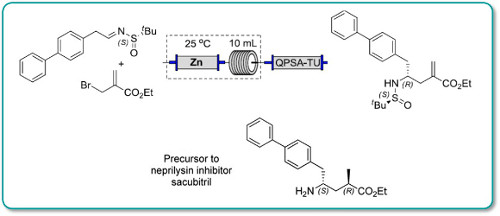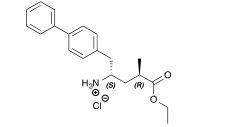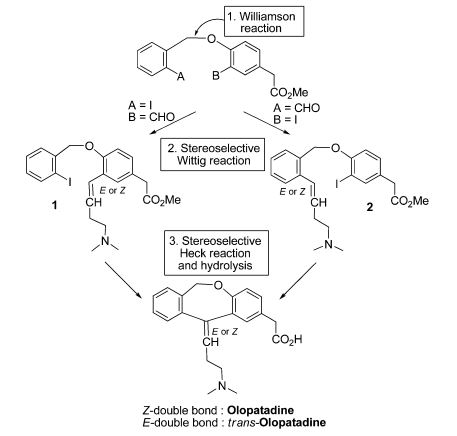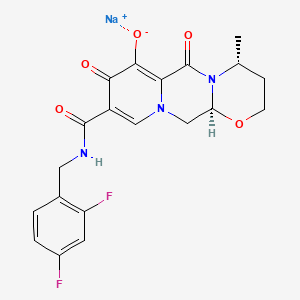


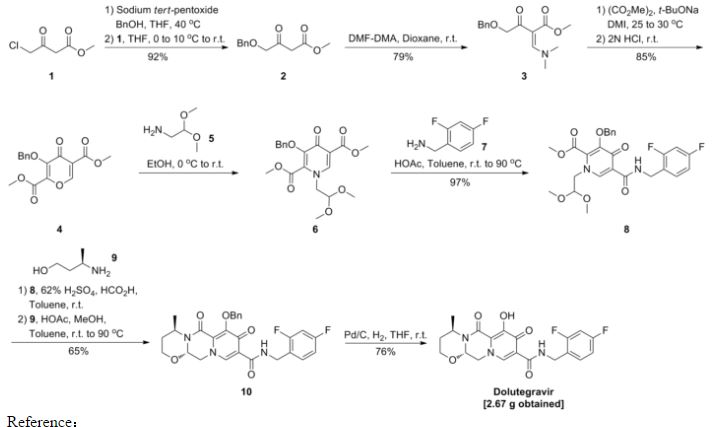
Dolutegravir
Soltegravir
2H-Pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazine-9-carboxamide, N-[(2,4-difluorophenyl)methyl]-3,4,6,8,12,12a-hexahydro-7-hydroxy-4-methyl-6,8-dioxo-, (4R,12aS)
(3R,11aS)—N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide
(4R,12aS)-N-(2,4-difluorobenzyl)-7-hydroxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazine-9-carboxamide
Trade Name:Tivicay
Synonym:GSK1349572, S-349572, GSK572
Date of Approval: August 12, 2013 (US)
Indication:HIV infection
Drug class: Integrase strand transfer inhibitor
Company: ViiV Healthcare,GlaxoSmithKline
INNOVATOR …ViiV Healthcare
CAS number: 1051375-16-6
1051375-19-9 (Dolutegravir Sodium)
MF:C20H19F2N3O5
MW:419.4
2H-Pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazine-9-carboxamide, N-[(2,4-difluorophenyl)methyl]-3,4,6,8,12,12a-hexahydro-7-hydroxy-4-methyl-6,8-dioxo-, (4R,12aS)- [ACD/Index Name]
Chemical Name: (4R,12aS)-N-[(2,4-difluorophenyl)methyl]-7-hydroxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a- hexahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazine-9-carboxamide
Patent: US8129385
Patent expiration date: Oct 5, 2027
PCT patent application: W02006116764
ドルテグラビルナトリウム
Dolutegravir Sodium

C20H18F2N3NaO5 : 441.36
[1051375-19-9]
Dolutegravir (DTG, GSK1349572) is an integrase inhibitor being developed for the treatment of human immunodeficiency virus (HIV)-1 infection by GlaxoSmithKline (GSK) on behalf of Shionogi-ViiV Healthcare LLC. DTG is metabolized primarily by uridine diphosphate glucuronyltransferase (UGT)1A1, with a minor role of cytochrome P450 (CYP)3A, and with renal elimination of unchanged drug being extremely low (< 1% of the dose).
Dolutegravir sodium was approved by the U.S. Food and Drug Administration (FDA) on Aug 12, 2013, then approved by European Medicine Agency (EMA) on Jan 16, 2014, and approved by Pharmaceuticals and Medical Devices Agency of Japan (PMDA) on Mar 24, 2014, then approved by Center For Drug Evaluation (CFDA) on Dec 30, 2015. It was co-developed by GlaxoSmithKline & ViiV Healthcare Corporation, then marketed as Tivicay® by ViiV Healthcare in the US and EU and by GlaxoSmithKline & ViiV Healthcare Corporation in JP.
Dolutegravir sodium is an integrase inhibitor which blocks HIV replication by preventing the viral DNA from integrating into the genetic material of human immune cells (T-cells). This step is essential in the HIV replication cycle and is also responsible for establishing chronic infection. It is in combination with other antiretroviral agents for the treatment of HIV-1 infection in adults and children aged 12 years and older and weighing at least 40 kg.
Tivicay® is available as film-coated tablet for oral use, containing 50 mg of free Dolutegravir. The recommended dose is 50 mg Dolutegravir once daily without regards to meals.

APPROVALS
| Approval Date |
Approval Type |
Trade Name |
Indication |
Dosage Form |
Strength |
Company |
Review Classification |
| 2013-08-12 |
Marketing approval |
Tivicay |
HIV infection |
Tablet, Film coated |
Eq. 50 mg Dolutegravir |
ViiV |
Priority |
| Approval Date |
Approval Type |
Trade Name |
Indication |
Dosage Form |
Strength |
Company |
Review Classification |
| 2014-01-16 |
Marketing approval |
Tivicay |
HIV infection |
Tablet, Film coated |
50 mg |
ViiV |
|
| Approval Date |
Approval Type |
Trade Name |
Indication |
Dosage Form |
Strength |
Company |
Review Classification |
| 2014-03-24 |
Marketing approval |
Tivicay |
HIV infection |
Tablet, Film coated |
50 mg |
ViiV, GlaxoSmithKline |
|
| Approval Date |
Approval Type |
Trade Name |
Indication |
Dosage Form |
Strength |
Company |
Review Classification |
| 2015-12-30 |
Marketing approval |
Tivicay/特威凯 |
HIV infection |
Tablet |
50 mg |
GlaxoSmithKline |
|
CLIP
The European Commission has on 21 January 2014 Dolutegravir (Tivicay, ViiV) permit as part of combination therapy for the treatment of HIV-infected persons over the age of 12 years.Dolutegravir (Tivicay, ViiV) is an integrase inhibitor, in combination with other antiretroviral drugs in adults and adolescents can be used from 12 years for the treatment of HIV infection.
Source: Communication from the European Commission
Dolutegravir[1] is a FDA-approved drug[2] for the treatment of HIV infection. Dolutegravir is an integrase inhibitor. Known as S/GSK1349572 or just “572” the drug is marketed as Tivicay[3] by GlaxoSmithKline (GSK). In February, 2013 the Food and Drug Administration announced that it would fast track dolutegravir’s approval process.[4] On August 13, 2013, dolutegravir was approved by the FDA. On November 4, 2013, dolutegravir was approved by Health Canada.[5]
The oral HIV integrase inhibitor S-349572 was originated by Shionogi-GlaxoSmithKline and Shionogi-ViiV Healthcare. In 2013, the product was approved and launched in the U.S. for the treatment of HIV-1 in adults and children aged 12 years and older, in combination with other antiretroviral agents. A positive opinion was received in the E.U for this indication and, in 2014, approval was attained in Europe for this indication. Registration is pending in Japan.
In 2013, orphan drug designation in Japan was assigned to the compound.
Dolutegravir is approved for use in a broad population of HIV-infected patients. It can be used to treat HIV-infected adults who have never taken HIV therapy (treatment-naïve) and HIV-infected adults who have previously taken HIV therapy (treatment-experienced), including those who have been treated with other integrase strand transfer inhibitors. Tivicay is also approved for children ages 12 years and older weighing at least 40 kilograms (kg) who are treatment-naïve or treatment-experienced but have not previously taken other integrase strand transfer inhibitors.[6]
Dolutegravir has also been compared head-to-head with a preferred regimen from the DHHS guidelines in each of the three classes (i.e. 1.) nuc + non-nuc, 2.) nuc + boosted PI, and 3.) nuc + integrase inhibitor).
SPRING-2 compared dolutegravir to another integrase inhibitor, raltegravir, with both coformulated with a choice of TDF/FTC orABC/3TC. After 48 weeks of treatment 88% of those on dolutegravir had less than 50 copies of HIV per mL compared to 85% in the raltegravir group, thus demonstrating non-inferiority.[9]
The FLAMINGO study has been presented at scientific meetings but as of early 2014 has not yet been published. It is an open-label trial of dolutegravir versus darunavir boosted with ritonavir. In this trial 90% of those on dolutegravir based regimens had viral loads < 50 at 48 weeks compared to 83% in the darunavir/r.[10] This 7% difference was statistically significant for superiority of the dolutegravir based regimens.
Another trial comparing dolutegravir to efavirenz, SINGLE, was the first trial to show statistical superiority to an efavirenz/FTC/TDF coformulated regimen for treatment naive patients.[11] After 48 weeks of treatment, 88% of the dolutegravir group had HIV RNA levels < 50 copies / mL versus 81% of the efavirenz group. This has led one commentator to predict that it may replace efavirenz as the first line choice for initial therapy as it can also be formulated in one pill, once-a-day regimens.[12]
Doultegravir has also been studied in patients who have been on previous antiretroviral medications. The VIKING trial looked at patients who had known resistance to the first generation integrase inhibitor raltegravir. After 24 weeks 41% of patients on 50mg dolutegravir once daily and 75% of patients on 50mg twice daily (both along with an optimized background regimen) achieved an HIV RNA viral load of < 50 copies per mL. This demonstrated that there was little clinical cross-resistance between the two integrase inhibitors. [13]
Dolutegravir (also known as S/GSK1349572), a second-generation integrase inhibitor under development by GlaxoSmithKline and its Japanese partner Shionogi for the treatment of HIV infection, was given priority review status from the US Food and Drug Administration (FDA) in February, 2013.
GlaxoSmithKline marketed the first HIV drug Retrovir in 1987 before losing out to Gilead Sciences Inc. (GILD) as the world’s biggest maker of AIDS medicines. The virus became resistant to Retrovir when given on its own, leading to the development of therapeutic cocktails.
The new once-daily drug Dolutegravir, which belongs to a novel class known as integrase inhibitors that block the virus causing AIDS from entering cells, is owned by ViiV Healthcare, a joint venture focused on HIV in which GSK is the largest shareholder.
Raltegravir (brand name Isentress) received approval by the U.S. Food and Drug Administration (FDA) on 12 October 2007, the first of a new class of HIV drugs, the integrase inhibitors, to receive such approval. it is a potent and well tolerated antiviral agent. However, it has the limitations of twice-daily dosing and a relatively modest genetic barrier to the development of resistance, prompting the search for agents with once-daily dosing.
Elvitegravir, approved by the FDA on August 27, 2012 as part of theelvitegravir/cobicistat/tenofovir disoproxil fumarate/emtricitabine fixed-dose combination pill (Quad pill, brand name Stribild) has the benefit of being part of a one-pill, once-daily regimen, but suffers from extensive cross-resistance with raltegravir.
 DOLUTEGRAVIR
DOLUTEGRAVIR
Gilead’s Atripla (Emtricitabine/Tenofovir/efavirenz), approved in 2006 with loss of patent protection in 20121, is the top-selling HIV treatment. The $3.2 billion medicine combines three drugs in one pill, two compounds that make up Gilead’s Truvada (Emtricitabine/Tenofovir) and Bristol- Myers Squibb Co.’s Sustiva (Efavirenz).
A three-drug combination containing dolutegravir and ViiV’s older two-in-one treatment Epzicom(Abacavir/Lamivudine, marketed outside US as Kivexa) proved better than Gilead’s market-leading Atripla in a clinical trial released in July, 2012 (See the Full Conference Report Here), suggesting it may supplant the world’s best-selling AIDS medicine as the preferred front-line therapy. In the latest Phase III study, after 48 weeks of treatment, 88% of patients taking the dolutegravir-based regimen had reduced viral levels to the goal compared with 81% of patients taking Atripla. More patients taking Atripla dropped out of the study because of adverse events compared with those taking dolutegravir — 10% versus just 2% — which was the main driver of the difference in efficacy. The result was the second positive final-stage clinical read-out for dolutegravir, following encouraging results against U.S. company Merck & Co’s rival Isentress in April, 2012 (See the Conference Abstract Here)..
Dolutegravir is viewed by analysts as a potential multibillion-dollar-a-year seller, as its once-daily dosing is likely to be attractive to patients. The FDA is scheduled to issue a decision on the drug’s approval by August 17。
TIVICAY contains dolutegravir, as dolutegravir sodium, an HIV INSTI. The chemical name of dolutegravir sodium is sodium (4R,12aS)-9-{[(2,4-difluorophenyl)methyl]carbamoyl}-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazin-7-olate. The empirical formula is C20H18F2N3NaO5 and the molecular weight is 441.36 g/mol. It has the following structural formula:
Dolutegravir sodium is a white to light yellow powder and is slightly soluble in water.
Each film-coated tablet of TIVICAY for oral administration contains 52.6 mg of dolutegravir sodium, which is equivalent to 50 mg dolutegravir free acid, and the following inactive ingredients: D-mannitol, microcrystalline cellulose, povidone K29/32, sodium starch glycolate, and sodium stearyl fumarate. The tablet film-coating contains the inactive ingredients iron oxide yellow, macrogol/PEG, polyvinyl alcohol-part hydrolyzed, talc, and titanium dioxide.

