
ROXADUSTAT
ASP1517; ASP 1517; ASP-1517; FG-4592; FG 4592; FG4592; Roxadustat.
CAS 808118-40-3
Chemical Formula: C19H16N2O5
Exact Mass: 352.10592
Fibrogen, Inc.
THERAPEUTIC CLAIM, Treatment of anemia
CHEMICAL NAMES
(4-hydroxy-1-methyl-7-phenoxyisoquinoline-3-carbonyl)glycine
1. Glycine, N-[(4-hydroxy-1-methyl-7-phenoxy-3-isoquinolinyl)carbonyl]-
2. N-[(4-hydroxy-1-methyl-7-phenoxyisoquinolin-3-yl)carbonyl]glycine
MF C19H16N2O5
MW 352.3
SPONSOR FibroGen
CODE FG-4592; ASP1517
CAS 808118-40-3
WHO NUMBER 9717
Roxadustat, also known as ASP1517 and FG-4592, is an HIF α prolyl hydroxylase inhibitor in a cell-free assay. It stabilizes HIF-2 and induces EPO production and stimulates erythropoiesis. Roxadustat transiently and moderately increased endogenous erythropoietin and reduced hepcidin
|
FG-4592 (also known as ASP1517), 2-(4-hydroxy-1-methyl-7-phenoxyisoquinoline-3-carboxamido)acetic acid,
is a potent small molecule inhibitor of hypoxia-inducible factor prolyl hydroxylase (HIF-PH),
an enzyme up-regulating the expression of endogenous human erythropoietin (Epo).
It is currently being investigated as an oral treatment for anemia associated with chronic kidney disease (CKD).
Unlike other anemia treating agents, erythropoiesis-stimulating agents (ESAs),
FG-4592 inhibits HIF, through a distinctive mechanism, by stabilization of HIF. According to previous studies,
FG-4592 is capable of correcting and maintaining hemoglobin levels in CKD patients not
receiving dialysis and in patients of end-stage renal disease
who receives dialysis but do not need intravenous iron supplement.
Reference
1. Luis Borges. Different modalities of erythropoiesis stimulating agents.
Port J Nephrol Hypert 2010; 24(2): 137-145
2. “FibroGen and Astellas announce initiation of phase 3 trial of FG-4592/ASP1517 for treatment
of anemia of chronic kidney disease” Fibrogen Press Release. Dec 11 2012
3. “FibroGen announces initiation of phase 2b studies of FG-4592,
an oral HIF prolyl hydroxylase inhibitor, for treatment of anemia”
|
- Originator FibroGen
- Developer Astellas Pharma; AstraZeneca; FibroGen
- Class Amides; Antianaemics; Carboxylic acids; Isoquinolines; Small molecules
- Mechanism of Action Basic helix loop helix transcription factor modulators; Hypoxia-inducible factor-proline dioxygenase inhibitors
- Phase III Anaemia
- Discontinued Sickle cell anaemia
Most Recent Events
- 09 Jun 2016 Phase-III clinical trials in Anaemia in Japan (PO)
- 20 May 2016 In collaboration with FibroGen, Astellas Pharma plans a phase III trial for Anaemia (In chronic kidney disease patients undergoing peritoneal dialysis) in Japan (PO) (NCT02780726)
- 19 May 2016 In collaboration with FibroGen, Astellas Pharma plans a phase III trial for Anaemia (In erythropoiesis stimulating agent-naive, chronic kidney disease patients undergoing haemodialysis) in Japan (PO) (NCT02780141)


Roxadustat (FG-4592) is a novel new-generation oral hypoxia-induciblefactor (HIF) prolyl hydroxylase inhibitor (PHI) for the treatment of ane-mia in patients with chronic kidney disease (CKD). HIF is a cytosolic tran-scription factor that induces the natural physiological response to lowoxygen conditions, by stimulating erythropoiesis and other protectivepathways. Roxadustat has been shown to stabilize HIF and induce ery-thropoiesis. Consequently, it corrects anemia and maintains hemoglo-bin levels without the need for intravenous iron supplementation in CKDpatients not yet receiving dialysis and in end-stage renal disease pa-tients receiving dialysis. There are many concerns about the use of ery-thropoiesis-stimulating agents (ESA) to treat anemia as they causesupra-physiologic circulating erythropoietin (EPO) levels and are asso-ciated with adverse cardiovascular effects and mortality. Available clin-ical data show that modest and transient increases of endogenous EPOinduced by HIF-PHI (10- to 40-fold lower than ESA levels) are sufficientto mediate erythropoiesis in CKD patients. Evidence suggests that rox-adustat is well tolerated and, to date, no increased risk of cardiovascu-lar events has been found. This suggests that roxadustat provides adistinct pharmacological and clinical profile that may provide a saferand more convenient treatment of CKD anemia
FG-4592 is a new-generation hypoxia-inducible factor prolyl hydroxylase inhibitor in early clinical trials at FibroGen for the oral treatment of iron deficiency anemia and renal failure anemia. Preclinical studies are ongoing for the treatment of sickle cell anemia.
The investigational therapy is designed to restore balance to the body’s natural process of erythropoiesis through mechanisms including: natural EPO production, suppression of the effects of inflammation, downregulation of the iron sequestration hormone hepcidin, and an upregulation of other iron genes, ensuring efficient mobilization and utilization of the body’s own iron stores. In April 2006, FG-4592 was licensed to Astellas Pharma by originator FibroGen in Asia, Europe and South Africa for the treatment of anemia. FibroGen retains rights in the rest of the world. In 2007, the FDA put the trial on clinical hold due to one case of death by fulminant hepatitis during a phase II clinical trial for patients with anemia associated with chronic kidney disease and not requiring dialysis. However, in 2008, the FDA informed the company that clinical trials could be resumed. Phase II/III clinical trials for this indication resumed in 2012. In 2013, the compound was licensed to AstraZeneca by FibroGen for development and marketing in US, CN and all major markets excluding JP, Europe, the Commonwealth of Independent States, the Middle East and South Africa, for the treatment of anemia associated with chronic kidney disease (CKD) and end-stage renal disease (ESRD).
