean body mass and enhance functional performance. Because of their highly-selective anabolic properties and demonstrated androgenic-sparing activities, SARMs should be useful for the prevention and/or treatment of many diseases in both men and women, including, but not limited to sarcopenia, cachexias (including those associated with cancer, heart failure, chronic obstructive pulmonary disease (COPD), and end stage renal disease (ESRD), urinary incontinence, osteoporosis, frailty, dry eye and other conditions associated with aging or androgen deficiency. See, for example, Ho et al. (2004) Curr Opin Obstet Gynecol. 16:405-9; Albaaj et al. (2006) Postgrad Med J 82:693-6; Caminti et al. (2009) J Am Coll Cardiol. 54(10):919-27; lellamo et al. (2010) J Am Coll Cardiol. 56(16): 1310-6; Svartberg (2010) Curr Opin Endocrinol Diabetes Obes. 17(3):257-61 , and Mammadov et al. (201 1 ) Int Urol Nephrol 43:1003-8. SARMS also show promise for use in promoting muscle regeneration and repair (see, for example, Serra et al. (Epub 2012 Apr 12)
doi:10.1093/Gerona/gls083),in the areas of hormonal male contraception and benign prostatic hyperplasia (BPH), and in wound healing (see, for example, Demling (2009) ePIasty 9:e9).
Preclinical studies and emerging clinical data demonstrate the therapeutic potential of SARMs to address the unmet medical needs of many patients. The demonstrated advantages of this class of compounds in comparison with steroidal androgens (e.g. , tissue-selective activity, oral administration, AR selectivity, and lack of androgenic effect) position SARMs for a bright future of therapeutic applications.
Although amorphous forms of SARMs may be developed for some uses, compounds having high crystallinity are generally preferred for pharmaceutical use due to their improved solubility and stability. Accordingly, there remains a need in the art for crystalline form of SARMs for therapeutic use.

Patent
EXAMPLES
Example 1 – Synthesis of (R)-1 -(1 -(methylsulfonyl)propan-2-yl)-4-(trifluoromethyl)- -indole-5-carbonitrile

(R)-1 -(1-(methylsulfonyl)propan-2-yl)^-(trifluoromethyl)-1 H-indole-5-carbonitrile
Method 1 :
![]()
A. (R)-1 -(Methylthio)propan-2 -amine
Step 1
To a solution of commercially available (R)-2-aminopropan-1 -ol (5 g, 66.6 mmol) in MeCN (20 mL), in an ice bath, is added very slowly, dropwise, chlorosulfonic acid (4.46 mL, 66.6 mmol) (very exothermic). The reaction mixture is kept in the cold bath for ~10 min, and then at rt for ~ 30 min. After stirring for another ~ 10 minutes, the solids are collected by filtration, washed sequentially with MeCN (40 mL) and hexanes (100 mL), and dried by air suction for ~ 40 min. to produce the intermediate ((R)-2-aminopropyl hydrogen sulfate.
Step 2:
To a solution of sodium thiomethoxide (5.60 g, 80 mmol) in water (20 mL) is added solid NaOH (2.66 g, 66.6 mmol) in portions over ~ 10 min. Then the intermediate from step 1 is added as a solid over ~ 5 min. The mixture is then heated at 90 °C for ~10 h. The reaction mixture is biphasic. Upon cooling, MTBE (20 mL) is added, and the organic phase (brownish color) is separated. The aqueous phase is extracted with MTBE (2 x 20 mL). The original organic phase is washed with 1 N NaOH (15 mL). The basic aqueous phase is re-extracted with MTBE (2 x 20 mL). All the ether phases are combined, dried over Na2S04, filtered, and concentrated (carefully, since the product is volatile) to afford the crude product as a light yellow oil.
Method 2
![]()
(R)-1-(methylthio)propan-2 -amine hydrochloride

A. (R)-2-((tert-Butoxycarbonyl)amino)propyl methanesulfonate
Step 1
Commercially available (R)-2-aminopropan-1 -ol (135 g, 1797 mmol) is dissolved in MeOH 1350 mL). The solution is cooled to 5°C with an icebath, then Boc20 (392 g, 1797 mmol) is added as a solution in MeOH (1000 mL). The reaction temperature is kept below 10°C. After the addition, the cooling bath is removed, and the mixture is stirred for 3 h. The MeOH is removed under vacuum (rotavap bath: 50°C). This material is used as is for the next step.
Step 2
The residue is dissolved in CH2CI2 (1200 mL) and NEt3 (378 mL, 2717 mmol) is added, then the mixture is cooled on an ice bath. Next, MsCI (166.5 mL, 2152 mmol) is added over ~2 h, while keeping the reaction temperature below 15°C. The mixture is stirred in an icebath for 1 h then the bath was removed. The mixture is stirred for 3 d, then washed with a 10% NaOH solution (500 mL 3 x), then with water. The organic phase is dried with MgS04, filtered, then stripped off (rota, 50°C waterbath. The impure residue is dissolved in a mix of 500mL EtOAc (500 mL) and MTBE (500 mL) and then extracted with water to remove all water-soluble salts. The organic phase is dried with MgS04, filtered, then stripped off to afford a white solid residue.