DOLUTEGRAVIR

http://blog.sina.com.cn/s/blog_de171b9b0101a1ah.html BELOW
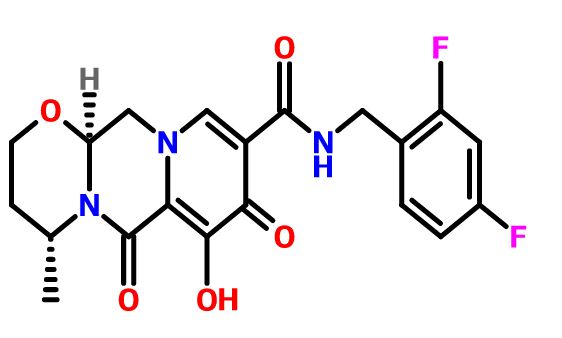
Dolutegravir Synthesis
Identifications:
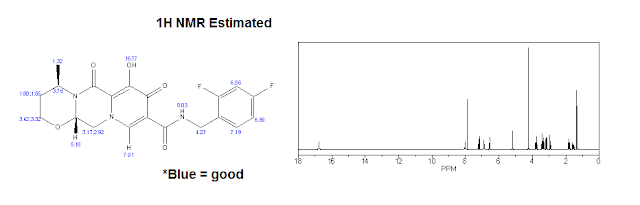 |
| 1H NMR (Estimated) for Dolutegravir |
Experimental: 1H NMR (CDCl3) δ 12.45 (s, 1H), 10.38 (br s, 1H), 8.30 (s, 1H), 7.40-7.30 (m, 1H), 6.85-6.75 (m, 2H), 5.26 (d, J = 5.8, 4.1 Hz, 2H), 5.05-4.95 (m, 1H), 4.64 (d, J = 5.9 Hz, 2H), 4.27 (dd, J = 13.4, 4.2 Hz, 1H), 4.12 (dd, J = 13.6, 6.0 Hz, 1H), 4.05 (t, J = 2.3 Hz, 1H), 4.02 (d, J = 2.2 Hz, 1H), 2.30-2.19 (m, 1H), 1.56 (dd, J = 14.0, 2.0 Hz, 1H), 1.42 (d, J = 7.0 Hz, 3H).
INTRODUCTION
Among viruses, human immunodeficiency virus (HIV), a kind of retrovirus, is known to cause acquired immunodeficiency syndrome (AIDS). The therapeutic agent for AIDS is mainly selected from a group of reverse transcriptase inhibitors (e.g., AZT, 3TC) and protease inhibitors (e.g., Indinavir), but they are proved to be accompanied by side effects such as nephropathy and the emergence of resistant viruses. Thus, the development of anti-HIV agents having the other mechanism of action has been desired.
On the other hand, a combination therapy is reported to be efficient in treatment for AIDS because of the frequent emergence of the resistant mutant. Reverse transcriptase inhibitors and protease inhibitors are clinically used as an anti-HIV agent, however agents having the same mechanism of action often exhibit cross-resistance or only an additional activity. Therefore, anti-HIV agents having the other mechanism of action are desired.
Under the circumstances above, an HIV integrase inhibitor has been focused on as an anti-HIV agent having a novel mechanism of action (Ref: Patent Documents 1 and 2). As an anti-HIV agent having such a mechanism of action, known are carbamoyl-substituted hydroxypyrimidinone derivative (Ref: Patent Documents 3 and 4) and carbamoyl-substituted hydroxypyrrolidione derivative (Ref: Patent Document 5). Further, a patent application concerning carbamoyl-substituted hydroxypyridone derivative has been filed (Ref: Patent Document 6, Example 8).
Other known carbamoylpyridone derivatives include 5-alkoxypyridine-3-carboxamide derivatives and γ-pyrone-3-carboxamide derivatives, which are a plant growth inhibitor or herbicide (Ref: Patent Documents 7-9).
Other HIV integrase inhibitors include N-containing condensed cyclic compounds (Ref: Patent Document 10).
- [Patent Document 1] WO03/0166275
- [Patent Document 2] WO2004/024693
- [Patent Document 3] WO03/035076
- [Patent Document 4] WO03/035076
- [Patent Document 5] WO2004/004657
- [Patent Document 6] JP Patent Application 2003-32772
- [Patent Document 7] JP Patent Publication 1990-108668
- [Patent Document 8] JP Patent Publication 1990-108683
- [Patent Document 9] JP Patent Publication 1990-96506
- [Patent Document 10] WO2005/016927
- Patent Document 1 describes compounds (I) and (II), which are useful as anti-HIV drugs and shown by formulae:
This document describes the following reaction formula as a method of producing compound (I).
Furthermore, Patent Documents 2 to 6 describe the following reaction formula as an improved method of producing compound (I).
[PATENT DOCUMENTS]
-
- [Patent Document 1] International publication No.2006/116764 pamphlet
- [Patent Document 2] International publication No.2010/011812 pamphlet
- [Patent Document 3] International publication No.2010/011819 pamphlet
- [Patent Document 4] International publication No.2010/068262 pamphlet
- [Patent Document 5] International publication No.2010/067176 pamphlet
- [Patent Document 6] International publication No.2010/068253 pamphlet
- [Patent Document 7] US Patent 4769380A
- [Patent Document 8] International applicationPCT/JP2010/055316
[NON-PATENT DOCUMENTS]
-
- [Non-Patent Document 1] Journal of Organic Chemistry, 1991, 56(16), 4963-4967
- [Non-Patent Document 2] Science of Synthesis, 2005, 15, 285-387
- [Non-Patent Document 3] Journal of Chemical Society Parkin Transaction. 1, 1997, Issue. 2, 163-169


A clip and its own references
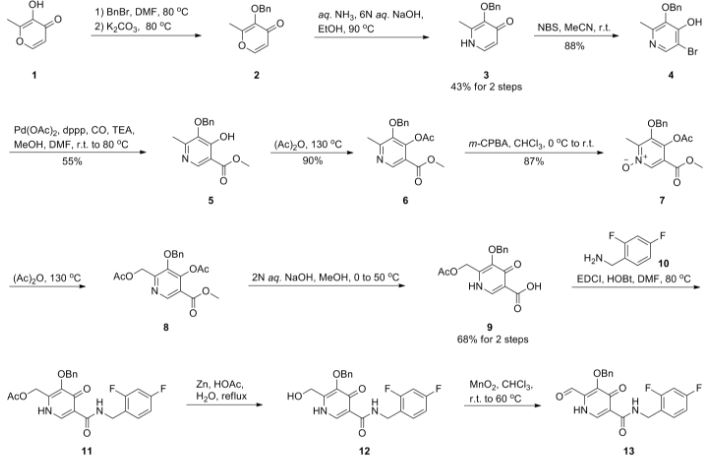
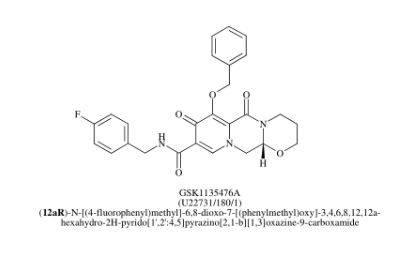
Dolutegravir sodium (Tivicay®), developed and marketed by GlaxoSmithKline,45 was approved by the FDA in August 2013 as a novel integrase inhibitor for the treatment of HIV infection.46 Dolutegravir was fast-tracked by the FDA in February 2012,47 and joins an important class of drugs known as Integrase Strand Transfer inhibitors (INSTi’s).48 INSTi’s are characterized by their two-metal-chelating scaffolds, which are known to chelate Mg2+ cofactors in the enzyme active site,49, 50 interrupting function of HIV-1 integrase, which is essential for replication of viral DNA into host chromatin.49-51,52 Other drugs in this class, raltegravir and elvitegravir, are known to require either high dosages53 or PK boosting agents,54 respectively, with raltegravir also exhibiting substantial loss of potency in several major HIV-1 integrase mutation pathways.55 Dolutegravir was pursued with the goal of developing a INSTi with a once-daily, low-dosage treatment with improved resistance profile and without the
need for the use of a PK boosting agent.51, 56 Dolutegravir sodium has been approved for treating a broad
population of HIV-infected patients, including adults undergoing their first treatment as well as those
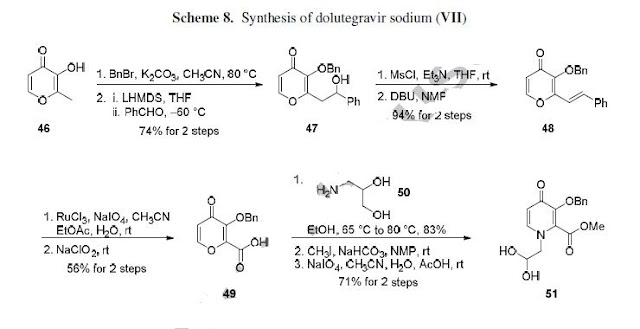
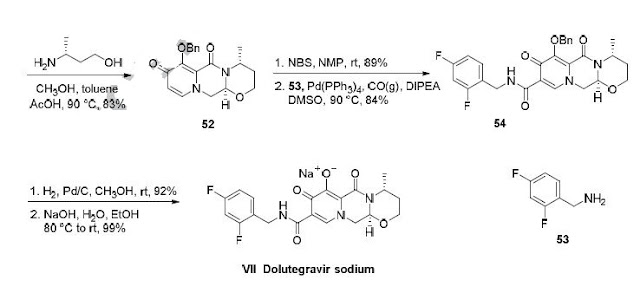
who have been treated with other integrase transfer strand inhibiting agents.46 The most likely process-scale synthesis of dolutegravir sodium, as described in Scheme 8, began with benzyl protection and alkylation of pyrone 46 with benzaldehyde, yielding alcohol 47 in 74% over 2 steps (Scheme 8).57, 58 Alcohol mesylation and in-situ elimination provided the styrenyl olefin 48 in 94% yield, which further underwent an oxidative cleavage of the olefin to generate 49 by sequential addition of RuCl3/NaIO4 and NaClO2 (56% overall yield). Treatment of pyranone 49 with 3-amino-propane-2-diol (50) in ethanol at elevated temperatures delivered the corresponding pyridinone in 83% yield, and this was followed by esterification and sodium periodate-mediated diol cleavage to furnish intermediate 51 in 71% overall yield across the two-step sequence.57, 58 Next, the key ring-forming step in the
synthesis of dolutegravir sodium consisted of cyclization of 51 with (R)-3-amino-butan-1-ol, a process which relies on substrate control to provide the desired tricyclic carbamoylpyridone system 52 in high stereoselectivity (20/1 in favor of the desired isomer).51 Previously, cyclization of systems such as 51 with unsubstituted amino alcohols were found to yield a mixture of diastereomeric products, therefore indicating the pivotal role of the chiral amino alcohol in influencing stereochemical bias during the overall cyclization step.51, 56 In practice, reaction of 51 with (R)-3-amino-butan-1-ol at 90 °C led to isolation of a single cyclization product 52, after recrystallization from EtOAc.57, 58 From 52, Nbromosuccinimide (NBS) bromination and subsequent treatment with amine 53 under palladiumcatalyzed amidocarbonylative conditions led to amide 54 in 75% yield over 2 steps. Finally, removal of the benzyl group and subsequent crystallization using sodium hydroxide in water and ethanol provided dolutegravir sodium (VII) in 99% yield.57, 58
45 Johns, B. A.; Kawasuji, T.; Taishi, T.; Taoda, Y. WO Patent 2006116764A1, 2006.
46. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm364744.htm.
47. http://newdrugapprovals.org/2013/07/16/dolutegravir-biggest-rival-to-worlds-best-selling-hivdrug-atripla-may-get-fda-approval-by-august-2013/.
48. Pendri, A.; Meanwell, N. A.; Peese, K. M.; Walker, M. A. Expert Opin. Ther. Pat. 2011, 21,1173.
49. Johns, B. A.; Svolto, A. C. Expert Opin. Ther. Pat. 2008, 18, 1225.60
50. Johns, B. A.; Weatherhead, J. G.; Allen, S. H.; Thompson, J. B.; Garvey, E. P.; Foster, S. A.;
Jeffrey, J. L.; Miller, W. H. Bioorg. Med. Chem. Lett. 2009, 19, 1802.
51. Johns, B. A.; Kawasuji, T.; Weatherhead, J. G.; Taishi, T.; Temelkoff, D. P.; Yoshida, H.;Akiyama, T.; Taoda, Y.; Murai, H.; Kiyama, R.; Fuji, M.; Tanimoto, N.; Jeffrey, J.; Foster, S.A.; Yoshinaga, T.; Seki, T.; Kobayashi, M.; Sato, A.; Johnson, M. N.; Garvey, E. P.; Fujiwara,
T. J. Med. Chem. 2013, 56, 5901.
52. Kawasuji, T.; Johns, B. A.; Yoshida, H.; Taishi, T.; Taoda, Y.; Murai, H.; Kiyama, R.; Fuji, M.;Yoshinaga, T.; Seki, T.; Kobayashi, M.; Sato, A.; Fujiwara, T. J. Med. Chem. 2012, 55, 8735.
53. Lennox, J. L.; De Jesus, E.; Lazzarin, A.; Pollard, R. B.; Valdez Ramalho Madruga, J.; Berger,D. S.; Zhao, J.; Xu, X.; Williams-Diaz, A.; Rodgers, A. J.; Barnard, R. J. O.; Miller, M. D.; DiNubile, M. J.; Nguyen, B.-Y.; Leavitt, R.; Sklar, P. Lancet 2009, 374, 796.
54. Ramanathan, S.; Mathias, A. A.; German, P.; Kearney, B. P. Clin. Pharmacokinet. 2011, 50,229.
55. Ceccherini-Silberstein, F.; Malet, I.; D’Arrigo, R.; Antinori, A.; Marcelin, A.-G.; Perno, C.-F.AIDS Rev. 2009, 11, 17.
56. Kawasuji, T.; Johns, B. A.; Yoshida, H.; Weatherhead, J. G.; Akiyama, T.; Taishi, T.; Taoda, Y.;Mikamiyama-Iwata, M.; Murai, H.; Kiyama, R.; Fuji, M.; Tanimoto, N.; Yoshinaga, T.; Seki, T.;Kobayashi, M.; Sato, A.; Garvey, E. P.; Fujiwara, T. J. Med. Chem. 2013, 56, 1124.
57. Johns, B. A.; Duan, M.; Hakogi, T. WO Patent 2010068262A1, 2010.
58. Yoshida, H.; Taoda, Y.; Johns, B. A. WO Patent 2010068253A1, 2010.
CLIPS
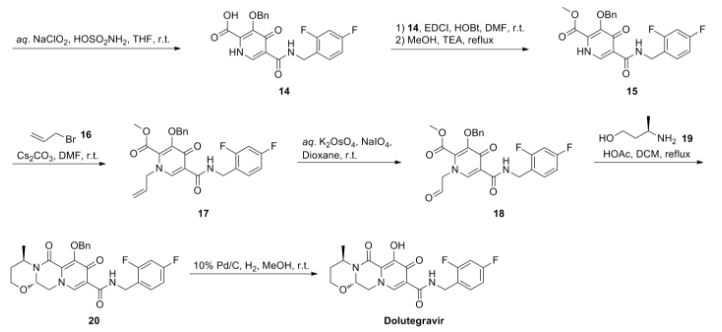
Dolutegravir synthesis (EP2602260, 2013). LiHMDS as the non-nucleophilic strong base pulling compound 1 carbonyl group proton alpha position with an acid chloride after 2 and ring closure reaction to obtain 3 , 3 via primary amine 4 ring opening ring closure to obtain 5 , NBS the bromine under acidic conditions to obtain aldehyde acetal becomes 6 , 6 of the aldehyde and amino alcohols 7 and turn off the condensation reaction obtained by the ring 8 , alkaline hydrolysis 8 of bromine into a hydroxyl group and hydrolyzable ester obtained 9 after the 10 occurred acid condensation Dolutegravir.