PATENTS
WO 2004108681
WO 2008042800
WO 2009058403
WO 2009075822
WO 2009075824
WO 2012037212
WO 2013013609
WO 2013070908
PATENT
CN 104892509
MACHINE TRANSLATED
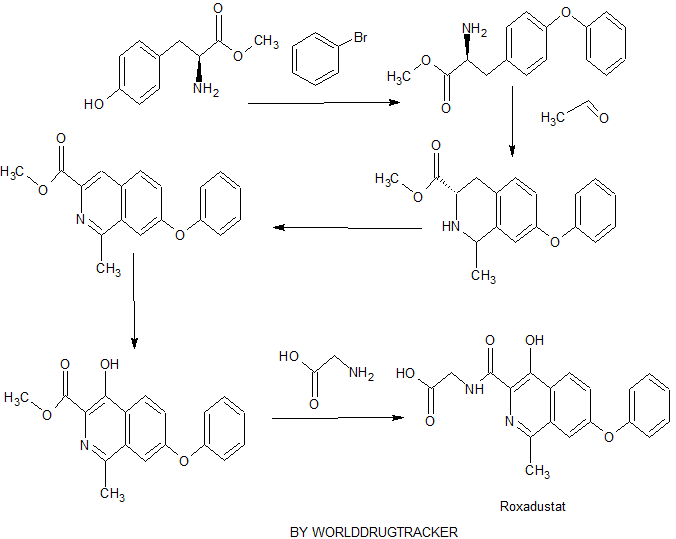
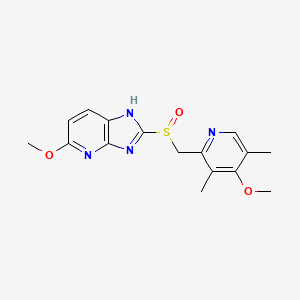
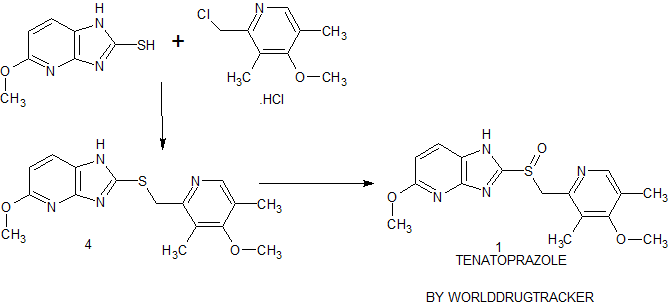
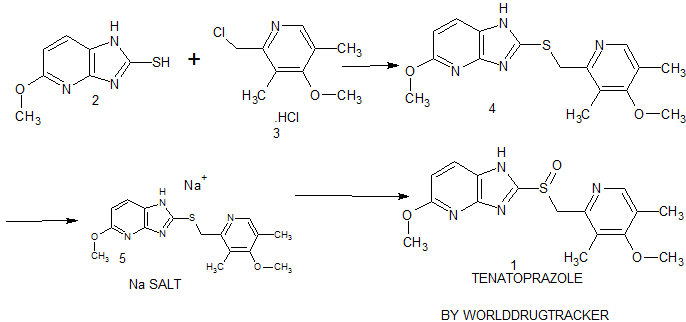
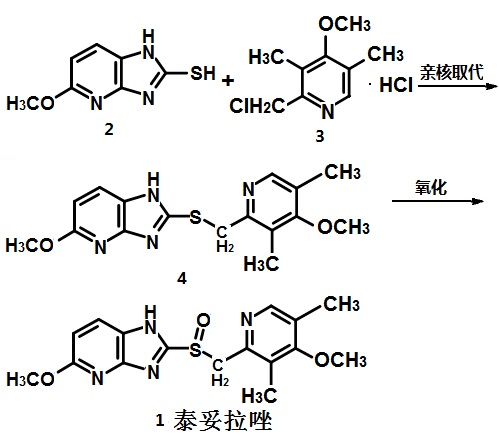
Connaught orlistat (Roxadustat) by the US company Phibro root (FibroGen) R & D, Astellas AstraZeneca and licensed by a hypoxia-inducible factor (HIF) prolyl hydroxylase small molecule inhibitors, codenamed FG-4592.As a first new oral drug, FG-4592 is currently in Phase III clinical testing stage, for the treatment of chronic kidney disease and end-stage renal disease related anemia. Because the drug does not have a standard Chinese translation, so the applicant where it is transliterated as “Connaught Secretary him.”
Connaught orlistat (Roxadustat, I) the chemical name: N_ [(4- hydroxy-1-methyl-7-phenoxy-3-isoquinolinyl) carbonyl] glycine, its structural formula is:
The original research company’s international patent W02004108681 Division provides a promise he was prepared from the intermediate and intermediate Connaught Secretary for his synthetic route:
Zhejiang Beida company’s international patent W02013013609 preparation and acylation of core intermediate was further optimized synthesis route is:
n PhO. eight XOOH
original research company’s international patent W02014014834 and W02014014835 also provides another synthetic route he Connaught Secretary prepared:
Analysis of the above synthetic route, although he continued to Connaught Division to improve and optimize the synthesis, but its essence rings manner that different form quinoline ring is basically the same mother. Especially methyl isoquinoline replaced either by way of introducing the Suzuki reaction catalyzed by a noble metal element, either through amine reduction achieved. Moreover, the above reaction scheme revelation raw materials are readily available, many times during the reaction need to be protected and then deprotected. Clearly, the preparation process is relatively complicated, high cost, industrial production has brought some difficulties.

Example One:
tyrosine was added to the reaction flask and dried (18. lg, 0.1 mmol) and methanol 250mL, cooling to ice bath 0_5 ° C, was added dropwise over 1 hour a percentage by weight of 98% concentrated sulfuric acid 10g. Drops Albert, heating to reflux. The reaction was stirred for 16-20 hours, TLC the reaction was complete. Concentrated under atmosphere pressure, the residue was added water 100mL, using 10% by weight sodium hydroxide to adjust the pH to 6. 5-7.0, precipitated solid was filtered, washed with methanol and water chloro cake (I: 1) and dried in vacuo tyrosine methyl ester as a white solid (11) 15.38, yield 78.5% out 1–] \ ^ 111/2: 196 [] \ 1 + 1] +!.