B. (R)-tert-Butyl (1 -(methylthio)propan-2-yl)carbamate
NaSMe (30 g, 428 mmol) is stirred with DMF (200 mL) to afford a suspension. Next, (R)-2-((tertbutoxycarbonyl)amino)propyl methanesulfonate (97 g, 383 mmol) is added portionwise while the temperature is kept below 45°C (exothermic). After the addition, the mixture is stirred for 2 h, then toluene (100 mL) is added. The mixture is washed with water (500 mL, 4 x), then dried with MgS04, and filtered. The filtrate is stripped off (rotavap) to a pale yellow oil.
![]()
C. (R)-1 -(Methylthio)propan-2 -amine hydrochloride
Acetyl chloride (150 mL,) is added to a stirred solution of MeOH (600 mL) cooled with an icebath. The mixture is stirred for 30 min in an icebath, then added to (R)-tert-butyl (1 -(methylthio)propan-2-yl)carbamate (78 g, 380 mmol). The mixture is stirred at rt for 2 h, (C02, (CH3)2C=CI-l2 evolution) and then stripped off to a white solid.

D. 4-Fluoro-3-iodo-2-(trifluoromethyl)benzonitrile
To a freshly prepared solution of LDA (1 19 mmol) in anhyd THF (250 mL) at -45°C is added a solution of commercially available 4-fluoro-2-(trifluoromethyl)benzonitrile (21 .5 g, 1 14 mmol) in THF (30 mL), dropwise at a rate such that the internal temperature remained < -40°C (became dark brown during addition). The mixture is stirred 30 min at -45°C, cooled to -70°C and iodine (31 .7 g, 125 mmol) is added in one portion (-70°C→ -52°C). The mixture is stirred for 1 h, removed from the cooling bath and quenched by addition of 10% Na2S203 (ca. 250 mL) and 1 N HCI (ca. 125 mL). The mixture is extracted with EtOAc (x3). Combined organics are washed (water, brine), dried over Na2S04 and concentrated in vacuo. The residue is purified by low pressure liquid chromatography (silica gel, EtOAc / hexanes, gradient elution) followed by
recrystallization from heptane (30 mL), twice, affording 4-fluoro-3-iodo-2-(trifluoromethyl)benzonitrile (15.79 g, 50.1 mmol, 44.1 % yield) as a pale yellow solid.

E. 4-Fluoro-2-(trifluoromethyl)-3-((trimethylsilyl)ethynyl)benzonitrile
A 20 mL vial is charged with 4-fluoro-3-iodo-2-(trifluoromethyl)benzonitrile,(0.315 g, 1 .00 mmol), Pd(PPh3)2CI2 (0.014 g, 0.020 mmol) and Cul (0.0076 g, 0.040 mmol), and sealed with a rubber septum. Anhyd PhMe (5 mL) and DIPA (0.210 mL, 1 .500 mmol) are added via syringe and the mixture is degassed 10 min by sparging with N2while immersed in an ultrasonic bath. Ethynyltrimethylsilane (0.155 mL, 1 .100 mmol) is added dropwise via syringe and the septum is replaced by a PTFE-faced crimp top. The mixture is stirred in a heating block at 60°C. Upon cooling the mixture is diluted with EtOAc and filtered through Celite. The filtrate is washed (satd NH4CI, water, brine), dried over Na2S04 and concentrated in vacuo. The residue is purified by low pressure liquid chromatography (silica gel, EtOAc / hexanes, gradient elution) affording 4-fluoro-2-(trifluoromethyl)-3-((trimethylsilyl)ethynyl)benzonitrile .