CLIPS
Synthesis of Dolutegravir (S/GSK1349572, GSK1349572)

SYNTHESIS
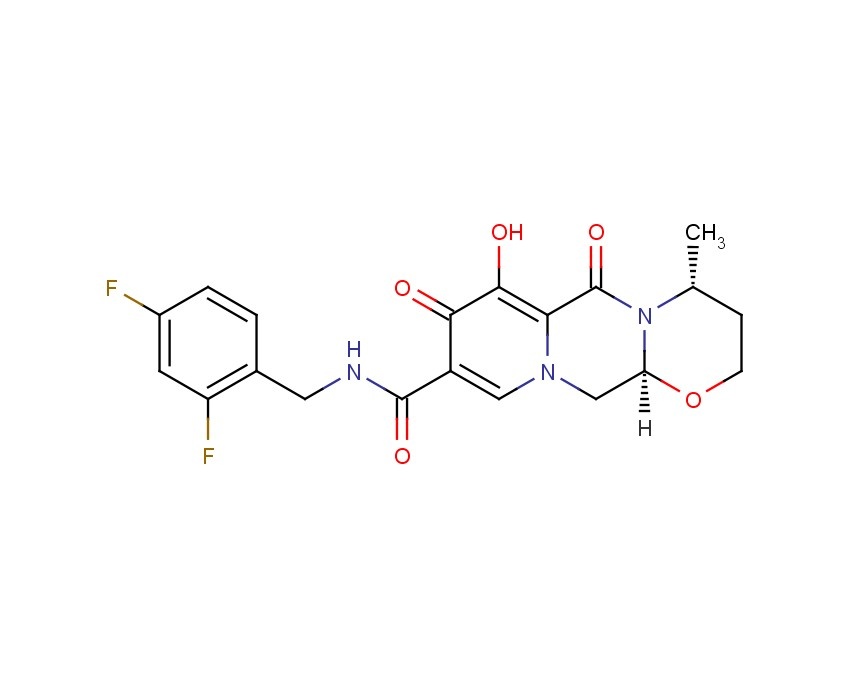
2H-Pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazine-9-carboxamide, N-[(2,4-difluorophenyl)methyl]-3,4,6,8,12,12a-hexahydro-7-hydroxy-4-methyl-6,8-dioxo-, (4R,12aS) ………..dolutegravir
PATENT
US8129385

Desired isomer
Example Z-1
(3R,11aS)—N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide sodium salt
a)
(3R,11aS)—N-[(2,4-Difluorophenyl)methyl]-3-methyl-5,7-dioxo-6-[(phenylmethyl)oxy]-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide. To a solution of 16a (409 mg, 0.87 mmol) in dichloroethane (20 mL) was added (2R)-2-amino-1-propanol (0.14 mL, 1.74 mmol) and 10 drops of glacial acetic acid. The resultant solution was heated at reflux for 2 h. Upon cooling, Celite was added to the mixture and the solvents removed in vacuo and the material was purified via silica gel chromatography (2% CH3OH/CH2Cl2 gradient elution) to give (3R,11aS)—N-[(2,4-difluorophenyl)methyl]-3-methyl-5,7-dioxo-6-[(phenylmethyl)oxy]-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide (396 mg, 92%) as a glass. 1H NMR (CDCl3) δ 10.38 (m, 1H), 8.42 (s, 1H), 7.54-7.53 (m, 2H), 7.37-7.24 (m, 4H), 6.83-6.76 (m, 2H), 5.40 (d, J=10.0 Hz, 1H), 5.22 (d, J=10.0 Hz, 1H), 5.16 (dd, J=9.6, 6.0 Hz, 1H), 4.62 (m, 2H), 4.41 (m, 1H), 4.33-4.30 (m, 2H), 3.84 (dd, J=12.0, 10.0 Hz, 1H), 3.63 (dd, J=8.4, 7.2 Hz, 1H), 1.37 (d, J=6.0 Hz, 3H); ES+MS: 496 (M+1).
b)
(3R,11aS)—N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide sodium salt. To a solution of (3R,11aS)—N-[(2,4-difluorophenyl)methyl]-3-methyl-5,7-dioxo-6-[(phenylmethyl)oxy]-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide (396 mg, 0.80 mmol) in methanol (30 mL) was added 10% Pd/C (25 mg). Hydrogen was bubbled through the reaction mixture via a balloon for 2 h. The resultant mixture was filtered through Celite with methanol and dichloromethane.
The filtrate was concentrated in vacuo to give (3R,11aS)—N-[(2,4-difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide , DOLUTEGRAVIR as a pink tinted white solid (278 mg, 86%).
1H NMR (CDCl3) δ 11.47 (m, 1H), 10.29 (m, 1H), 8.32 (s, 1H), 7.36 (m, 1H), 6.82 (m, 2H), 5.31 (dd, J=9.6, 3.6 Hz, 1H), 4.65 (m, 2H), 4.47-4.38 (m, 3H), 3.93 (dd, J=12.0, 10.0 Hz, 1H), 3.75 (m, 1H), 1.49 (d, J=5.6 Hz, 3H); ES+ MS: 406 (M+1).
DOLUTEGRAVIR NA SALT
The above material (278 mg, 0.66 mmol) was taken up in ethanol (10 mL) and treated with 1 N sodium hydroxide (aq) (0.66 ml, 0.66 mmol). The resulting suspension was stirred at room temperature for 30 min. Ether was added and the liquids were collected to provide the sodium salt of the title compound as a white powder (291 mg, 99%). 1H NMR (DMSO-d6) δ 10.68 (m, 1H), 7.90 (s, 1H), 7.35 (m, 1H), 7.20 (m, 1H), 7.01 (m, 1H), 5.20 (m, 1H), 4.58 (m, 1H), 4.49 (m, 2H), 4.22 (m, 2H), 3.74 (dd, J=11.2, 10.4 Hz, 1H), 3.58 (m, 1H), 1.25 (d, J=4.4 Hz, 3H).
UNDESIRED ISOMER
Example Z-9
(3S,11aR)—N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide sodium salt

The title compound was made in two steps using a similar process to that described in example Z-1. 16a (510 mg, 1.08 mmol) and (25)-2-amino-1-propanol (0.17 mL, 2.17 mmol) were reacted in 1,2-dichloroethane (20 mL) with acetic acid to give (3S,11aR)—N-[(2,4-difluorophenyl)methyl]-3-methyl-5,7-dioxo-6-[(phenylmethyl)oxy]-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide (500 mg, 93%). This material was hydrogenated in a second step as described in example Z-1 to give (3S,11aR)—N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide (386 mg, 94%) as a tinted white solid. 1H NMR (CDCl3) δ 11.46 (m, 1H), 10.28 (m, 1H), 8.32 (s, 1H), 7.35 (m, 1H), 6.80 (m, 2H), 5.30 (dd, J=10.0, 4.0 Hz, 1H), 4.63 (m, 2H), 4.48-4.37 (m, 3H), 3.91 (dd, J=12.0, 10.0 Hz, 1H), 3.73 (m, 1H), 1.48 (d, J=6.0 Hz, 3H); ES+ MS: 406 (M+1). This material (385 mg, 0.95 mmol) was treated with sodium hydroxide (0.95 mL, 1.0 M, 0.95 mmol) in ethanol (15 mL) as described in example Z-1 to provide its corresponding sodium salt (381 mg, 94%) as a white solid. 1H NMR (DMSO-d6) δ 10.66 (m, 1H), 7.93 (s, 1H), 7.33 (m, 1H), 7.20 (m, 1H), 7.01 (m, 1H), 5.19 (m, 1H), 4.59 (m, 1H), 4.48 (m, 2H), 4.22 (m, 2H), 3.75 (m, 1 H), 3.57 (m, 1H), 1.24 (d, J=5.6 Hz, 3H).
SYNTHESIS OF INTERMEDIATES