Example Two:
[0041] a nitrogen atmosphere and ice bath, was added to the reaction flask tyrosine methyl ester (II) (9. 8g, 50mmol), potassium methoxide (3. 5g, 50mmol) and methanol 50mL, until no gas generation after, was heated to reflux, the reaction was stirred for 2 hours. Concentrated under atmosphere pressure to remove the solvent, the residue was added dimethylsulfoxide 25mL, freshly prepared copper powder (0.2g, 3. Lmmol), was slowly warmed to 150-155 ° C, for about half an hour later, a solution of bromobenzene ( 7. 9g, 50mmol), continue to heat up to 170-175 ° C, the reaction was stirred for 3 hours, TLC detection of the end of the reaction. Was cooled to 60 ° C, and methanol was added to keep micro-boiling, filtered while hot, the filter cake washed three times with hot ethanol, and the combined organic phases, was cooled to square ° C, filtered, and dried in vacuo to give a white solid of 2-amino-3- ( 4-phenoxyphenyl) propanoate (111) 8 11.5, yield 84.9% as 1 -] \ ^ 111/2:! 272 [] \ 1 + 1] +.
Example Three:
in the reaction flask was added 2-amino-3- (4-phenoxyphenyl) propionic acid methyl ester (III) (10. 8g, 40mmol), 40% by weight acetaldehyde (20g, 0. 2mol ) and the percentage by weight of 35% concentrated hydrochloric acid 50mL, refluxed for 1 hour. Continue 40% by weight was added acetaldehyde (10g, 0.1mol), and the percentage by weight of 35% concentrated hydrochloric acid 25mL, and then the reaction was refluxed for 3-5 hours. Was cooled to 4-7 ° C, ethyl acetate was added, and extracted layers were separated. The aqueous layer was adjusted with sodium hydroxide solution to pH 11-12, extracted three times with ethyl acetate. The combined organic phase was dried over anhydrous sodium sulfate, and concentrated under reduced pressure to give a white solid of 1-methyl-3-carboxylate -7- phenoxy-1,2,3,4-tetrahydroisoquinoline (IV) 8 4g, 70.7% yield; Mass spectrum (EI): EI-MS m / z: 298 [M + H] + .
Example Four:
Under ice bath, the reaction flask was added methyl 3-carboxylate I- -7- phenoxy-1,2, 3,4-tetrahydro-isoquinoline (IV) (5. 9g, 20mmol) and dichloromethane 100mL, 0 ° C and under stirring added potassium carbonate (13. 8g, 0. lmol), p-toluenesulfonyl chloride (11. 4g, 60mmol), the addition was completed, the ice bath was removed and stirred at room temperature 3 hour. Water was added 30mL, after stirring standing layer, the organic phase was washed with dilute hydrochloric acid, water and saturated brine, and concentrated, the resulting product was added a 30% by weight sodium hydroxide solution (8. 0g, 60mmol) and dimethyl sulfoxide 60mL, gradually warming to 120-130 ° C, the reaction was stirred for 2-4 hours to complete the reaction by TLC. Cooled to room temperature, water was added lOOmL, extracted three times with ethyl acetate, the combined organic phase was successively washed with water and saturated brine, dried over anhydrous magnesium sulfate, and concentrated, the resulting oil was treated with ethyl acetate and n-hexane (1: 3) recrystallization, vacuum dried to give an off-white solid 1-methyl-3-carboxylate 7-phenoxyheptanoic isoquinoline (V) 5. 25g, yield 89. 6%; EI-MS m / z: 294 [M + H] VH NMR (DMS0-d6) δ 2. 85 (s, 3H), 3 · 97 (s, 3H), 7 · 16-7. 24 (m, 3H), 7 · 49-7. 60 (m, 4Η), 8 · 35 (d, J = 9 · 0,1Η), 8 · 94 (s, 1Η).
Example five:
[0047] added 1-methyl-3-carboxylic acid methyl ester 7-phenoxyheptanoic isoquinoline (V) (2. 93g, IOmmol) and glacial acetic acid 50mL reaction flask, stirring solution of 30% by weight hydrogen peroxide 5mL, warmed to 60-70 ° C, was slowly added dropwise within 10 hours the percentage by weight of a mixture of 30% hydrogen peroxide 2mL and 12mL of glacial acetic acid, a dropping was completed, the reaction was continued for 20-24 hours. Concentrated under reduced pressure, ethanol was added, distillation is continued to be divisible remaining glacial acetic acid. The residue was dissolved with dichloromethane, washed with 5% by weight of sodium bicarbonate, the organic phase was separated, dried over anhydrous sodium sulfate. Filtered and the resulting solution was added p-toluenesulfonyl chloride (3. 8g, 20mmol), was heated to reflux, the reaction was stirred for 3-4 hours, TLC detection completion of the reaction. The solvent was distilled off under reduced pressure, cooled to room temperature, methanol was added, the precipitated solid, cooled to square ° C, allowed to stand overnight. Filtered, the filter cake washed twice with cold methanol and vacuum dried to give an off-white solid 1- methyl-3-methyl-4-hydroxy-phenoxy-isoquinoline -7- (VI) I. 86g, yield 60.2 %; EI-MS m / z:.. 310 [M + H] +, 1H NMR (DMS0-d6) δ 2.90 (s, 3H), 4.05 (s, 3H), 7 17-7 26 (m, 3H ), 7. 49-7. 61 (m, 4H), 8. 38 (d, J = 9. 0,1H), 11. 7 (s, 1H) 〇
Example VI:
in the reaction flask with magnetic stirring and pressure to join I- methyl-3-methyl-4-hydroxy-7-phenoxyheptanoate isoquinoline (VI) (1.55g, 5mmol), glycine (I. 13g, 15mmol) and sodium methoxide (3. 25g, 6mmol) in methanol (30mL).Sealed, slowly heated to 120 ° C, the reaction was stirred for 8-10 hours to complete the reaction by TLC. Cooled to room temperature, solid precipitated. Filtration, and the resulting solid was recrystallized from methanol, acetone and then beating the resulting solid was dried under vacuum to give a white solid Connaught orlistat 1.40g, yield 79.5%;
EI-MS m / z: 353 [M + H] +,
1H NMR (DMS0-d6) S2.72 (s, 3H), 3 · 99 (d, J = 6 · 0, 2H), 7 · 18-7. 28 (m, 3H), 7 · 49-7. 63 (m, 4H), 8 · 31 (d, J = 8 · 8,1H), 9 · 08 (s, lH), 13.41 (brs, lH).