F. (R)-1 -(1 -(methylthio)propan-2-yl)-4-(trifluoromethyl)-1 H-indole-5-carbonitrile
A mixture of 4-fluoro-2-(trifluoromethyl)-3-((trimethylsilyl)ethynyl)benzonitrile (1 .16 g, 4.07 mmol), (R)-1 -(methylthio)propan-2-amine (0.599 g, 5.69 mmol) and DIEA (1 .42 mL, 8.13 mmol) in DMSO (7 mL) is heated (sealed tube) at 100°C for 50 min. Upon cooling, the reaction mixture is diluted with EtOAc (50 mL) and washed with water (30 mL). The organic phase is washed with water and brine, dried over Na2S04, filtered and concentrated to give the intermediate aniline. This intermediate is dissolved in NMP (7 mL), treated with KOtBu (1 M in THF) (5.69 mL, 5.60 mmol) and heated at 50°C. The reaction is monitored by LCMS, and deemed complete after 40 min. Upon cooling, the reaction mixture is diluted with EtOAc (40 mL) and washed with water (30 mL). The organic phase is washed with more water and brine, dried over Na2S04, filtered and concentrated. The residue is chromatographed over silica gel using a 5-40% EtOAc-hexane gradient to give the thioether intermediate:

G. (R)-1 -(1-(methylsulfonyl)propan-2-yl)-4-(trifluoromethyl)-1 H-indole-5-carbonitrile
To an ice-cold solution of (R)-1 -(1 -(methylthio)propan-2-yl)-4-(trifluoromethyl)-1 H-indole-5-carbonitrile (0.560 g, 1.88 mmol) in MeOH (10 mL) is added a solution of Oxone (4.04 g, 6.57 mmol) in water (10 mL). After 50 min, the reaction mixture is diluted with water (30 mL) and extracted with EtOAc (50 mL). The organic phase is washed with brine, dried over Na2S04, filtered and concentrated. The residue is chromatographed over silica gel using 100% CH2CI2 to give (R)-1-(1 -(methylsulfonyl)propan-2-yl)-4-(trifluoromethyl)-l H-indole-5-carbonitrile as a white foam that is crystallized from
CH2CI2/hexanes to afford a white solid.
Example 2- Preparation of crystalline form 1 of (R)-1 -(1-(methylsulfonyl)propan-2-yl)-4-(trifluoromethyl)indoline-5-carbonitrile
(R)-1 -(1-(methylsulfonyl)propan-2-yl)-4-(trifluoromethyl)indoline-5-carbonitrile (1 .74kg, 1wt) was dissolved in ethyl acetate (12.0 Kg, 6.9 wt) at 20-30°C. The solution was transferred into a clean reaction vessel via an in-line cartridge filter. The solution was concentrated to ~3.0-5.0 volumes under reduced pressure, keeping the temperature below 50°C. The solution was cooled to 20-30°C, and n-heptane (23.0 Kg, 13.2 wt) was added slowly over ~1 hour. The solution was stirred 1 -2 hrs at 20-30°C, heated to 50-55°C for 2-3 hours, cooled back to 20-30°C and stirred for 1 -2 hours. The slurry was sampled and analyzed by XRPD. The solid was collected by filtration, washed with n-heptane (1 .4 Kg, 0.8 wt), and dried in vacuo at 40-50 °C to provide crystalline
(R)-1 -(1-(methylsulfonyl)propan-2-yl)-4-(trifluoromethyl)indoline-5-carbonitrile (1 .54 Kg, Form 1 ; 88.5 % yield, 99.5% purity) as a slightly colored solid.
Example 3- Preparation of crystalline form 2 of (R)-1 -(1-(methylsulfonyl)propan-2-yl)-4-(trifluoromethyl)indoline-5-carbonitrile
Crude (R)-1 -(1 -(methylsulfonyl)propan-2-yl)-4-(trifluoromethyl)indoline-5-carbonitrile (1 .54 g [theoretical], 1 wt) was dissolved in dichloromethane (5mL, 3.25 vol) and loaded onto a 12-g ISCO column (Si02). The column was eluted with DCM (-500 mL, 325 vol) and the product-containing fractions were combined and concentrated in vacuo. The resulting residue was triturated in n-heptane. The solid was collected by filtration, air-dried, and placed under high vacuum for 3 h to provide GSK2881078A (1 .009 g, Form 2; 65.1 % yield, 100% AUC HPLC-UV) as a white solid.