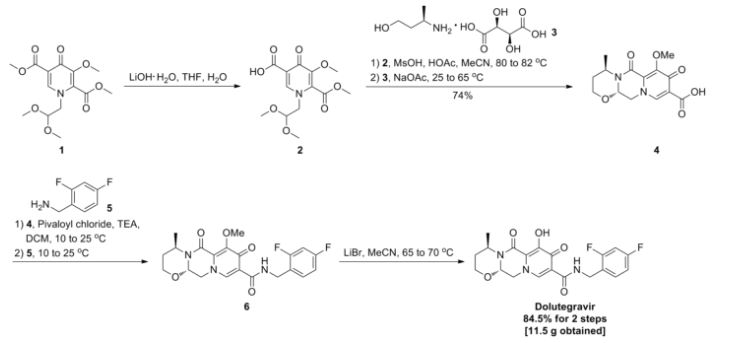
IN ABOVE SCHEME SYNTHESIS UPTO COMPD 9 MAY BE USEFUL IN SYNTHESIS BUT READERS DISCRETION IS SOUGHT IN THIS ?????????????????
1) Maltol 1 (189 g, 1.5 mol) was dissolved in dimethylformamide (1890 ml), and benzyl bromide (184 ml, 1.5 mol) was added. After the solution was stirred at 80° C. for 15 minutes, potassium carbonate (228 g, 1.65 mol) was added, and the mixture was stirred for 1 hour. After the reaction solution was cooled to room temperature, an inorganic salt was filtered, and the filtrate was distilled off under reduced pressure. To the again precipitated inorganic salt was added tetrahydrofuran (1000 ml), this was filtered, and the filtrate was distilled off under reduced pressure to obtain the crude product (329 g, >100%) of 3-benzyloxy-2-methyl-pyran-4-one 2 as a brown oil.
NMR (CDCl3) δ: 2.09 (3H, s), 5.15 (2H, s), 6.36 (1H, d, J=5.6 Hz), 7.29-7.41 (5H, m), 7.60 (1H, d, J=5.6 Hz).
2) The compound 2 (162.2 g, 750 mmol) was dissolved in ethanol (487 ml), and aqueous ammonia (28%, 974 ml) and a 6N aqueous sodium hydroxide solution (150 ml, 900 mmol) were added. After the reaction solution was stirred at 90° C. for 1 hour, this was cooled to under ice-cooling, and ammonium chloride (58 g, 1080 mmol) was added. To the reaction solution was added chloroform, this was extracted, and the organic layer was washed with an aqueous saturated sodium bicarbonate solution, and dried with anhydrous sodium sulfate. The solvent was distilled off under reduced pressure, isopropyl alcohol and diethyl ether were added to the residue, and precipitated crystals were filtered to obtain 3-benzyloxy-2-methyl-1H-pyridine-4-one 3 (69.1 g, 43%) as a pale yellow crystal.
NMR (DMSO-d6) δ: 2.05 (3H, s), 5.04 (2H, s), 6.14 (1H, d, J=7.0 Hz), 7.31-7.42 (5H, m), 7.46 (1H, d, J=7.2 Hz), 11.29 (1H, brs).
3) The above compound 3 (129 g, 699 mmol) was suspended in acetonitrile (1300 ml), and N-bromosuccinic acid imide (117 g, 659 mmol) was added, followed by stirring at room temperature for 90 minutes. Precipitated crystals were filtered, and washed with acetonitrile and diethyl ether to obtain 3-benzyloxy-5-bromo-2-methyl-pyridine-4-ol 4 (154 g, 88%) as a colorless crystal.
NMR (DMSO-d6) δ: 2.06 (3H, s), 5.04 (2H, s), 7.32-7.42 (5H, m), 8.03 (1H, d, J=5.5 Hz), 11.82 (1H, brs).
4) To a solution of the compound 4 (88 g, 300 mmol), palladium acetate (13.4 g, 60 mmol) and 1,3-bis(diphenylphosphino)propane (30.8 g, 516 mmol) in dimethylformamide (660 ml) were added methanol (264 ml) and triethylamine (210 ml, 1.5 mol) at room temperature. The interior of a reaction vessel was replaced with carbon monoxide, and the material was stirred at room temperature for 30 minutes, and stirred at 80 degree for 18 hours. A vessel to which ethyl acetate (1500 ml), an aqueous saturated ammonium chloride solution (1500 ml) and water (1500 ml) had been added was stirred under ice-cooling, and the reaction solution was added thereto. Precipitates were filtered, and washed with water (300 ml), ethyl acetate (300 ml) and diethyl ether (300 ml) to obtain 5-benzyloxy-4-hydroxy-6-methyl-nicotinic acid methyl ester 5 (44.9 g, 55%) as a colorless crystal.
NMR (DMSO-d6) δ: 2.06 (3H, s), 3.72 (3H, s), 5.02 (2H, s), 7.33-7.42 (5H, m), 8.07 (1H, s).
5) After a solution of the compound 5 (19.1 g, 70 mmol) in acetic anhydride (134 ml) was stirred at 130° C. for 40 minutes, the solvent was distilled off under reduced pressure to obtain 4-acetoxy-5-benzyloxy-6-methyl-nicotinic acid methyl ester 6 (19.9 g, 90%) as a flesh colored crystal.
NMR (CDCl3) δ: 2.29 (3H, s), 2.52 (3H, s), 3.89 (3H, s), 4.98 (2H, s), 7.36-7.41 (5H, m), 8.85 (1H, s).
6) To a solution of the compound 6 (46.2 g, 147 mmol) in chloroform (370 ml) was added metachloroperbenzoic acid (65%) (42.8 g, 161 mmol) in portions under ice-cooling, and this was stirred at room temperature for 90 minutes. To the reaction solution was added a 10% aqueous potassium carbonate solution, and this was stirred for 10 minutes, followed by extraction with chloroform. The organic layer was washed with successively with a 10% aqueous potassium carbonate solution, an aqueous saturated ammonium chloride solution, and an aqueous saturated sodium chloride solution, and dried with anhydrous sodium sulfate. The solvent was distilled off under induced pressure, and the residue was washed with diisopropyl ether to obtain 4-acetoxy-5-benzyloxy-6-methyl-1-oxy-nicotinic acid methyl ester 7 (42.6 g, 87%) as a colorless crystal.
NMR (CDCl3) δ: 2.30 (3H, s), 2.41 (3H, s), 3.90 (3H, s), 5.02 (2H, s), 7.37-7.39 (5H, m), 8.70 (1H, s).
7) To acetic anhydride (500 ml) which had been heated to stir at 130° C. was added the compound 7 (42.6 g, 129 mmol) over 2 minutes, and this was stirred for 20 minutes. The solvent was distilled off under reduced pressure to obtain 4-acetoxy-6-acetoxymethyl-5-benzyloxy-nicotinic acid methyl ester 8 (49.6 g, >100%) as a black oil.
NMR (CDCl3) δ: 2.10 (3H, s), 2.28 (3H, s), 3.91 (3H, s), 5.07 (2H, s), 5.20 (2H, s), 7.35-7.41 (5H, m), 8.94 (1H, s).
8) To a solution of the compound 8 (46.8 g, 125 mmol) in methanol (140 ml) was added a 2N aqueous sodium hydroxide solution (376 ml) under ice-cooling, and this was stirred at 50° C. for 40 minutes. To the reaction solution were added diethyl ether and 2N hydrochloric acid under ice-cooling, and precipitated crystals were filtered. Resulting crystals were washed with water and diethyl ether to obtain 5-benzyloxy-4-hydroxy-6-hydroxymethyl-nicotinic acid 9 (23.3 g, 68%) as a colorless crystal.
NMR (DMSO-d6) δ: 4.49 (2H, s), 5.19 (2H, s), 5.85 (1H, brs), 7.14-7.20 (2H, m), 7.33-7.43 (7H, m), 8.30 (1H, s), 10.73 (1H, t, J=5.8 Hz), 11.96 (1H, brs).
9) To a solution of the compound 9 (131 g, 475 mmol), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (219 g, 1140 mmol) and 1-hydroxybenzotriazole (128 g, 950 mmol) in dimethylformamide (1300 ml) was added 4-fluorobenzylamine (109 ml, 950 mmol), and this was stirred at 80° C. for 1.5 hours. After the reaction solution was cooled to room temperature, hydrochloric acid was added, followed by extraction with ethyl acetate. The extract was washed with a 5% aqueous potassium carbonate solution, an aqueous saturated ammonium chloride solution, and an aqueous saturated sodium chloride solution, and dried with anhydrous sodium sulfate. The solvent was distilled off under reduced pressure to obtain a mixture (175 g) of 10 and 11. the resulting mixture was dissolved in acetic acid (1050 ml) and water (1050 ml), and zinc (31.1 g, 475 mmol) was added, followed by heating to reflux for 1 hour. After the reaction solution was cooled to room temperature, a 10% aqueous potassium carbonate solution was added, followed by extraction with ethyl acetate. The extract was washed with an aqueous saturated ammonium chloride solution, and an aqueous saturated sodium chloride solution, and dried with anhydrous sodium sulfate. After the solvent was distilled off under reduced pressure, this was washed with diethyl ether to obtain 5-benzyloxy-N-(4-fluoro-benzyl)-4-hydroxy-6-hydroxymethyl-nicotinic acid amide 10 (107 g, 59%) as a colorless crystal.
NMR (DMSO-d6) δ: 4.45 (2H, d, J=4.3 Hz), 4.52 (2H, d, J=5.8 Hz), 5.09 (2H, s), 6.01 (1H, brs), 7.36-7.43 (5H, m), 8.31 (1H, s), 12.63 (1H, brs).
PATENT
SYNTHESIS
EP2602260A1
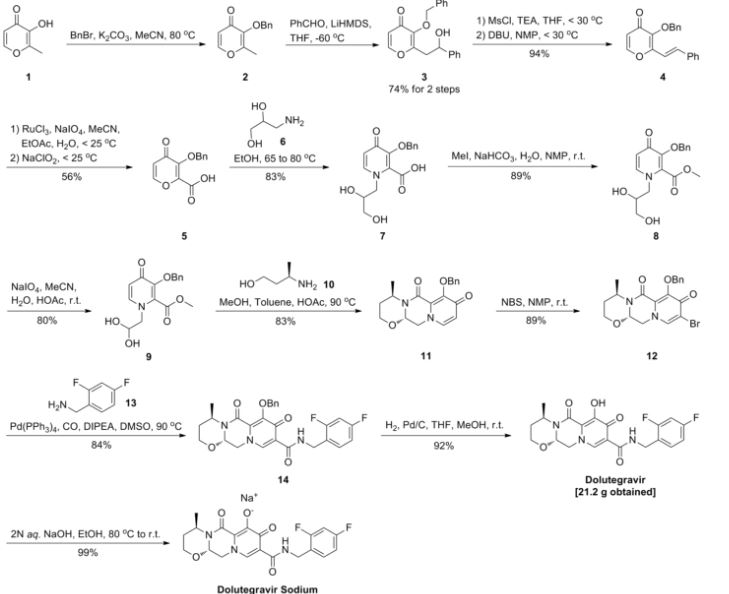
Example 3

3H IS DOLUTEGRAVIR
Step 1
N,N-dimethylformamide dimethyl acetal (4.9 ml, 36.5 mmol) was added dropwise to compound 3A (5.0 g, 30.4 mmol) under cooling at 0°C. After stirring at 0°C for 1 hour, 100 ml of ethyl acetate was added to the reaction solution, and the organic layer was washed with a 0.5 N aqueous hydrochloric acid solution (50 ml). The aqueous layer was separated, followed by extraction with ethyl acetate (50 ml). The organic layers were combined, washed with a saturated aqueous solution of sodium bicarbonate and saturated saline in this order, and then dried over anhydrous sodium sulfate. The solvent was distilled off, and the obtained residue was purified by silica gel column chromatography (n-hexane-ethyl acetate: 1:1 (v/v) → ethyl acetate) to obtain 4.49 g (yield: 67%) of compound 3B as an oil.
1H-NMR (CDCl3)δ:1.32 (3H, t, J = 7.1 Hz), 2.90 (3H, br s), 3.29 (3H, br s), 4.23 (2H, q, J = 7.1 Hz), 4.54 (2H, s), 7.81 (1H, s).
Step 2
Lithium hexamethyldisilazide (1.0 M solution in toluene, 49 ml, 49.0 mmol) was diluted with tetrahydrofuran (44 ml). A tetrahydrofuran (10 ml) solution of compound 3B (4.49 g, 20.4 mmol) was added dropwise thereto under cooling at -78°C, and a tetrahydrofuran (10 ml) solution of ethyl oxalyl chloride (3.35 g, 24.5 mmol) was then added dropwise to the mixture. The mixture was stirred at -78°C for 2 hours and then heated to 0°C. 2 N hydrochloric acid was added to the reaction solution, and the mixture was stirred for 20 minutes, followed by extraction with ethyl acetate (200 ml x 2). The organic layer was washed with a saturated aqueous solution of sodium bicarbonate and saturated saline and then dried over anhydrous sodium sulfate. The solvent was distilled off, and the obtained residue was purified by silica gel column chromatography (n-hexane-ethyl acetate: 7:3 → 5:5 → 0:10 (v/v)) to obtain 1.77 g (yield: 31%) of compound 3C as a white solid.
1H-NMR (CDCl3)δ:1.36-1.46 (6H, m), 4.35-4.52 (8H, m), 8.53 (1H, s).
Step 3
Aminoacetaldehyde dimethyl acetal (0.13 ml, 1.20 mmol) was added to an ethanol (6 ml) solution of compound 3C (300 mg, 1.09 mmol) at 0°C, and the mixture was stirred at 0°C for 1.5 hours, then at room temperature for 18 hours, and at 60°C for 4 hours. The solvent in the reaction solution was distilled off under reduced pressure, and the obtained residue was then purified by silica gel column chromatography (n-hexane-ethyl acetate: 5:5 → 0:10 (v/v)) to obtain 252 mg (yield: 64%) of compound 3D as an oil.
1H-NMR (CDCl3)δ:1.36-1.47 (6H, m), 3.42 (6H, s), 3.90 (2H, d, J = 5.2 Hz), 4.37 (3H, q, J = 7.2 Hz), 4.50 (2H, q, J = 7.2 Hz), 8.16 (1H, s).
Step 4
62% H2SO4 (892 mg, 5.64 mmol) was added to a formic acid (10 ml) solution of compound 3D (1.02 g, 2.82 mmol), and the mixture was stirred at room temperature for 16 hours. The formic acid was distilled off under reduced pressure. To the residue, methylene chloride was added, and the mixture was pH-adjusted to 6.6 by the addition of a saturated aqueous solution of sodium bicarbonate. The methylene chloride layer was separated, while the aqueous layer was subjected to extraction with methylene chloride. The methylene chloride layers were combined and dried over anhydrous sodium sulfate. The solvent was distilled off to obtain 531.8 mg of compound 3E as a yellow oil.
1H-NMR (CDCl3) δ: 1.28-1.49 (6H, m), 4.27-4.56 (4H, m), 4.84 (2H, s), 8.10 (1H, s), 9.72 (1H, s).
Step 5
Methanol (0.20 ml, 5.0 mmol), (R)-3-amino-butan-1-ol (179 mg, 2.0 mmol), and acetic acid (0.096 ml, 1.70 mmol) were added to a toluene (5 ml) solution of compound 3E (531 mg, 1.68 mmol), and the mixture was heated to reflux for 4 hours. The reaction solution was cooled to room temperature, then diluted with chloroform, and then washed with a saturated aqueous solution of sodium bicarbonate. The aqueous layer was subjected to extraction with chloroform. The chloroform layers were combined, washed with saturated saline, and then dried over anhydrous sodium sulfate. The solvent was distilled off, and the obtained residue was purified by silica gel column chromatography (chloroform-methanol: 100:0 → 90:10) to obtain 309.4 mg of compound 3F as a brown oil.
1H-NMR (CDCl3) δ: 1.40 (3H, t, J = 7.1 Hz), 1.40 (3H, d, J = 7.1 Hz), 1.55-1.61 (1H, m), 2.19-2.27 (1H, m), 4.00 (1H, d, J = 1.5 Hz), 4.03 (1H, d, J = 2.5 Hz), 4.10 (1H, dd, J = 13.2, 6.3 Hz), 4.26 (1H, dd, J = 13.2, 3.8 Hz), 4.38 (2H, q, J = 7.1 Hz), 5.00-5.05 (1H, m), 5.31 (1H, dd, J = 6.4, 3.9 Hz), 8.10 (1H, s).
Step 6
Potassium trimethylsilanolate (333 mg, 2.34 mmol) was added to a 1,2-dimethoxyethane (2 ml) solution of compound 3F (159 mg, 0.47 mmol), and the mixture was stirred at room temperature for 7 hours. 1 N hydrochloric acid and saturated saline were added to the reaction solution, followed by extraction with chloroform. The chloroform layers were combined and dried over anhydrous sodium sulfate. The solvent was distilled off to obtain 34.4 mg (yield: 25%) of compound 3G as an orange powder.
1H-NMR (CDCl3) δ: 1.46 (3H, d, J = 3.5 Hz), 1.58-1.65 (1H, m), 2.26-2.30 (1H,m), 4.06-4.10 (2H, m), 4.31 (1H, dd, J = 13.8, 5.6 Hz), 4.48 (1H, dd, J = 13.6, 3.9 Hz), 5.03 (1H, t, J = 6.4 Hz), 5.36 (1H, dd, J = 5.5, 4.0 Hz), 8.44 (1H, s), 12.80 (1H, s), 14.90 (1H, s).
Step 7
Compound 3G (16 mg, 0.054 mmol) and 2,4-difluorobenzylamine (17 mg, 0.12 mmol) were dissolved in N,N-dimethylformamide (1 ml). To the solution, N,N,N’,N’-tetramethyl-O-(7-aza-benzotriazol-1-yl)uronium hexafluorophosphate (HATU) (53 mg, 0.14 mmol) and N-methylmorpholine (0.031 ml, 0.28 mmol) were added, and the mixture was stirred at room temperature for 16 hours. 2,4-difluorobenzylamine (17 mg, 0.12 mmol), HATU (64 mg, 0.17 mmol), and N-methylmorpholine (0.037 ml, 0.34 mmol) were further added thereto, and the mixture was stirred at room temperature for additional 16 hours. 0.5 N hydrochloric acid was added to the reaction solution, followed by extraction with ethyl acetate. The ethyl acetate layers were combined, washed with 0.5 N hydrochloric acid and then with saturated saline, and then dried over anhydrous sodium sulfate. The solvent was distilled off, and the obtained residue was purified by preparative high-performance liquid chromatography to obtain 12.5 mg (yield: 55%) of compound 3H as an orange solid.
DOLUTEGRAVIR
1H-NMR (DMSO-d6) δ: 1.36 (3H, d, J = 6.9 Hz), 1.55-1.60 (1H, m), 2.01-2.05 (1H, m), 3.92-3.94 (1H, m), 4.04 (1H, t, J = 12.6 Hz), 4.38-4.41 (1H, m), 4.57-4.60 (1H, m), 4.81-4.83 (1H, m), 5.46-5.49 (1H, m), 7.08-7.11 (1H, m), 7.25-7.30 (1H, m), 7.41 (1H, dd, J = 15.3, 8.7 Hz), 8.53 (1H, s), 10.38 (1H, s), 12.53 (1H, s).
ISOMERS OF DOLUTEGRAVIR
Reference Example 1