PATENT
WO 2014014835

Example 10. Preparation of Compound A
a) 5-Phenoxyphthalide
[0200] A reactor was charged with DMF (68 Kg), and stirring was initiated. The reactor was then charged with phenol (51 Kg), acetylacetone (8 Kg), 5-bromophthalide (85 Kg), copper bromide (9 Kg), and potassium carbonate (77 Kg). The mixture was heated above 85 °C and maintained until reaction completion and then cooled. Water was added. Solid was filtered and washed with water. Solid was dissolved in dichloromethane, and washed with aqueous HCl and then with water. Solvent was removed under pressure and methanol was added. The mixture was stirred and filtered. Solid was washed with methanol and dried in an oven giving 5- phenoxyphthalide (Yield: 72%, HPLC: 99.6%). b) 2-Chloromethyl-4-phenoxybenzoic acid methyl ester
[0201] A reactor was charged with toluene (24 Kg), and stirring was initiated. The reactor was then charged with 5-phenoxyphthalide (56 Kg), thionyl chloride (41 Kg), trimethyl borate (1
Kg), dichlorotriphenylphosphorane (2.5 Kg), and potassium carbonate (77 Kg). The mixture was heated to reflux until reaction completion and solvent was removed leaving 2-chloromethyl-4- phenoxybenzoyl chloride. Methanol was charged and the mixture was heated above 50 °C until reaction completion. Solvent was removed and replaced with DMF. This solution of the product methyl 2-chloromethyl-4-phenoxybenzoic acid methyl ester in DMF was used directly in the next step (HPLC: 85%). c) 4-Hydroxy-7-phenoxyisoquinoline-3-carboxylic acid methyl ester (la)

[0202] A reactor was charged with a solution of 2-chloromethyl-4-phenoxybenzoic acid methyl ester (~68 Kg) in DMF, and stirring was initiated. The reactor was then charged with p- toluenesulfonylglycine methyl ester (66 Kg), potassium carbonate (60 Kg), and sodium iodide (4 Kg). The mixture was heated to at least 50 °C until reaction completion. The mixture was cooled. Sodium methoxide in methanol was charged and the mixture was stirred until reaction completion. Acetic acid and water were added, and the mixture was stirred, filtered and washed with water. Solid was purified by acetone trituration and dried in an oven giving la (Yield from step b): 58%; HPLC: 99.4%). 1H NMR (200 MHz, DMSO-d6) δ 11.60 (s, 1 H), 8.74 (s, 1H),
8.32 (d, J = 9.0 Hz, 1 H), 7.60 (dd, J = 2.3 & 9.0 Hz, 1H), 7.49 (m, 3 H), 7.24 (m, 3 H), 3.96 (s, 3 H); MS-(+)-ion M+l = 296.09 d) Methyl l-((dimethylamino)methyl)-4-hydroxy-7-phenoxyisoquinoline-3-carboxylate
(lb)
[0203] A flask was charged with la (29.5 g) and acetic acid (44.3 g ± 5%), and then stirred. Bis-dimethylaminomethane (12.8 g ± 2%) was slowly added. The mixture was heated to 55 ± 5 °C and maintained until reaction completion. The reaction product was evaluated by MS, HPLC and 1H NMR. 1H NMR (200 MHz, DMSO-d6) δ 11.7 (s, 1 H), 8.38 (d, J = 9.0 Hz, 1 H), 7.61 (dd, J = 9.0, 2.7 Hz, 1 H), 7.49 (m, 3 H), 7.21 (m, 3 H), 5.34 (s, 2 H), 3.97 (s, 3 H), 1.98 (s, 3 H); MS-(+)-ion M+l = 368.12. e) Methyl l-((acetoxy)methyl)-4-hydroxy-7-phenoxyisoquinoline-3-carboxylate (lc)
[0204] The solution of lb from a) above was cooled below 25 °C, at which time acetic anhydride (28.6 g ± 3.5 %) was added to maintain temperature below 50 °C. The resulting mixture was heated to 100 ± 5 °C until reaction completion.
[0205] The solution of lc and Id from above was cooled to less than 65 ± 5 °C. Water (250 mL) was slowly added. The mixture was then cooled to below 20 ± 5 °C and filtered. The wet cake was washed with water (3 x 50 mL) and added to a new flask. Dichloromethane (90 mL) and water (30 mL) were added, and the resulting mixture was stirred. The dichloromethane layer was separated and evaluated by HPLC.