PATENT
https://www.google.com/patents/WO2014013309A1?cl=en22
Example 26

1-(1-(Methylsulfonyl)propan-2-yl)-4-(trifiuoromethyl)-1H-indole-5-carbonitrile Synthesized in a manner similar to Example 9 using 1-(1-(methylthio)propan-2-yl)-4-(trifluoromethyl)-1 H-indole-5-carbonitrile (Example 25): MS (ESI): m/z 331 (MH+).
Example 27

(R)-1 -(1 -(Methylsulfonyl)propan-2-yl)-4-(trifluoromethyl)-1 H-indole-5-carbonitrile
![]()
A. (R)-1-(Methylthio)propan-2-amine
Step l
To a solution of commercially available (R)-2-aminopropan-1-ol (5 g, 66.6 mmol) in MeCN (20 mL), in an ice bath, was added very slowly, dropwise, chlorosulfonic acid (4.46 mL, 66.6 mmol) (very exothermic). A gummy beige precipitate formed. The reaction mixture was kept in the cold bath for -10 min, and then at rt for ~ 30 min. The reaction mixture was scratched with a spatula to try to solidify the gummy precipitate. After a few minutes, a beige solid formed. After stirring for another ~ 10 minutes, the solids were collected by filtration, washed sequentially with MeCN (40 mL) and hexanes (100 mL), and dried by air suction for ~ 40 min. The intermediate ((R)-2-aminopropyl hydrogen sulfate, weighed 0.46 g (~ 96% yield).
Step 2:
To a solution of sodium thiomethoxide (5.60 g, 80 mmol) in water (20 mL) was added solid NaOH (2.66 g, 66.6 mmol) in portions over – 10 min. Then the intermediate from step 1 was added as a solid over ~ 5 min. The mixture was then heated at 90 °C for -10 h. The reaction mixture was biphasic. Upon cooling, MTBE (20 mL) was added, and the organic phase (brownish color) was separated. The aqueous phase was extracted with MTBE (2 x 20 mL). The original organic phase is washed with 1 NaOH (15 mL) (this removes most of the color). The basic aqueous phase was re-extracted with MTBE (2 x 20 mL). All the ether phases are combined, dried over Na2S04, filtered, and
concentrated (carefully, since the product is volatile) to afford the crude product as a light yellow oil: 1H NMR (400 MHz, DMSO-cf6) δ 2.91-2.87 (m, 1 H), 2.43-2.31 (m, 2 H), 2.04 (s, 3 H), 1.50 (bs, 2 H), 1.01 (d, J = 6.3 Hz, 3 H).
Alternative synthesis of example 27A:
![]()
(R)-1 -(Methylthio)propan-2 -amine hydrochloride

A. (R)-2-((tert-Butoxycarbonyl)amino)propyl methanesulfonate
Step 1
Commercially available (R)-2-aminopropan-1-ol (135 g, 1797 mmol) was dissolved in MeOH 1350 mL). The solution was cooled to 5°C with an icebath, then Boc20 (392 g, 1797 mmol) was added as a solution in MeOH (1000 mL). The reaction temperature was kept below 10°C. After the addition, the cooling bath was removed, and the mixture was stirred for 3 h. The MeOH was removed under vacuum (rotavap bath: 50°C). The resulting residue was a colorless oil that solidified overnight to a white solid. This material was used as is for the next step.
Step 2
The residue was dissolved in CH2CI2 (1200 mL) and NEt3 (378 mL, 2717 mmol) was added, then the mixture was cooled on an ice bath. Next, MsCI (166.5 mL, 2152 mmol) was added over ~2 h, while keeping the reaction temperature below 15°C. The mixture was stirred in an icebath for 1 h then the bath was removed. The mixture was stirred for 3 d, then washed with a 10% NaOH solution (500 mL 3 x), then with water. The organic phase was dried with MgS0 , filtered, then stripped off (rota, 50°C waterbath. The impure residue was dissolved in a mix of 500mL EtOAc (500 mL) and MTBE (500 mL) and then, extracted with water to remove all water-soluble salts.The organic phase was dried with MgS04, filtered, then stripped off to afford a white solid residue: 1H NMR (400 MHz, DMSO-ds) δ 6.94-6.92 (m, 1 H), 4.02 (d, J = 5.8 Hz, 2 H), 3.78-3.71 (m, 1 H), 3.16 (s, 3 H), 1.38 (s, 9 H), 1.06 (d, J = 6.8 Hz, 3 H).