Step 1
Acetic acid (180 mg, 3.00 mmol) was added to a toluene (90 ml) solution of compound A-1 (4.39 g, 9.33 mmol) and (R)-3-aminobutan-1-ol (998 mg, 11.2 mmol), and the mixture was stirred at 50°C for 90 minutes. The reaction solution was allowed to cool to room temperature and then poured to a saturated aqueous solution of sodium bicarbonate. The organic layer was separated, while the aqueous layer was subjected to extraction three times with ethyl acetate. The combined extracts were washed with saturated saline and then dried over sodium sulfate. The solvent was distilled off to obtain 4.29 g of crude product A-2.
Step 2
The crude product A-2 obtained in the preceding step was dissolved in ethanol (40 ml). To the solution, a 2 N aqueous sodium hydroxide solution (20 ml) was added at room temperature, and the mixture was stirred at the same temperature for 2 hours. The reaction solution was neutralized to pH 7 using a 2 N aqueous hydrochloric acid solution. The solvent was directly distilled off. The obtained crude product A-3 was subjected to azeotropy with toluene (100 ml) and used in the next step without being purified.
Step 3
HOBt (1.65 g, 12.2 mmol) and WSC HCl (2.34 g, 12.2 mmol) were added at room temperature to a DMF (100 ml) solution of the crude product A-3 obtained in the preceding step, and the mixture was stirred at the same temperature for 15 hours. Water was added to the reaction solution, followed by extraction three times with ethyl acetate. The combined extracts were washed with water three times and then dried over sodium sulfate. The solvent was distilled off, and the obtained oil was subjected to silica gel column chromatography for purification. Elution was performed first with n-hexane-ethyl acetate (3:7, v/v) and then with only ethyl acetate. The fraction of interest was concentrated, and the obtained oil was then dissolved in ethyl acetate. The solution was crystallized with diisopropyl ether as a poor solvent. The obtained crystals were collected by filtration and dissolved again in ethyl acetate. The solution was recrystallized to obtain 1.84 g of compound A-4.
1HNMR (CDCl3) δ: 1.49 (3H, d, J = 6.6 Hz), 1.88-1.96 (1H, m), 2.13-2.26 (1H, m), 3.90-4.17 (4H, m), 4.42-4.47 (1H, m), 4.63 (2H, d, J = 6.0 Hz), 5.12-5.17 (1H, m), 5.17 (1H, d, J = 9.9 Hz), 5.33 (1H, d, J = 9.9 Hz), 6.77-6.87 (2H, m), 7.27-7.42 (4H, m), 7.59-7.62 (2H, m), 8.35 (1H, s), 10.41 (1H, t, J = 5.7 Hz).
Step 4
The compound A-4 was subjected to the hydroxy deprotection reaction described in Step F of the paragraph [0088] to obtain compound A-5.
1HNMR (DMSO-d6) δ:1.41 (3H, d, J = 6.3 Hz), 1.85-1.92 (1H, m), 1.50-1.75 (1H, m), 4.02-4.09 (3H, m), 4.28-4.34 (1H, m), 4.53 (2H, d, J = 5.7 Hz), 4.64 (1H, dd, J = 3.9 Hz, 12.6 Hz), 5.45 (1H, dd, J = 3.6 Hz, 9.3 Hz), 7.06 (1H, ddd, J = 2.7 Hz, 8.4 Hz, 8.4 Hz), 7.20-7.28 (1H, m), 7.35-7.42 (1H, m), 8.43 (1H, s),10.37 (1H, t, J = 6.0 Hz),12.37 (1H, brs).
Reference Example 2

Compound A-1 was reacted with (S)-3-aminobutan-1-ol in Step 1. Compound B-5 was obtained in the same way as in Reference Example 1.
-
1HNMR (DMSO-d6) δ:1.41 (3H, d, J = 6.3 Hz), 1.85-1.92 (1H, m), 1.50-1.75 (1H, m), 4.02-4.09 (3H, m), 4.28-4.34 (1H, m), 4.53 (2H, d, J = 5.7 Hz), 4.64 (1H, dd, J = 3.9 Hz, 12.6 Hz), 5.45 (1H, dd, J = 3.6 Hz, 9.3 Hz), 7.06 (1H, ddd, J = 2.7 Hz, 8.4 Hz, 8.4 Hz), 7.20-7.28 (1H, m), 7.35-7.42 (1H, m), 8.43 (1H, s),10.37 (1H, t, J = 6.0 Hz),12.37 (1H, brs).
PATENT
W02006116764

ENTRY 68
PATENT
WO 2010068262
PATENT
WO 2010068253
PATENT
WO 2011119566
PATENT
Synthesis
WO 2012018065
Example 3

I was under cooling added dropwise at 0 ℃ (4.9 ml, 36.5 mmol) and N, N-dimethylformamide dimethyl acetal (5.0 g, 30.4 mmol) in the first step compound 3A. After stirring for 1 hour at 0 ℃, ethyl acetate was added to 100ml, the reaction mixture was washed with 0.5N aqueous hydrochloric acid (50 ml). Was extracted with ethyl acetate (50ml) and solution was separated and the aqueous layer. The organic layers were combined, washed successively with saturated aqueous sodium bicarbonate solution and saturated brine, and then dried over anhydrous sodium sulfate. After the solvent was distilled off, silica gel column chromatography and the residue obtained was – and purified by (n-hexane (v / v) → ethyl acetate 1:1) to an oil (67% yield) of Compound 3B 4.49 g I got a thing.
1 H-NMR (CDCl 3)δ: 1.32 (3H, t, J = 7.1 Hz), 2.90 (3H, br s), 3.29 (3H, br s), 4.23 (2H, q, J = 7.1 Hz), 4.54 (2H, s), 7.81 (1H, s).
Diluted with tetrahydrofuran (44 ml) (1.0M toluene solution, 49 ml, 49.0 mmol) the second step lithium hexamethyldisilazide, under cooling at -78 ℃, compound 3B (4.49 g, 20.4 mmol) in this After dropwise tetrahydrofuran (10 ml) was added dropwise tetrahydrofuran (3.35 g, 24.5 mmol) of ethyl oxalyl chloride and (10 ml) solution. After stirring for 2 hours at -78 ℃, I was warmed to 0 ℃. After washing (200 ml x 2), saturated aqueous sodium bicarbonate solution and the organic layer with saturated brine After stirring for 20 minutes, extracted with ethyl acetate by adding 2N hydrochloric acid, the reaction solution was dried over anhydrous sodium sulfate. After removal of the solvent, silica gel column chromatography and the residue obtained – was purified (n-hexane (v / v) ethyl acetate 7:3 → 5:5 → 0:10), compound 3C 1.77 g (yield I as a white solid 31%).
1 H-NMR (CDCl 3)δ :1.36-1 .46 (6H, m), 4.35-4.52 (8H, m), 8.53 (1H, s).
Was added at 0 ℃ (0.13 ml, 1.20 mmol) the aminoacetaldehyde dimethyl acetal ethanol (300 mg, 1.09 mmol) of the third step compound 3C to (6 ml) solution, 1 hour and 30 minutes at 0 ℃, 18 hours at room temperature , then I was stirred for 4 hours at 60 ℃. After the solvent was evaporated under reduced pressure and the reaction mixture by silica gel column chromatography and the residue obtained was – and purified by (n-hexane (v / v) ethyl acetate 5:5 → 0:10), compound 3D 252 mg (yield: I got as an oil 64%) rate.
1 H-NMR (CDCl 3)δ :1.36-1 .47 (6H, m), 3.42 (6H, s), 3.90 (2H, d, J = 5.2 Hz), 4.37 (3H, q, J = 7.2 Hz), 4.50 (2H, q, J = 7.2 Hz), 8.16 (1H, s).
Was added (892 mg, 5.64 mmol) and 2 SO 4 62-H% formic acid (1.02 g, 2.82 mmol) in a fourth step the compound for 3D (10 ml) solution was stirred at room temperature for 16 hours. Methylene chloride was added to the residue Shi distilled off under reduced pressure and formic acid was adjusted to pH = 6.6 by addition of saturated aqueous sodium bicarbonate. The solution was separated methylene chloride layer was extracted with methylene chloride and the aqueous layer. I was dried over anhydrous sodium sulfate combined methylene chloride layers. The solvent was then distilled off and was obtained as a yellow oil 531.8 mg compound 3E.
1H-NMR (CDCl3) δ: 1.28-1.49 (6H, m), 4.27-4.56 (4H, m), 4.84 (2H, s), 8.10 (1H, s), 9.72 (1H, s).
Amino – – butane – 1 – ol (179 mg, 2.0 mmol), methanol (0.20 ml, 5.0 mmol), (R) -3 toluene (531 mg, 1.68 mmol) in the fifth step to compound 3E (5 ml) solution was added (0.096 ml, 1.70 mmol) acetic acid was heated under reflux for 4 hours. After dilution with chloroform, cooled to room temperature, the reaction mixture was washed with a saturated aqueous sodium bicarbonate solution, and the aqueous layer was extracted with chloroform. After washing with saturated brine combined chloroform layer was dried over anhydrous sodium sulfate. The solvent was then distilled off, silica gel column chromatography and the residue obtained – and (chloroform methanol 100:0 → 90:10), was obtained as a brown oil 309.4 mg compound 3F.
1H-NMR (CDCl3) δ: 1.40 (3H, t, J = 7.1 Hz), 1.40 (3H, d, J = 7.1 Hz), 1.55-1.61 (1H, m), 2.19-2.27 (1H, m), 4.00 (1H, d, J = 1.5 Hz), 4.03 (1H, d, J = 2.5 Hz), 4.10 (1H, dd, J = 13.2, 6.3 Hz), 4.26 (1H, dd, J = 13.2, 3.8 Hz ), 4.38 (2H, q, J = 7.1 Hz), 5.00-5.05 (1H, m), 5.31 (1H, dd, J = 6.4, 3.9 Hz), 8.10 (1H, s).
1,2 (159 mg, 0.47 mmol) in the sixth step compound 3F – was added (333 mg, 2.34 mmol) and potassium trimethylsilanolate dimethoxyethane (2 ml) solution was stirred for 7 hours at room temperature. Brine was added to the 1N-hydrochloric acid to the reaction mixture, followed by extraction with chloroform. The combined chloroform layer was dried over anhydrous sodium sulfate. The solvent was removed by distillation, and I as an orange powder (25% yield) of compound 3G 34.4 mg.
1H-NMR (CDCl3) δ: 1.46 (3H, d, J = 3.5 Hz), 1.58-1.65 (1H, m), 2.26-2.30 (1H, m), 4.06-4.10 (2H, m), 4.31 (1H , dd, J = 13.8, 5.6 Hz), 4.48 (1H, dd, J = 13.6, 3.9 Hz), 5.03 (1H, t, J = 6.4 Hz), 5.36 (1H, dd, J = 5.5, 4.0 Hz) , 8.44 (1H, s), 12.80 (1H, s), 14.90 (1H, s).
2,4 (16 mg, 0.054 mmol) and the seventh step compound 3G – was dissolved in N, N-dimethylformamide (1 ml) (17 mg, 0.12 mmol) difluorobenzyl amine, N, N, N ‘, N was added (0.031 ml, 0.28 mmol) and N-methylmorpholine uronium hexafluorophosphate (HATU) (53 mg, 0.14 mmol), and ‘- tetramethyl-O-(yl 7 – aza – – benzo triazolopyrimidine -1) I was stirred at room temperature for 16 h. 2,4 – was added (0.037 ml, 0.34 mmol) and N-methylmorpholine (64 mg, 0.17 mmol) and (17 mg, 0.12 mmol), HATU difluorobenzylamine, and the mixture was stirred for 16 hours at room temperature. I was extracted with ethyl acetate addition of 0.5N-hydrochloric acid to the reaction mixture. 0.5N-hydrochloric acid and then was washed with saturated brine, and dried over anhydrous sodium sulfate and combined ethyl acetate layer. The solvent was then distilled off, and purified by preparative high performance liquid chromatography residue was obtained as an orange solid (55% yield) of compound 3H 12.5 mg.
1H-NMR (DMSO-d6) δ: 1.36 (3H, d, J = 6.9 Hz), 1.55-1.60 (1H, m), 2.01-2.05 (1H, m), 3.92-3.94 (1H, m), 4.04 (1H, t, J = 12.6 Hz), 4.38-4.41 (1H, m), 4.57-4.60 (1H, m), 4.81-4.83 (1H, m), 5.46-5.49 (1H, m), 7.08-7.11 (1H, m), 7.25-7.30 (1H, m), 7.41 (1H, dd, J = 15.3, 8.7 Hz), 8.53 (1H, s), 10.38 (1H, s), 12.53 (1H, s)
PAPER
http://pubs.acs.org/doi/abs/10.1021/jm400645w
Carbamoyl Pyridone HIV-1 Integrase Inhibitors 3. A Diastereomeric Approach to Chiral Nonracemic Tricyclic Ring Systems and the Discovery of Dolutegravir (S/GSK1349572) and (S/GSK1265744)
Brian A. Johns*†, Takashi Kawasuji‡, Jason G. Weatherhead†, Teruhiko Taishi‡, David P. Temelkoff†, Hiroshi Yoshida‡, Toshiyuki Akiyama‡, Yoshiyuki Taoda‡, Hitoshi Murai‡, Ryuichi Kiyama‡, Masahiro Fuji‡, Norihiko Tanimoto‡, Jerry Jeffrey†, Scott A. Foster†, Tomokazu Yoshinaga‡, Takahiro Seki‡, Masanori Kobayashi‡, Akihiko Sato‡, Matthew N. Johnson†, Edward P. Garvey†, and Tamio Fujiwara‡
† GlaxoSmithKline Research & Development, Infectious Diseases Therapeutic Area Unit, Five Moore Drive, Research Triangle Park, North Carolina 27709, United States
‡ Shionogi Pharmaceutical Research Center, Shionogi & Co., Ltd., 3-1-1 Futaba-cho, Toyonaka-shi, Osaka 561-0825, Japan
J. Med. Chem., 2013, 56 (14), pp 5901–5916
DOI: 10.1021/jm400645w
J. Med. Chem. 2013, 56, 5901-5916.