[0206] The organic layer was added to a flask and cooled 5 ± 5 °C. Morpholine was added and the mixture was stirred until reaction completion. Solvent was replaced with acetone/methanol mixture. After cooling, compound lc precipitated and was filtered, washed and dried in an oven (Yield: 81%, HPLC: >99.7%). 1H NMR (200 MHz, DMSO-d6) δ 11.6 (S, 1 H), 8.31 (d, J = 9.0 Hz, 1 H), 7.87 (d, J = 2.3 Hz, 1 H), 7.49 (m, 3 H), 7.24 (m, 3 H), 3.95 (s, 3 H), 3.68 (s, 2H), 2.08 (s, 6 H); MS-(+)-ion M+l = 357.17. f) Methyl 4-hydroxy-l-methyl-7-phenoxyisoquinoline-3-carboxylate (le)

[0207] A reactor was charged with lc (16.0 g), Pd/C (2.08 g), anhydrous Na2C03 (2.56 g) and ethyl acetate (120 mL). The flask was vacuum-purged with nitrogen (3X) and vacuum-purged with hydrogen (3X). The flask was then pressurized with hydrogen and stirred at about 60 °C until completion of reaction. The flask was cooled to 20-25 °C, the pressure released to ambient, the head space purged with nitrogen three times and mixture was filtered. The filtrate was concentrated. Methanol was added. The mixture was stirred and then cooled. Product precipitated and was filtered and dried in an oven (Yield: 90%, HPLC: 99.7%). g) [(4-Hydroxy-l-methyl-7-phenoxy-isoquinoline-3-carbonyl)-amino]-acetic acid
(Compound A)
[0208] A pressure flask was charged with le (30.92 g), glycine (22.52 g), methanol (155 mL), sodium methoxide solution (64.81 g) and sealed (as an alternative, sodium glycinate was used in place of glycine and sodium methoxide). The reaction was heated to about 110 °C until reaction was complete. The mixture was cooled, filtered, washed with methanol, dried under vacuum, dissolved in water and washed with ethyl acetate. The ethyl acetate was removed and to the resulting aqueous layer an acetic acid (18.0 g) solution was added. The suspension was stirred at room temperature, filtered, and the solid washed with water (3 x 30 mL), cold acetone (5-10 °C, 2 x 20 mL), and dried under vacuum to obtain Compound A (Yield: 86.1%, HPLC: 99.8%). Example 11. Biological Testing
[0209] The solid forms provided herein can be used for inhibiting HIF hydroxylase activity, thereby increasing the stability and/or activity of hypoxia inducible factor (HIF), and can be used to treat and prevent HIF-associated conditions and disorders (see, e.g., U.S. Patent No. 7,323,475, U.S. Patent Application Publication No. 2007/0004627, U.S. Patent Application Publication No. 2006/0276477, and U.S. Patent Application Publication No. 2007/0259960, incorporated by reference herein).
SYNTHESIS……..

Condensation of 5-bromophthalide (I) with phenol (II) in the presence of K2CO3, CuBr and acetylacetone in DMF gives 5-phenoxyphthalide (III), which upon lactone ring opening using SOCl2, Ph3PCl2, B(OMe)3 and K2CO3 in refluxing toluene yields 2-chloromethyl-4-phenoxybenzoyl chloride (IV). Esterification of acid chloride (IV) with MeOH at 50 °C furnishes the methyl ester (V), which is then condensed with methyl N-tosylglycinate (VI) in the presence of K2CO3 and NaI in DMF at 50 °C to afford N-substituted aminoester (VII). Cyclization of the intermediate diester (VII) using NaOMe in MeOH leads to methyl 4-hydroxy-7-phenoxyisoquinoline-3-carboxylate (VIII), which is submitted to Mannich reaction with bis-dimethylaminomethane (IX) in the presence of AcOH at 57 °C to provide the dimethylaminomethyl compound (X). Treatment of amine (X) with Ac2O at 103 °C, followed by selective hydrolysis of the phenolic acetate with morpholine leads to methyl 1-acetoxymethyl-4-hydroxy-7-phenoxyisoquinoline-3-carboxylate (XI). Hydrogenolysis of the benzylic acetate (XII) in the presence of Pd/C and Na2CO3 in EtOAc yields methyl 4-hydroxy-1-methyl-7-phenoxyisoquinoline-3-carboylate (XII), which finally couples with glycine (XIII) in the presence of NaOMe in MeOH at 110 °C to afford the target roxadustat (1-3).

Cyclization of 4-phenoxyphthalic acid (I) with glycine (II) at 215 °C gives the phthalimide (III), which upon esterification with MeOH and H2SO4 at reflux yields methyl ester (IV). Subsequent rearrangement of phthalimidoacetate (IV) by means of Na in BuOH at 97 °C, followed by flash chromatography provides the isoquinoline-2-carboxylate (V). Bromination of intermediate (V) using POBr3 and NaHCO3 in acetonitrile leads to butyl 8-bromo-3-hydroxy-6-phenoxy-isoquinoline-2-carboxylate (VI), which upon hydrolysis with NaOH in refluxing H2O/EtOH furnishes carboxylic acid (VII). Substitution of bromine in intermediate (VII) using MeI and BuLi in THF at -78 °C, followed by alkylation with PhCH2Br in the presence of K2CO3 in refluxing acetone affords the 2-methyl isoquinoline (VIII). Ester hydrolysis in intermediate (VIII) using KOH in MeOH gives the corresponding carboxylic acid (IX), which is then activated with i-BuOCOCl and Et3N in CH2Cl2, followed by coupling with benzyl glycinate hydrochloride (X) to yield benzylated roxadustat (XI). Finally, debenzylation of intermediate (XI) with H2 over Pd/C in EtOAc/MeOH provides the title compound (1).

Condensation of 4-nitro-ortho-phthalonitrile (I) with phenol (II) in the presence of K2CO3 in DMSO gives 4-phenoxy-ortho-phthalonitrile (III) (1), which upon hydrolysis with NaOH (1) or KOH (2) in refluxing MeOH yields 4-phenoxyphthalic acid (IV) (1,2). Dehydration of dicarboxylic acid (IV) using Ac2O and AcOH at reflux furnishes the phthalic anhydride (V), which is then condensed with methyl 2-isocyanoacetate (VI) using DBU in THF to provide oxazole derivative (VII). Rearrangement of intermediate (VII) with HCl in MeOH at 60 °C leads to isoquinoline derivative (VIII), which is partially chlorinated by means of POCl3 at 70 °C to afford 1-chloro-isoquinoline derivative (IX). Substitution of chlorine in intermediate (IX) using Me3B, Pd(PPh3)4 and K2CO3 in refluxing dioxane gives methyl 4-hydroxy-1-methyl-7-phenoxy-3-carboxylate (X), which is then hydrolyzed with aqueous NaOH in refluxing EtOH to yield the carboxylic acid (XI). Coupling of carboxylic acid (XI) with methyl glycinate hydrochloride (XII) by means of PyBOP, (i-Pr)2NH and Et3N in CH2Cl2 yields roxadustat methyl ester (XII), which is finally hydrolyzed with aqueous NaOH in THF to afford the target roxadustat (1).
CLIPS
SAN FRANCISCO, Nov 12, 2013 (BUSINESS WIRE) — FibroGen, Inc. (FibroGen), today announced that data from a China-based Phase 2 study of roxadustat (FG-4592), a first-in-class oral compound in late stage development for the treatment of anemia associated with chronic kidney disease (CKD) and end-stage renal disease (ESRD), were presented in an oral session at the 2013 American Society of Nephrology (ASN) Kidney Week in Atlanta, Georgia.