B. (R)-tert-Butyl (1-(methylthio)propan-2-yl)carbamate
NaSMe (30 g, 428 mmol) was stirred with DMF (200 mL) to afford a suspension. Next, (R)-2-((tertbutoxycarbonyl)amino)propyl methanesulfonate (97 g, 383 mmol) was added
portionwise while the temperature was kept below 45°C (exothermic).. After the addition, the mixture was stirred for 2 h, then toluene (100 ml_) was added. The mixture was washed with water (500 ml_, 4 x), then dried with MgS04, and filtered. The filtrate was stripped off (rotavap) to a pale yellow oil: 1H NMR (400 MHz, DMSO-d6) δ 6.77-6.75 (m, 1 H), 3.60-3.54 (m, 1 H), 2.54-2.50 (m, 1 H), 2.43-2.38 (m, 1 H), 2.05 (s, 3 H), 1.38 (s, 9 H), 1.08 (d, J = 7.8 Hz, 3 H).
![]()
C. (R)-1-(Methylthio)propan-2-amine hydrochloride
Acetyl chloride (150 mL,) was added to a stirred solution of MeOH (600 mL) cooled with an icebath. The mixture was stirred for 30 min in an icebath, then added to (R)-tert-butyl (1-(methylthio)propan-2-yl)carbamate (78 g, 380 mmol). The mixture was stirred at rt for 2 h, (C02, (CH3)2C=CH2 evolution) and then stripped off to a white solid: 1H NMR (400 MHz, DMSO-d6) δ 8.22 (bs, 3 H), 3.36-3.29 (m, 1 H), 2.80-2.75 (m, 1 H), 2.64-2.59 (m, 1 H (d, J = 6.6 Hz, 3 H).

D. (R)-1 -(1 -(Methylthio)propan-2-yl)-4-(trif luoromethy l)-1 H-indole-5-carbonitrile
A mixture of 4-fluoro-2-(trifluoromethyl)-3-((trimethylsilyl)ethynyl)benzonitrile (Example 21 D,1.16 g, 4.07 mmol), (R)-1-(methylthio)propan-2-amine (0.599 g, 5.69 mmol) and DIEA (1.42 mL, 8.13 mmol) in DMSO (7 mL) was heated (sealed tube) at 100°C for 50 min. Upon cooling, the reaction mixture was diluted with EtOAc (50 mL) and washed with water (30 mL). The organic phase was washed with water and brine, dried over Na2S04, filtered and concentrated to give the intermediate aniline. This intermediate was dissolved in NMP (7 mL), treated with KOtBu (1 M in THF) (5.69 mL, 5.60 mmol) and heated at 50°C. The reaction was monitored by LCMS, and deemed complete after 40 min. Upon cooling, the reaction mixture was diluted with EtOAc (40 mL) and washed with water (30 mL). The organic phase was washed with more water and brine, dried over Na2S04, filtered and concentrated. The residue was chromatographed over silica
gel using a 5-40% EtOAc-hexane gradient to give the thioether intermediate: MS (ESI):

E. (R)-1-(1-(Methylsulfonyl)propan-2-yl)-4-(trifluoromethyl)-1H-indole-5-carbonitrile
To an ice-cold solution of (R)-1-(1-(methylthio)propan-2-yl)-4-(trifluoromethyl)-1 H-indole-5-carbonitrile (0.560 g, 1.88 mmol) in MeOH (10 mL) was added a solution of Oxone (4.04 g, 6.57 mmol) in water (10 mL). After 50 min, the reaction mixture was diluted with water (30 mL) and extracted with EtOAc (50 mL). The organic phase was washed with brine, dried over Na2S04, filtered and concentrated. The residue was chromatographed over silica gel using 100% CH2CI2 to give (R)-1-(1-(methylsulfonyl)propan-2-yl)-4-(trifluoromethyl)-l H-indole-5-carbonitrile as a white foam that was crystallized from CH2CI2/hexanes to afford a white solid (0.508 g, 79% yield): 1H NMR (400 MHz, DMSO-d6) δ 8.17 (d, J = 8.6 Hz, 1 H), 8.12 (d, J = 3.5 Hz, 1 H), 7.81 (d, J – 8.5 Hz, 1 H), 6.87-6.84 (m, 1 H), 5.43-5.35 (m, 1 H), 4.01 (dd, J = 14.8, 8.6 Hz, 1 H), 3.83 (dd, J = 14.8, 4.9 Hz, 1 H), 2.77 (s, 3 H), 1.59 (d, J = 6.8 Hz, 3 H); MS (ESI): m/z 331 (M+H).


Philip Turnbull
Director of Chemistry
https://www.linkedin.com/in/philip-turnbull-21266a8
Experience

Director of Chemistry
Receptos, a wholly-owned subsidiary of Celgene
– Present (1 year 1 month)Greater San Diego Area

Section Head
GSK
– (3 years 1 month)RTP
Group Manager
GlaxoSmithKline
– (4 years 1 month)RTP
Investigator
GSK
– (4 years 11 months)RTP
Research Associate
Biophysica Foundation
– (3 years 8 months)La Jolla, Ca
Education

////////GSK-2881078, 1539314-06-1, Phase 1, clinical trials, Cachexia , GlaxoSmithKline