Roxadustat is an orally administered, small molecule inhibitor of hypoxia-inducible factor (HIF) prolyl hydroxylase. HIF is a protein that responds to oxygen changes in the cellular environment and meets the body’s demands for oxygen by inducing erythropoiesis, the process by which red blood cells are produced and iron is incorporated into hemoglobin (Hb).
The randomized, double-blind, placebo-controlled study was designed to evaluate the efficacy, safety, and tolerability of roxadustat in the correction of anemia in patients (N=91) with chronic kidney disease who had not received dialysis treatment, were not receiving erythropoiesis-stimulating agents (ESAs), and had Hb levels less than 10 g/dL. The correction study randomized patients 2:1 between roxadustat and placebo for 8 weeks of dosing, and included a low-dose cohort (n=30) and high-dose cohort (n=31). Intravenous (IV) iron was not allowed. The study also evaluated iron utilization, changes in serum lipids, and other biomarkers during treatment with roxadustat.
Data from this study suggest that roxadustat effectively corrected hemoglobin levels in anemic CKD patients in a dose-dependent manner as compared to placebo, and did so in the absence of IV iron supplementation regardless of degree of iron repletion at baseline. At the end of the 8-week treatment period, subjects showed mean maximum Hb increases from baseline of 2.6 g/dL in the high dose cohort and 1.8 g/dL in the low dose cohort, as compared to 0.7 g/dL in the placebo group (p < 0.0001) from mean baseline Hb of 8.8 g/dL, 8.8 g/dL, and 8.9 g/dL in the high dose, low dose, and placebo groups, respectively. 87% of patients in the high-dose cohort, 80% of patients in the low-dose cohort, and 23% of patients in the placebo group experienced a hemoglobin increase of 1 g/dL or greater from baseline (p < 0.0001). Similarly, 71% of patients in the high-dose cohort, 50% of patients in the low-dose cohort, and 3% of patients in the placebo group achieved target hemoglobin of 11 g/dL or greater (p < 0.0001). Serum iron levels remained stable in subjects randomized to roxadustat while the subjects underwent brisk erythropoiesis.
Study data also suggest that roxadustat may lower cholesterol. Dyslipidemia is highly prevalent in chronic kidney disease patients and a major cardiovascular risk factor in this population. Patients treated with roxadustat experienced a statistically significant reduction in total cholesterol (p <0.0001) and low-density lipoprotein (LDL) cholesterol (p <0.0001) at the end of the treatment period. The relative proportion of high density lipoprotein (HDL) cholesterol to LDL cholesterol increased significantly (p <0.02). Overall LDL cholesterol levels declined by a mean of 26% and median of 23% from a mean baseline value of 103 mg/dL.
Roxadustat was well tolerated by patients in the study with incidence of adverse events similar across all groups. In contrast to the exacerbation of hypertension observed in studies in which patients received currently available ESA therapies, subjects who received roxadustat in the present study showed small decreases in blood pressure that were similar to blood pressure changes in the placebo group. No cardiovascular serious adverse events were reported in patients treated with roxadustat.
The efficacy and safety of roxadustat are currently being investigated in a global pivotal Phase 3 development program.
“There is a global need for effective, safe, and accessible anemia therapies,” said Thomas B. Neff, Chief Executive Officer of FibroGen. “Side effects associated with current treatments include exposure to supra-physiological levels of erythropoietin and depletion of iron stores. Preliminary clinical findings show that oral administration of roxadustat (FG-4592) is able to correct anemia and maintain hemoglobin levels in patients with chronic kidney disease, to do so with peak erythropoietin levels within physiological range, and to achieve these effects without the administration of intravenous iron. These results suggest roxadustat, as an oral agent, has the potential to overcome the treatment barriers and inconveniences of current ESA therapies, including administration by injection and IV iron supplementation, in treating anemia in CKD patients.”
About Chronic Kidney Disease (CKD) and Anemia
Diabetes, high blood pressure, and other conditions can cause significant damage to the kidneys. If left untreated, those can result in chronic kidney disease and progress to kidney failure. Such deterioration can lead to patients needing a kidney transplant or being placed on dialysis to remove excess fluid and toxins that build up in the body. The progression of CKD also increases the prevalence of anemia, a condition associated with having fewer of the red blood cells that carry oxygen through the body, and/or lower levels of hemoglobin, the protein that enables red blood cells to carry oxygen. As hemoglobin falls, the lower oxygen-carrying capacity of an anemic patients’ blood results in various symptoms including fatigue, loss of energy, breathlessness, and angina. Anemia in CKD patients has been associated with increased hospitalization rates, increased mortality, and reduced quality of life.
Chronic kidney disease is a worldwide critical healthcare problem that affects millions of people and drives significant healthcare cost. In the US, prevalence of CKD has increased dramatically in the past 20 years, from 10 percent of the adult population (or approximately 20 million U.S. adults) as stated in the National Health and Nutrition Evaluation Survey (NHANES) 1988-1994, to 15 percent (or approximately 30 million U.S. adults) in NHANES 2003-2006. In 2009, total Medicare costs for CKD patients were $34 billion. China has an estimated 145 million CKD patients, or approximately five times the number of CKD patients in the U.S. (Lancet April 2012).
About Roxadustat / FG-4592
Roxadustat (FG-4592) is an orally administered small molecule inhibitor of hypoxia-inducible factor (HIF) prolyl hydroxylase activity, in development for the treatment of anemia in patients with chronic kidney disease (CKD). HIF is a protein transcription factor that induces the natural physiological response to conditions of low oxygen, “turning on” erythropoiesis (the process by which red blood cells are produced) and other protective pathways. Roxadustat has been shown to correct anemia and maintain hemoglobin levels without the need for supplementation with intravenous iron in CKD patients not yet receiving dialysis and in end-stage renal disease patients receiving dialysis. An Independent Data Monitoring Committee has found no signals or trends to date to suggest that treatment with roxadustat is associated with increased risk of cardiovascular events, thrombosis, or increases in blood pressure requiring initiation or intensification of antihypertensive medications.
About FibroGen
FibroGen is a privately-held biotechnology company focused on the discovery, development, and commercialization of therapeutic agents for treatment of fibrosis, anemia, cancer, and other serious unmet medical needs. FibroGen’s FG-3019 monoclonal antibody is in clinical development for treatment of idiopathic pulmonary fibrosis and other proliferative diseases, including pancreatic cancer and liver fibrosis. Roxadustat (FG-4592), FibroGen’s small molecule inhibitor of hypoxia-inducible factor (HIF) prolyl hydroxylase, is currently in clinical development for the treatment of anemia. FibroGen is also currently pursuing the use of proprietary recombinant human type III collagens in synthetic corneas for treatment of corneal blindness. For more information please visit:
www.fibrogen.com .

References
1: Besarab A, Provenzano R, Hertel J, Zabaneh R, Klaus SJ, Lee T, Leong R, Hemmerich S, Yu KH, Neff TB. Randomized placebo-controlled dose-ranging and pharmacodynamics study of roxadustat (FG-4592) to treat anemia in nondialysis-dependent chronic kidney disease (NDD-CKD) patients. Nephrol Dial Transplant. 2015 Oct;30(10):1665-73. doi: 10.1093/ndt/gfv302. Epub 2015 Aug 3. PubMed PMID: 26238121; PubMed Central PMCID: PMC4569392.
2: Forristal CE, Levesque JP. Targeting the hypoxia-sensing pathway in clinical hematology. Stem Cells Transl Med. 2014 Feb;3(2):135-40. doi: 10.5966/sctm.2013-0134. Epub 2013 Dec 26. PubMed PMID: 24371328; PubMed Central PMCID: PMC3925058.
3: Bouchie A. First-in-class anemia drug takes aim at Amgen’s dominion. Nat Biotechnol. 2013 Nov;31(11):948-9. doi: 10.1038/nbt1113-948b. PubMed PMID: 24213751.
4: Flight MH. Deal watch: AstraZeneca bets on FibroGen’s anaemia drug. Nat Rev Drug Discov. 2013 Oct;12(10):730. doi: 10.1038/nrd4135. PubMed PMID: 24080688.
5: Beuck S, Schänzer W, Thevis M. Hypoxia-inducible factor stabilizers and other small-molecule erythropoiesis-stimulating agents in current and preventive doping analysis. Drug Test Anal. 2012 Nov;4(11):830-45. doi: 10.1002/dta.390. Epub 2012 Feb 24. Review. PubMed PMID: 22362605.
6: Cases A. The latest advances in kidney diseases and related disorders. Drug News Perspect. 2007 Dec;20(10):647-54. PubMed PMID: 18301799.
//////////ASP1517, ASP 1517, ASP-1517, FG-4592, FG 4592, FG4592, Roxadustat, PHASE 3, ASTELLAS, FibroGen, 808118-40-3
O=C(O)CNC(C1=C(O)C2=C(C(C)=N1)C=C(OC3=CC=CC=C3)C=C2)=O




















































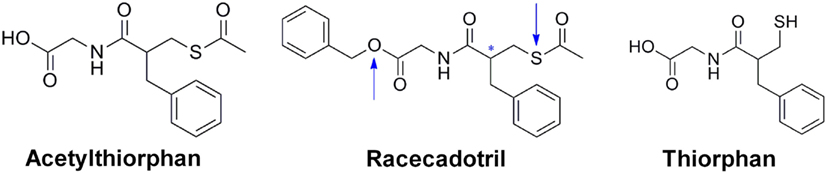














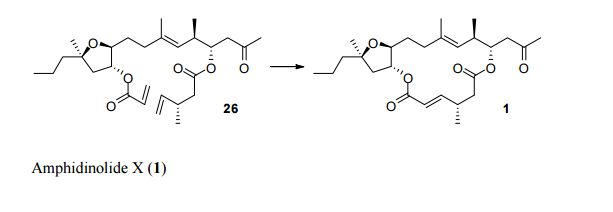



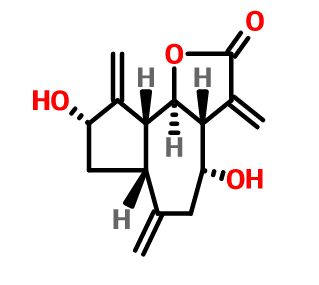
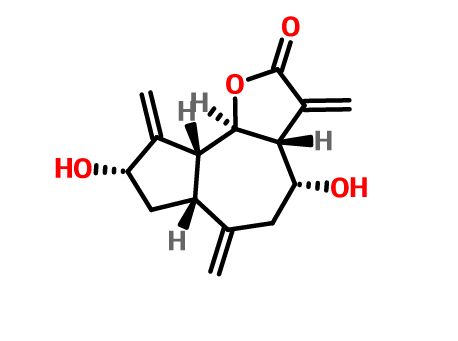 Integrifolin
Integrifolin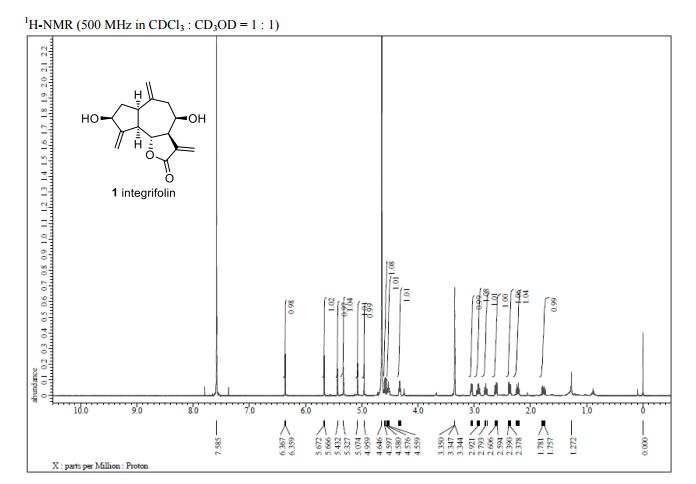
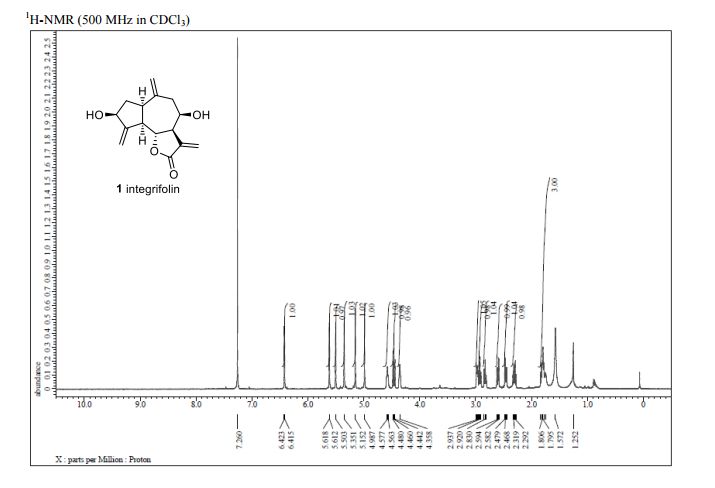
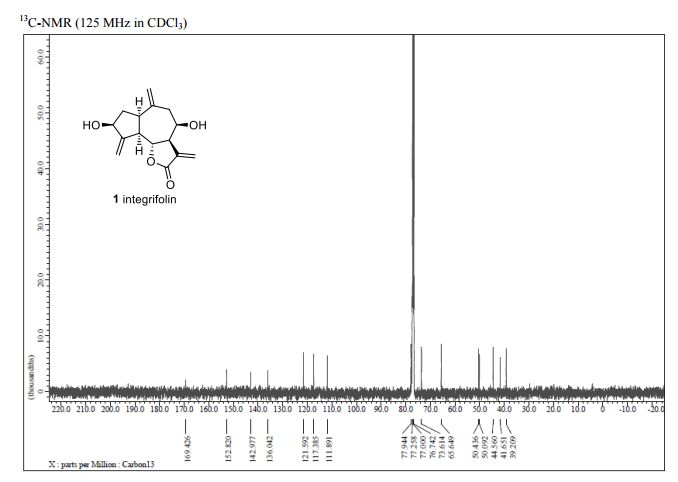
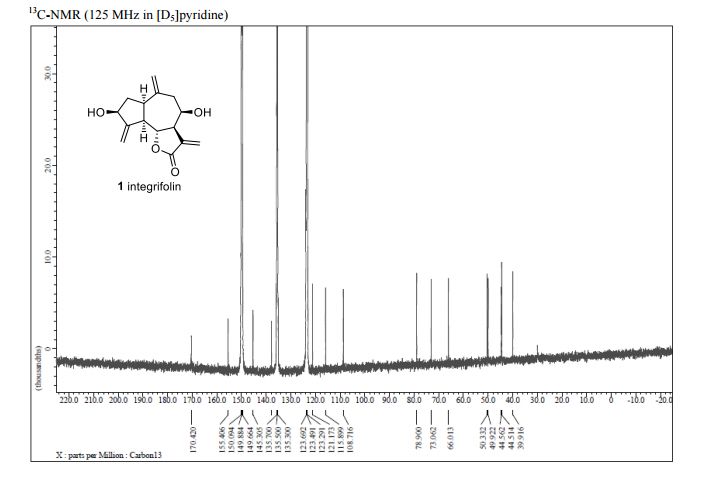
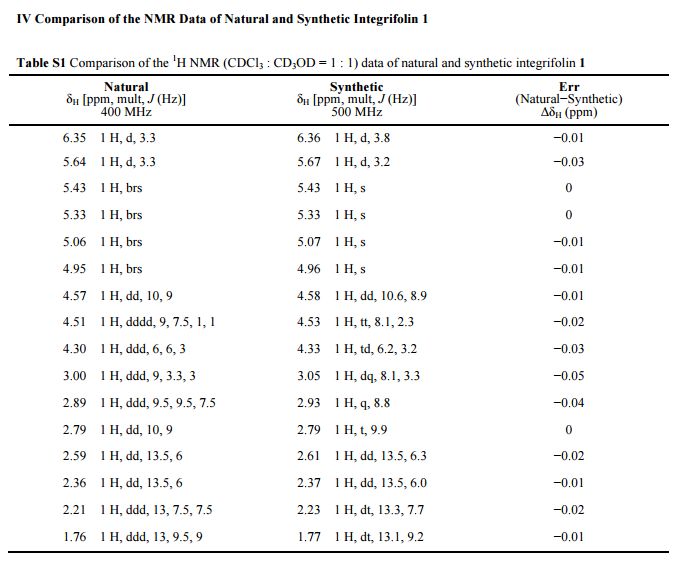
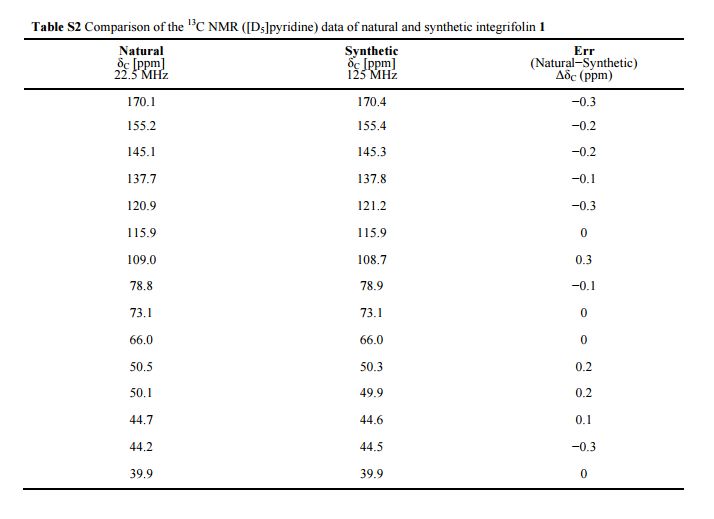

 Journal title :
Journal title :