We report herein the discovery of the human immunodeficiency virus type-1 (HIV-1) integrase inhibitors dolutegravir (S/GSK1349572) (3) and S/GSK1265744 (4). These drugs stem from a series of carbamoyl pyridone analogues designed using a two-metal chelation model of the integrase catalytic active site. Structure–activity studies evolved a tricyclic series of carbamoyl pyridines that demonstrated properties indicative of once-daily dosing and superior potency against resistant viral strains. An inherent hemiaminal ring fusion stereocenter within the tricyclic carbamoyl pyridone scaffold led to a critical substrate controlled diastereoselective synthetic strategy whereby chiral information from small readily available amino alcohols was employed to control relative and absolute stereochemistry of the final drug candidates. Modest to extremely high levels of stereochemical control were observed depending on ring size and position of the stereocenter. This approach resulted in the discovery of 3 and 4, which are currently in clinical development.
(4R,12aS)-N-(2,4-Difluorobenzyl)-7-hydroxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[1′,2′:4,5]pyrazino-
[2,1-b][1,3]oxazine-9-carboxamide (3). 1H NMR (CDCl3) δ 12.45 (s, 1H),10.38 (br s, 1H), 8.30 (s, 1H), 7.40−7.30 (m, 1H), 6.85−6.75 (m, 2H),5.26 (d, J = 5.8, 4.1 Hz, 2H), 5.05−4.95 (m, 1H), 4.64 (d, J = 5.9 Hz,2H), 4.27 (dd, J = 13.4, 4.2 Hz, 1H), 4.12 (dd, J = 13.6, 6.0 Hz, 1H), 4.05(t, J = 2.3 Hz, 1H), 4.02 (d, J = 2.2 Hz, 1H), 2.30−2.19 (m, 1H), 1.56(dd, J = 14.0, 2.0 Hz, 1H), 1.42 (d, J = 7.0 Hz, 3H). ES+ LC/MS: m/zcalcd 419.13; found 420.13 (M + 1)+.
(4R,12aS)-N-(2,4-Difluorobenzyl)-7-hydroxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[1′,2′:4,5]pyrazino-
[2,1-b][1,3]oxazine-9-carboxamide (3) sodium salt.
1H NMR(DMSO-d6) δ 10.70 (t, J = 6.0 Hz, 1H), 7.89 (s, 1 H), 7.40−7.30 (m, 1H), 7.25−7.16 (m, 1H), 7.06−6.98 (m, 1H), 5.22−5.12 (m, 1H), 4.87−4.74 (m, 1H), 4.51 (d, J = 5.4 Hz, 2H), 4.35−4.25 (m, 1 H), 4.16 (dd, J =1.8, 14.1 Hz, 1 H), 4.05−3.90 (m, 1H), 3.86−3.74 (m, 1 H), 2.00−1.72(m, 1 H), 1.44−1.32 (m, 1 H), 1.24 (d, J = 6.9 Hz, 3H).
MORE UPDATES……………………………
Process for preparing integrase inhibitors such as dolutegravir and cabotegravir and their analogs, useful for treating viral infections eg HIV infection. Also claims a process for preparing intermediates of dolutegravir and cabotegravir.
(4R, 12aS)-N-[(2,4-Difluorophenyl)methyl]-3 ,4,6,8, 12, 12a-hexahydro-7-hydroxy-4-methyl-6,8-dioxo-2H-pyrido[1 ‘,2’:4,5]pyrazino[2, 1-b][1 ,3]oxazine-9-carboxamide (Formula A):

Formula A
known by the INN name dolutegravir, is a new efficient antiviral agent from the group of HIV integrase inhibitors which is used in combination with some other antiviral agents for treatment of HIV infections, such as AIDS. The compound, which belongs to condensed polycyclic pyridines and was first disclosed in WO2006/1 16764, is marketed.
Another compound disclosed in WO2006/1 16764 is (3S, 1 1 aR)-N-[(2,4-difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7, 1 1 ,1 1 a-hexahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-d]pyrazine-8-carboxamide (Formula

Formula C
known by the INN name cabotegravir.
The complex structures of dolutegravir and cabotegravir present a synthetic challenge. The first description of the synthesis in WO2006/1 16764 shows a 16-steps synthesis (see Scheme A), which is industrially impractical due to its length and low overall yield.

Scheme A
WO 2010/068253 and WO 2006/1 16764 describe an alternative synthesis. The 1 1 -step synthesis, shown in Scheme B1 and Scheme B2, is based on bromination of the 9-position for further introduction of the carboxylic group. The synthesis relies on the use of expensive palladium catalysts and toxic selenium compounds. Furthermore, some variations of these approaches involve pyrone intermediates in several steps. In some cases pyrones are liquids which can complicate purification, while further reactions form complex mixtures.


 doiutegravir
doiutegravir
Scheme B2
In further alternative syntheses, acetoacetates were used as starting materials. Such an approach is challenging in terms of introducing the hydroxy group in the 7-position. The variation in Scheme C1 , described in WO2012/018065, starts from 4-benzyloxyacetoacetate. The procedure requires 9 steps, but use expensive reagents like palladium catalysts. Moreover, there is described a possibility of formation a co-crystal between an intermediate and hydroquinone, wherein however the additional step may diminish yields and make the process longer and time consuming.

Scheme C1
The variation in Scheme C2, described in WO2012/018065, starts from 4-chloroacetoacetate. The process is not optimal because of problems in steps which include pyrones and because of problems with conversion of 7-chloro to 7-hydroxy group which includes a disadvantageous use of silanolates with low yield (25%).

Scheme C2
The variation in Scheme C3, described in WO201 1/1 19566, starts from unsubstituted acetoacetate. For the introduction of the 7-hydroxy group, bromination is used and substitution of bromo with hydroxy is performed by a use of silanolates. The substitution of the bromine is achieved in a 43% yield.

Scheme C3
The variation in Scheme C4, described in WO201 1/1 19566, starts from 4-methoxyacetoacetate aiming at preparing dolutegravir or cabotegravir. The process uses lithium bases to affect a difficult to control selective monohydrolysis of a diester.

PATENT
WO 2016113372
Carbotegravir, New Patent, WO 2016113372, Lek Pharmaceutical and Chemical Co DD
LEK PHARMACEUTICALS D.D. [SI/SI]; Verovskova 57 1526 Ljubljana (SI)
MARAS, Nenad; (SI).
SELIC, Lovro; (SI).
CUSAK, Anja; (SI)
ViiV Healthcare is developing cabotegravir (first disclosed in WO2006088173), which in July 2016, was reported to be in phase 2 clinical development.
WO-2016113372
The object of the present invention is to provide short, simple, cost-effective, environmentally friendly and industrially suitable processes for beneficially providing dolutegravir and analogues thereof and cabotegravir and analogues thereof, in particular dolutegravir.
Scheme 1

According to an embodiment of the process of the invention the building block 3-aminobutanol can suitably be substituted with other aminoalcohols to give dolutegravir analogues. For example, using (S)-alaninol gives cabotegravir as the final product. Similarly, using amines other than 2,4-difluorobenzylamine in the amidation step results in the synthesis of other dolutegravir analogues.
According to the another preferred embodiment cabotegravir or a pharmaceutically acceptable salt thereof is prepared by the analogue process, which comprises providing a compound of formula (5c)

5c
converting the compound of formula (5c) to a compound of formula (6c)

6c
by carrying out a chlorination reaction, and converting the compound of formula (6c) to cabotegravir and/or a pharmaceutically acceptable salt thereof.
The compound of formula (5c) can preferably be provided by converting a compound of formula (3) to a compound of formula (4c)

Scheme 2

1. ) EtOCOCI, Et3N / Me2CO
2. ) 2,4-difiuorobenzylamine

Scheme 3

Analogous compound of formula 7c is a useful intermediate in the synthesis of cabotegravir. Scheme 3a

Scheme 4

Examples
The following examples are merely illustrative of the present invention and they should not be considered as limiting the scope of the invention in any way. The examples and modifications or other equivalents thereof will become apparent to those versed in the art in the light of the present entire disclosure. Particularly, all Examples related to the preparation of dolutegravir and intermediates thereof can be used by the analogy for the preparation of cabotegravir and intermediates thereof.
Example 1 :

Methyl acetoacetate (1 , 25.22 g) and dimethylformamide dimethyl acetal (DMFDMA, 35 mL) was heated at 50-55°C for 2 h, then methanol (60 mL), aminoacetaldehyde dimethyl acetal (24 mL) and acetic acid (4 mL) was added an the mixture was heated under reflux for one hour, then concentrated. MTBE (100 mL) was added and the mixture was kept at 5 °C overnight to crystallize. Upon filtration 46 g (92%) of product 2 was recovered.
1H NMR (DMSO-d6): δ 2.31 (s, 3H), 3.30 (s, 6H), 3.49 (m, 2H), 3.61 (s, 3H), 4.43 (m, 1 H), 8.02 (d, 1 H), 10.8 (bs, 1 H). 13C NMR (DMSO-d6): δ 30.52, 35.48, 50.53, 54.23, 98.99, 102.47, 160.70, 166.92, 197.21 .
Example 2:

Compound 2 (5.00 g) was dissolved in 2-propanol, dimethyl oxalate (7.02 g) was added and heated to 40 °C. Sodium methylate (25% in methanol; 20 mL) was slowly (10 min) added, the mixture was then heated to 50-55 °C and stirred at that temperature for 2-2.5 h. The mixture was cooled to ambient temperature, then sodium hydroxide solution (1 M, 65 mL) was added to the mixture and stirred for another 2 h, followed by addition of concentrated hydrochloric acid (1 1 mL) and stirred for another 2 h. The precipitate was filtered and dried to give 8.08 g (NMR assay 47%; 65% yield) of compound 3.
1H NMR (DMSO-d6): δ 2.50 (m, 2H), 3.30 (s. 6H), 4.49 (m, 1 H), 7.06 (s, 1 H); 8.70 (s, 1 H). 13C NMR (DMSO-d6): δ 55.23, 55.37, 102.34, 1 15.47, 120.24, 145.17, 162.71 , 165.22, 178.55.
Example 3:

Compound 2 (158.37 g) was dissolved in methanol (548 mL), followed by the addition of dimethyl oxalate (202.2 g). While keeping the temperature below 30°C, potassium ferf-butoxide (192.1 g) was added and reaction mixture was heated at 50 °C overnight. The suspension was then filtered and the filter cake washed with methanol. The filtrate was concentrated (approximately to 680 mL), then water (680 mL) was added, followed by addition of lithium hydroxide hydrate (143.7 g) while keeping the temperature below 40 °C. The suspension was then stirred at ambient temperature overnight and filtered. To the obtained filtrate, concentrated hydrochloric acid (339 mL) was added while keeping the temperature below 30 °C. The suspension was aged for 2 h and filtered to give 4 as a white powder (95.6 g, NMR assay 100%; 52% yield).
Example 4:

Compound 2 (5.00 g) was dissolved in 2-propanol, dimethyl oxalate (7.02 g) was added and heated to 40 °C. Sodium methylate (25% in methanol; 15 mL) was slowly (10 min) added then the mixture was heated to 50-55 °C and stirred at that temperature for 72 h. The mixture was concentrated and components were separated by flash column chromatography (ethyl acetate/methanol 9:1 to 6:4). Early fractions gave compound 22 upon concentration, late fractions gave compound 23.
Compound 22: 1H NMR (DMSO-d6): δ 2.49 (m, 2H), 3.28 (s, 6H), 3.73 (s, 3H), 3.85 (s, 3H), 4.41 (m, 1 H), 4.50 (m, 1 H), 6.65 (s, 1 H), 8.36 (s, 1 H). 13C NMR (DMSO-d6): δ 51.63, 53.36, 54.25, 55.47, 102.71 , 1 18.24, 123.60, 140.81 , 150.21 , 162.44, 164.49, 173.43.
Compound 23: 1H NMR (DMSO-d6): δ 2.49 (m, 2H), 3.26 (s, 6H); 3.70 (s, 3H); 4.33 (d, 1 H); 4.60 (m, 1 H), 6.19 (s, 1 H), 8.12 (s, 1 H). 13C NMR (DMSO-d6): δ 50.03, 51.34, 54.59, 54.85, 102.91 , 1 16.04, 1 18.19, 148.32, 152.12, 163.46, 165.24, 174.99
Example 5:

Compound 3 (5.5 g; assay 53%) was suspended in acetonitrile, acetic acid (6 mL) and methanesulfonic acid (2.5 mL) were added followed by the heating of mixture to 70 °C for 4 h. The suspension was filtered and filtrate cooled to ambient temperature. Triethylamine (6.6 mL) and (R)-3-amino-butan-1 -ol (1.24 mL) was added followed by heating the mixture at reflux temperature for 20-24 h. The mixture was filtered, filtrate concentrated and 1 M HCI (100 mL) was added, followed by extraction with dichloromethane (3 x 50 mL). Combined organic fractions were concentrated, 2-propanol was added (10 mL) and suspension was stirred at 70-80 °C for 10 min, left to cool to ambient temperature then filtered to give 2.19 g of compound 4 (73%).
1H NMR (DMSO-de): δ 1.31 (d, 3H), 1.52 (m, 1 H), 1 .97 (m, 1 H), 3.89 (m, 1 H), 4.01 (m, 1 H), 4.46 (m, 1 H), 4.64 (m, 1 H), 4.78 (m, 1 H), 5.50 (m, 1 H), 7.29 (s, 1 H), 8.88 (s, 1 H), 15.83 (s, 1 H). 13C NMR (DMSO-d6): δ 15.22, 29.14, 45.26, 51.13, 62.09, 76.03, 1 16.31 , 1 18.79, 140.53, 146.79, 155.36, 165.24, 178.75.
Example 6:

Compound 3 (14.55 g; assay 49%) was suspended in acetonitrile (125 mL), acetic acid (15 mL) and methanesulfonic acid (6.25 mL) were added followed by the heating of mixture to 70 °C for 4 h. The suspension was filtered and filtrate cooled to ambient temperature. Triethylamine (16.5 mL) and (S)-2-aminopropanol (2.45 mL) was added followed by heating the mixture at reflux temperature for 24 h. The insoluble product was filtered, washed with 2-propanol (20 mL) and dried to give (3S, 1 1 aR)-3-methyl-5,7-dioxo-2,3,5,7, 1 1 ,1 1 a-hexahydrooxazolo[3,2-a]pyrido[1 ,2-d]pyrazine-8-carboxylic acid (5.2 g, 75%).
1H NMR (DMSO-d6): δ 1.31 (d, J = 6.3 Hz, 3H), 3.65 (dd, J = 8.6, 6.8 Hz, 1 H), 4.13 (dd, J = 1 1.7, 10.3 Hz, 1 H), 4.28 (m, 1 H), 4.39 (dd, J = 8.6, 6.8 Hz, 1 H), 4.92 (dd, J = 12.3, 4.2 Hz, 1 H), 5.45 (dd, J = 10.2, 4.1 Hz, 1 H), 7.16 (s, 1 H), 8.84 (s, 1 H), 15.74 (s, 1 H).
Example 7:

Compound 4 (0.63 g) was dissolved in dichloromethane (15 mL), cooled to 5°C, then triethylamine (0.31 mL) was added, followed by ethyl chloroformate (0.26 mL), followed by slow (30 min) addition of 2,4-difluorobenzylamine. The mixture was then stirred at ambient temperature for 24 h. Water (10 mL) was added, organic phase was separated and washed with 1 M HCI (15 mL) and water (15 mL), concentrated and treated with 2-propanol to give the product 5 in a quantitative yield.
1H NMR (CDCI3): δ 1.39 (d, 3H), 1.52 (s, 1 H), 2.19 (m, 1 H), 4.00 (m, 2H), 4.16 (m, 1 H), 4.31 (m, 1 H), 4.62 (d, 2H), 5.00 (m, 1 H), 5.27 (m, 1 H), 6.80 (m 2H), 7.33 (m, 2H), 8.49 (s, 1 H), 10.48 (s, 1 H). 13C NMR (CDCI3): 15.50, 29.22, 36.43, 45.19, 51.83, 62.79, 103.71 , 103.91 , 1 1 1 .0, 1 1 1 .18, 120.59, 123.04, 130.40, 137.41 , 144.58, 156.27, 163. 87, 177.83.
Example 8:

To a suspension of 4 (2.84 g, 10 mmol) in a mixture of triethylamine (2.24 mL, 16 mmol) and acetone (50 mL) stirring on an ice bath was added ethyl chloroformate (1 .20 mL, 12 mmol). After stirring for 10 min, 2,4-difluorobenzylamine (1.21 mL, 10 mmol) was added and the mixture left stirring at room temperature for 1 h. The product was isolated by slowly diluting the reaction mixture with water (50 mL), partial concentration, filtration, washing with water (2 50 mL) and drying. There was obtained 5 as a white powder (3.48 g, 86%): mp 181.0-184.7 °C.1H NMR (DMSO-d6): δ 1.29 (d, J = 7.0 Hz, 3H), 1 .56 (dd, J = 13.9, 2.0 Hz, 1 H), 1 .93-2.06 (m, 1 H), 3.90 (ddd, J = 1 1.6, 5.0, 2.1 Hz, 1 H), 3.98 (td, J = 12.0, 2.2 Hz, 1 H), 4.45 (dd, J = 13.6, 6.6 Hz, 1 H), 4.72 (dd, J = 13.6, 3.8 Hz, 1 H), 4.74-4.81 (m, 1 H), 5.44 (dd, J = 6.6, 3.8 Hz, 1 H), 8.93 (s, 1 H), 15.14 (s, 1 H). 13C NMR (DMSO-d6): δ 15.78, 29.13, 44.89, 52.88, 61 .63, 75.61 , 1 13.54, 128.49, 136.42, 145.64, 154.62, 164.58, 174.58
Example 9:

To a suspension of 4 (1 1.36 g, 40 mmol) in acetonitrile (80 mL) stirring at room temperature was added TCCA (9.29 g, 38 mmol) and DABCO (0.23 g, 5 mol%). After stirring at room temperature for 1 h, the reaction was quenched with a mixture of DMSO (5.26 mL) and water (1.33 mL). The insoluble cyanuric acid was removed by filtration and the filtrate evaporated under reduced pressure to give viscous oil. This was triturated in methanol (20 mL) to induce crystallization. The product was filtered, washed with cold methanol (10 mL) and dried to give 7 as a yellowish powder (5.13 g, 41 %): mp 191 .3-198.7 °C.
Example 10:

Attempted chlorination of 23: Compound 23 (0.54g) was suspended in acetonitrile (10 mL) and trichlorocyanuric acid (0.44 g) was added and the solution was stirred at ambient temperature overnight. Precipitate was filtered. Only traces of a product corresponding to the compound 26 could be detected in the reaction mixture by LC-MS analysis. Conversion did not improve with time.
Example 11 :

Attempted chlorination of 3: Compound 3 (0.30 g) was suspended in acetonitrile (5 mL) and trichlorocyanuric acid (0.13 g) was added. The suspension was stirred at ambient temperature overnight. Only traces of a product corresponding to the compound 24 could be detected in the reaction mixture by LC-MS analysis.
Example 12:

9 10
Trichloroisocyanuric acid (0.23 g) was added in a single portion to a stirred solution of the diethyl 1 -(2,2-dimethoxyethyl)-4-oxo-1 ,4-dihydropyridine-2,5-dicarboxylate (9, 0.66 g) in dry acetonitrile (4 mL) at room temperature. The resulting suspension was stirred at room temperature for ca. 24 h. The reaction mixture was diluted with dichloromethane and filtrated. The filtrate was then concentrated in vacuo to afford crude oil (0.86 g). Purification by flash chromatography (eluting ethyl acetate/cyclohexane) furnished diethyl 3-chloro-1 -(2,2-dimethoxyethyl)-4-oxo-1 ,4-dihydropyridine-2,5-dicarboxylate, 10 as a yellow semi-solid (0.38 g). 1H NMR (CDCI3): δ 1.28 (t, J=7A Hz, 3H), 1 .37 (t, J=7.2 Hz, 3H), 3.35 (s, 6H), 3.89 (d, J=5.0 Hz, 2H), 4.27 (q, J=l A Hz, 2H), 4.43 (q, J=l A Hz, 2H), 4.48 (t, J=4.9 Hz, 1 H), 8.15 (s, 1 H). 13C NMR (CDCI3): δ 13.83, 14.13, 55.82, 57.09, 61.41 , 63.72, 102.52, 1 17.35, 126.90, 140.22, 146.92, 160.67, 164.13, 168.95.
Example 13:

Diethyl 1 -(2,2-dimethoxyethyl)-4-oxo-1 ,4-dihydropyridine-2,5-dicarboxylate (9, 0.64 g) was dissolved in anhydrous acetonitrile (6 mL) and treated sequentially with acetic acid (560 μί) and methanesulfonic acid (40 μί). The resulting mixture was heated to 62 °C and stirred for 4 h and more methanesulfonic acid (40 μΙ_) was added. After additional 2 h, more methanesulfonic acid (80 μΙ_) was added. This was repeated after additional 2 h, when more methanesulfonic acid (80 μΙ_) was added. The reaction mixture was stirred additional 17 h at 62 °C then was treated with a mixture of (R)-3-aminobutanol (0.22 g), triethylamine (0.5 mL) and acetonitrile (0.7 mL). The reaction mixture was stirred additional 22 h at 62 °C and then concentrated in vacuo. The crude material was partitioned between dichloromethane and 1 M HCI solution (15 mL). The combined organic phases were dried (Na2S04), filtered and concentrated in vacuo to afford the crude (4R, 12aS)-ethyl 4-methyl-6,8-dioxo-3,4,6,8, 12,12a-hexahydro-2H-pyrido[1 ‘,2’:4,5]pyrazino[2, 1 -b][1 ,3]oxazine-9-carboxylate (11 ) as a brownish oil (0.61 g).
1H NMR (CD3OD): δ 8.44 (s, 1 H), 7.16 (m, 1 H), 5.48 (t, J=4.8 Hz, 1 H), 4.86 (m, 1 H), 4.49 (dd, J=13.6, 4.0 Hz, 1 H), 4.30-4.25 (m, 3H), 4.09 (dt, J=12.1 , 2.3 Hz, 1 H), 3.96 (ddd, J=1 1.7, 5.0, 2.1 Hz, 1 H), 2.18-2.10 (m, 1 H), 1.60-1 .56 (m, 1 H) 1 .39 (d, J=7A Hz, 3H), 1.33 (t, J=7A Hz, 3H). 13C NMR (CDCI3): δ 8.45, 14.08, 15.39, 29.17, 45.04, 45.72, 51 .56, 60.86, 62.61 , 76.33, 1 19.54, 123.72, 136.96, 145.67, 156.26, 163.68, 175.43
Example 14:

10
Diethyl 3-chloro-1 -(2,2-dimethoxyethyl)-4-oxo-1 ,4-dihydropyridine-2,5-dicarboxylate (10, 1.23 g) was dissolved in 85% formic acid (25 mL) at room temperature. The mixture was warmed to 40 °C and stirred for 23 h. The reaction mixture was concentrated in vacuo, and then partitioned between dichloromethane and aqueous NaHC03 solution. The combined organic phases were dried (Na2S04), filtered and concentrated in vacuo to afford brownish oil (0.49 g). The crude oil was dissolved in anhydrous toluene (5 mL) and treated sequentially with (R)-3-aminobutanol (0.19 g), methanol (0.2 mL) and acetic acid (96 μί). The resulting mixture was heated to 90 °C and stirred for 20 h. The reaction mixture was cooled to room temperature and then partitioned between dichloromethane and aqueous NaHC03 solution. The combined organic phases were dried (Na2S04), filtered and concentrated in vacuo to afford the crude (4R,12aS)-Ethyl 7-chloro-4-methyl-6,8-dioxo-3,4,6,8,12, 12a-hexahydro-2H-pyrido[1 ‘,2’:4,5] pyrazino [2, 1-b][1 ,3]oxazine-9-carboxylate (12) as a brownish oil (0.24 g).
Example 15:

To a solution of 4 (5.68 g, 20 mmol) in dichloromethane (50 mL) stirring in an ice bath was added triethylamine (5.6 mL, 40 mmol), followed by ethyl chloroformate (2.61 mL, 26 mmol). After 20 min, ethanol (50 mL) was added. The mixture was then left stirring 24 h at room temperature and concentrated under reduced pressure. The residue was triturated in acetone (80 mL). The insoluble salt (triethylamine hydrochloride) was removed by filtration. The filtrate was evaporated under reduced pressure to give 11 as an amorphous solid in a quantitative yield (6.1 g).
Example 16:

To a stirring solution of 11 (0.94 g, 3.0 mmol) in acetonitrile (8 mL) heated at 40 °C was added TCCA in portions during 1 h (0.44 g, 1 .8 mmol). After an additional 1 h, the reaction mixture was diluted with a solution of NaHS03 (0.60 g) in water (60 mL), extracted with dichloromethane (50 mL) and the extract evaporated under reduced pressure to give a crude product which was purified by flash chromatography (CH2CI2 : MeOH, from 98 : 2 to 80 : 20) to give 12 (0.45 g, 44%).
1H NMR (CDCI3): δ 1.37 (t, J = 7.1 Hz, 3H), 1.38 (d, J = 7.0 Hz, 3H), 1 .56 (dq, J = 13.9, 2.2 Hz, 1 H), 2.21 (m, 1 H), 3.99 (d, J = 2.3 Hz, 1 H), 4.00 (t, J = 1.8 Hz, 1 H), 4.10 (dd, J = 13.2, 6.6 Hz, 1 H), 4.37-4.27 (m, 3H), 4.98 (m, 1 H), 5.35 (dd, J = 6.6, 3.8 Hz, 1 H), 8.07 (s, 1 H).
13C NMR (CDCI3): δ 14.20, 16.09, 29.34, 44.87, 53.73, 61.49, 62.29, 76.01 , 1 16.22, 133.1 1 , 134.18, 144.52, 155.48, 163.88, 169.98.
Example 17:

To a mixture of 7 (3.89 g, 12.2 mmol) in methanol (12 mL) was added sodium methylate (22.3 mL, 97.6 mmol). The reaction mixture was stirred for 24 h at 30 °C and then quenched with a slow addition of 3M hydrochloric acid (35 mL) while stirring in an ice bath. The mixture was concentrated under reduced pressure to remove most of the methanol, then extracted with dichloromethane (2 30 mL), the combined extracts washed with water (30 mL) and evaporated under reduced pressure. Methanol (20 mL) was added to the obtained amorphous residue and removed under reduced pressure to yield the solid 8 (3.69 g, 98%).
1H NMR (CDCI3): δ 15.04 (s, 1 H), 8.42 (s, 1 H), 5.29 (dd, J=5.6, 3.9 Hz, 1 H), 5.01 -4.96 (m, 1 H), 4.42 (dd, J=13.6, 3.6 Hz, 1 H), 4.25 (dd, J=13.6, 6.0 Hz, 1 H), 4.05 (s, 3H), 4.00-3.97 (m, 2H), 2.21 -2-14 (m, 1 H), 1.53 (dd, J=14.1 , 1.9 Hz, 1 H), 1.36 (d, J=7 Hz, 3H). 13C NMR (CDCI3): δ 176.35, 165.94, 155.03, 153.70, 143.08, 130.90, 1 15.94, 76.05, 62.65, 61.45, 53.86, 44.96, 29.43, 16.06.
Example 18:

To a suspension of 7 (2.55 g, 8.0 mmol) in a mixture of triethylamine (1 .46 mL, 10.4 mmol) and acetone (32 mL) stirring on an ice bath was added ethyl chloroformate (0.88 mL, 8.8 mmol). After stirring for 10 min, 2,4-difluorobenzylamine (1.07 mL, 8.8 mmol) was added and the mixture left stirring at room temperature for 1 h. The product was isolated by slowly diluting the reaction mixture with water (40 mL), filtration, washing with water (2 30 mL) and drying. There was obtained 2.91 g of 6 as a white powder (83%).
1H NMR (CDCI3): δ 1.30 (d, J = 7.0 Hz, 3H), 1 .49 (dd, J = 14.0, 2.2 Hz, 1 H), 2.14 (ddd, J = 14.6, 1 1.1 , 6.4 Hz, 1 H), 3.89-3.95 (m, 2H), 4.09-4.15 (m, 1 H), 4.26 (dd, J = 13.4, 3.8 Hz, 1 H), 4.55 (d, J = 5.8 Hz, 2H), 4.89-4.98 (m, 1 H), 5.18 (dd, J = 6.2, 3.8 Hz, 1 H), 6.68-6.79 (m, 2H), 7.23-7.31 (m, 1 H), 8.41 (s, 1 H), 10.24 (t, J = 5.8 Hz, 1 H). 13C NMR (CDCI3): δ 16.09, 26.95, 29.30, 36.79, 45.1 1 , 45.28, 53.86, 62.47, 75.93, 103.87 (t, J = 25.4 Hz), 1 1 1 .21 (dd, J = 21 .0, 3.4 Hz), 1 17.32, 130.58 (dd, J = 9.3, 5.8 Hz), 133.40, 143.54, 155.34, 163.16, 163.25, 163.35, 172.88.
Example 19:

To a suspension of 5 (1 .67 g, 4 mmol) in acetonitrile (20 mL) was added DABCO (23 mg, 5 mol%) and TCCA (0.62 g, 2.52 mmol). The mixture was stirred 18 h at 40 °C protected from light and then quenched with a mixture of DMSO (0.48 mL) and water (0.12 mL). The insoluble cyanuric acid was removed by filtration and washed with acetonitrile (5 mL). The filtrate was evaporated under reduced pressure to give viscous oil that was crystallized from a mixture of methanol (6 mL) and water (3 mL), by slowly cooling the solution from 60 °C to room
temperature. The product 6 was filtered, washed with cold methanol (5 mL) and dried to give an off-white powder (1.07 g, 61 %).
1H NMR (CDCI3): δ 1.30 (d, J = 7.0 Hz, 3H), 1 .49 (dd, J = 14.0, 2.2 Hz, 1 H), 2.14 (ddd, J = 14.6, 1 1.1 , 6.4 Hz, 1 H), 3.89-3.95 (m, 2H), 4.09-4.15 (m, 1 H), 4.26 (dd, J = 13.4, 3.8 Hz, 1 H), 4.55 (d, J = 5.8 Hz, 2H), 4.89-4.98 (m, 1 H), 5.18 (dd, J = 6.2, 3.8 Hz, 1 H), 6.68-6.79 (m, 2H), 7.23-7.31 (m, 1 H), 8.41 (s, 1 H), 10.24 (t, J = 5.8 Hz, 1 H). 13C NMR (CDCI3): δ 16.09, 26.95, 29.30, 36.79, 45.1 1 , 45.28, 53.86, 62.47, 75.93, 103.87 (t, J = 25.4 Hz), 1 1 1 .21 (dd, J = 21.0, 3.4 Hz), 1 17.32, 130.58 (dd, J = 9.3, 5.8 Hz), 133.40, 143.54, 155.34, 163.16, 163.25, 163.35, 172.88.
Example 20:

To a suspension of 6 (0.44 g) in anhydrous methanol (1 mL) was added a 25% methanolic solution of sodium methylate (1 .14 mL) and the mixture stirred for 4 h at 40 °C. The reaction was quenched with acetic acid (0.4 mL), diluted with water (8 mL), extracted with 2-methyltetrahydrofuran (12 mL), the extract washed with 1 M NaOH(aq) (8 mL), water (8 mL) and evaporated under reduced pressure. To the oily residue was added methanol (8 mL) and evaporated under reduced pressure to give 27 as a white solid (0.38 g, 88%).
Example 21 :

The suspension of (4R, 12aS)-7-chloro-N-(2,4-difluorobenzyl)-4-methyl-6,8-dioxo-3,4,6,8,12, 12a-hexahydro-2H-pyrido[1 ‘,2’:4,5]pyrazino[2, 1 -b][1 ,3]oxazine-9-carboxamide (6, 0.44 g) and solid sodium hydroxide (0.20 g) in absolute ethanol (2 mL) was stirred at room temperature for 24 h. The reaction was quenched with 2M H2S04 (1 .18 mL) and left stirring for 2 h at room temperature. The reaction mixture was filtered through fitted funnel rinsing with water (2 x 2 mL). The obtained white precipitate (0.38 g) was suspended in THF-water (1 :1 , 4.5 mL) and stirred at room temperature for ca. 2 h. The reaction mixture was filtered through fitted funnel rinsing with water (2 χ 1 mL) and dried in vacuo at 40°C to afford pure dolutegravir as a white solid (0.33 g, HPLC purity: 99.38%).
1H NMR (DMSO-d6): δ 12.51 (s, 1 H), 10.36 (t, J=5.9 Hz, 1 H), 8.50 (s, 1 H), 7.41-7.36 (m, 1 H), 7.26-7.21 (m, 1 H), 7.07-7.03 (m, 1 H), 5.45 (dd, J=5.4, 4.3 Hz, 1 H), 4.81 -4.76 (m, 1 H), 4.59-4.53 (m, 3H), 4.36 (dd, J=13.8, 5.8 Hz, 1 H), 4.05-4.00 (m, 1 H), 3.91-3.88 (m, 1 H), 2.05-1 .97 (m, 1 H), 1.55-1.52 (m, 1 H), 1 .33 (d, J=7.1 Hz, 3H). 13C NMR (DMSO-d6): δ 170.27, 163.68, 162.29, 161 .78 (dd), 159.82 (dd), 154.61 , 140.64, 130.74 (d), 130.67 (d), 122.37 (d), 1 16.73, 1 15.38, 1 1 1 .33 (d), 103.80 (t), 62.01 , 51 .16, 44.69, 35.74, 29.13, 15.21.
Example 22:

A suspension of dolutegravir (0.31 g) in methanol (4 mL) was cooled to 0 °C.25% Solution of sodium methoxide in methanol was added to the mixture and the resulting suspension was stirred at 0 °C for 2 h, then at room temperature for 23 h. The reaction mixture was then filtered through fitted funnel rinsing with methanol (3 x 10 mL). The white precipitate was dried overnight at room temperature to afford pure dolutegravir sodium as a white solid (0.26 g, HPLC purity: 99.84%).
1H NMR (DMSO-d6): δ 10.70 (t, J=5.8, 1H), 7.89 (s, 1H), 7.37-7.30 (m, 1H), 7.23-7.19 (m, 1H), 7.04-7.01 (m, 1H), 5.17 (m, 1H), 4.81 (t, J=6.4Hz, 1H), 4.51 (d, J=5.5Hz, 2H), 4.32-4.29 (m, 1H), 4.16 (dd, J=14.1, 4.8 Hz, 1H), 3.99-3.94 (m, 1H), 3.82-3.80 (m, 1H), 1.89-1.84 (m, 1H), 1.38 (d, J=12.9 Hz, 1H), 1.24 (d, J=7.0Hz, 3H).13C NMR (DMSO-d6): δ 177.93, 167.12, 166.08, 161.59 (dd), 161.13, 159.63 (dd), 134.26, 130.44 (d), 130.38 (d), 122.90 (d), 114.95, 111.23 (d), 108.78, 103.64 (t), 75.59, 61.95, 53.11, 43.01, 35.32, 29.22, 15.30.
Example 23:

The suspension of 6 (0.44 g) and solid sodium hydroxide (0.20 g) in absolute ethanol (2 mL) was stirred at room temperature for 24 h. The reaction was diluted with absolute ethanol (10 mL) and left stirring for ca. 30 min at room temperature. The reaction mixture was filtered through fitted funnel rinsing with absolute ethanol (3 x 10 mL) and dried in vacuo at room temperature to afford dolutegravir sodium as a pale yellow solid (0.43 g, HPLC purity: 98.80%). 1H NMR (DMSO-d6): δ 10.70 (t, J = 5.8 Hz, 1H), 7.89 (s, 1H), 7.37-7.30 (m, 1H), 7.23-7.19 (m, 1H), 7.04-7.01 (m, 1H), 5.17 (m, 1H), 4.81 (t, J = 6.4 Hz, 1H), 4.51 (d, J = 5.5 Hz, 2H), 4.32-4.29 (m, 1H), 4.16 (dd, J= 14.1, 4.8 Hz, 1H), 3.99-3.94 (m, 1H), 3.82-3.80 (m, 1H), 1.89-1.84 (m, 1H), 1.38 (d, J = 12.9 Hz, 1H), 1.24 (d, J = 7.0 Hz, 3H).13C NMR (DMSO-d6): δ 177.93, 167.12, 166.08, 161.59 (dd), 161.13, 159.63 (dd), 134.26, 130.44 (d), 130.38 (d), 122.90 (d), 114.95, 111.23 (d), 108.78, 103.64 (t), 75.59, 61.95, 53.11, 43.01, 35.32, 29.22, 15.30.
Example 24:

The suspension of (4R,12aS)-N-(2,4-difluorobenzyl)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12, 12a-hexahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-6][1,3]oxazine-9-carboxamide (27, 0.43 g) and solid sodium hydroxide (0.20 g) in absolute ethanol (2.5 mL) was stirred at room temperature for ca.24 h. The reaction was diluted with mixture of water/ethanol (5 mL, 1:1) and left stirring for ca. 1.5 h at room temperature. The reaction mixture was filtered through fitted funnel rinsing with mixture of water/ethanol (3 x 5 mL, 1:1) and dried in vacuo at room temperature to afford 15 as a pale yellow solid (0.41 g, HPLC purity: 98.87%).
1H NMR (DMSO-de): δ 10.70 (t, J = 5.8 Hz, 1H), 7.89 (s, 1H), 7.37-7.30 (m, 1H), 7.23-7.19 (m, 1H), 7.04-7.01 (m, 1H), 5.17 (m, 1H), 4.81 (t, J = 6.4 Hz, 1H), 4.51 (d, J = 5.5 Hz, 2H), 4.32-4.29 (m, 1H), 4.16 (dd, J = 14.1, 4.8 Hz, 1H), 3.99-3.94 (m, 1H), 3.82-3.80 (m, 1H), 1.89-1.84 (m, 1H), 1.38 (d, J = 12.9 Hz, 1H), 1.24 (d, J = 7.0 Hz, 3H).13C NMR (DMSO-d6): δ 177.93, 167.12, 166.08, 161.59 (dd), 161.13, 159.63 (dd), 134.26, 130.44 (d), 130.38 (d), 122.90 (d), 114.95, 111.23 (d), 108.78, 103.64 (t), 75.59, 61.95, 53.11, 43.01, 35.32, 29.22, 15.30.
Example 25:

The suspension of {4R, 12aS)-7-chloro-4-methyl-6,8-dioxo-3,4, 6,8, 12,12a-hexahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-6][1,3]oxazine-9-carboxylic acid (7, 0.31 g) and solid sodium hydroxide (0.20 g) in absolute ethanol (2.5 mL) was stirred at 50 °C for 3 days. The reaction was quenched with 2M H2S04 (1.2 mL) and left stirring for 7 h at room temperature. The reaction mixture was filtered through fitted funnel rinsing with water (3×5 mL) and ethanol (5 mL) dried in vacuo at 40°C to afford 28 as a pale yellow solid (0.17 g).
1H NMR (DMSO-d6): δ 15.37 (s, 1H), 12.76 (s, 1H), 8.66 (s, 1H), 5.51-5.49 (m, 1H), 4.80-4.78 (m, 1H), 4.65 (dd, J=13.8, 3.7 Hz, 1H), 4.43 (dd, J=13.8, 5.9 Hz, 1H), 4.05 (t, J^^.b Hz, 1H), 3.91 (dd, J=11.4, 3.1 Hz, 1H), 2.07-2.00 (m, 1H), 1.56 (d, J=13.8 Hz, 1H), 1.34 (d, J=7.0 Hz, 3H).13C NMR (DMSO-de): δ 172.21, 165.39, 161.73, 153.61, 141.11, 118.66, 112.99, 75.95, 62.03, 51.50, 44.90, 29.08, 15.18.
Example 26:

The suspension of (4R,12aS)-N-(2,4-difluorobenzyl)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8, 12, 12a-hexahydro-2H-pyrido[1 ‘,2’:4,5]pyrazino[2, 1 ,3]oxazine-9-carboxamide (27, 0.88 g) and solid sodium hydroxide (0.24 g) in absolute ethanol (20 mL) was stirred at 30 °C for 1.5 h. The reaction was quenched with 2M H2S04 (1 .5 mL) and left stirring for 3 hours at room temperature. The reaction mixture was filtered through fritted funnel and rinsed with water (3 x 2 mL) and ethanol (4 mL), and dried in vacuo at 40 °C to afford O-ethyl dolutegravir (29) as a pale yellow solid (0.25 g). The filtrate was extracted with ethyl acetate (3 x 5 mL). The combined organic layers were dried over MgS04, filtered and concentrated, then dried in vacuo at 40 °C to afford more 29 as a pale yellow solid (0.27 g).
1H NMR (CDCI3): δ 10.37 (t, J = 5.8 Hz, 1 H), 8.36 (s, 1 H), 7.37-7.32 (m, 1 H), 6.83-6.77 (m, 2H), 5.19 (dd, J = 5.9, 3.8 Hz, 1 H), 5.04-4.98 (m, 1 H), 4.61 (d, J = 6Hz, 2H), 4.26-4.22 (m, 3H), 4.1 1 (dd, J = 13.4, 5.9 Hz, 1 H), 3.97 (t, J = 2.4 Hz, 1 H), 3.96 (d, J = 2.4 Hz, 1 H), 2.21-2.14 (m, 1 H), 1.51 (dq, J = 14.0, 2.3 Hz, 1 H), 1 .47 (t, J = 7.0 Hz, 3H), 1 .35 (d, J = 7.1 Hz, 3H).
13C NMR (CDCI3): δ 174.78, 164.17, 162.49 (dd), 160.51 (dd), 155.72, 154.08, 142.32, 130.60 (dd), 129.33, 121 .51 (dd), 1 18.67, 1 1 1 .23 (dd), 103.78 (t), 76.15, 69.74, 62.58, 53.42, 44.58, 36.50 (d), 29.44, 16.04, 15.64.
Example 27:

The suspension of (4R, 12aS)-7-(benzyloxy)-4-methyl-3,4, 12,12a-tetrahydro-2H-pyrido[1 ‘,2’:4,5]pyrazino[2, 1-b][1 ,3]oxazine-6,8-dione (30, 0.68 g, prepared according to prior art) and solid sodium hydroxide (0.40 g) in absolute ethanol (5 mL) was stirred at 50 °C for 14 h. The reaction was quenched with formic acid (0.35 mL), water (2 mL) was added and mixture was left stirring for additional 1 h at room temperature. The reaction mixture was extracted with ethyl acetate (3 x 5 mL) and the combined organic layers concentrated to afford a crude oil. Purification by flash chromatography (eluting with CH2CI2/methanol) afforded 32 as an orange solid (0.26 g, 52 %).
The above procedure if done at room temperature in same time period, affords 31 as orange oil (0.24 g, 43 %).
Compound 32: 1H NMR (DMSO-d6): δ 7.64 (d, J = 7.4 Hz, 1 H), 6.20 (d, J = 7.3 Hz, 1 H), 5.40 (dd, J = 5.1 , 4.2 Hz, 1 H), 4.83-4.78 (m, 1 H), 4.35 (dd, J = 13.6, 3.9 Hz, 1 H), 4.13 (dd, J = 13.6, 5.4 Hz, 1 H), 4.05-4.00 (m, 1 H), 3.90-3.85 (m, 1 H), 2.03-1.95 (m, 1 H), 1.52 (dd, J = 13.9, 1 .9 Hz, 1 H), 1.33 (d, J = 7.1 Hz, 3H). 13C NMR (DMSO-d6): δ 170.96, 163.01 , 153.48, 137.96, 1 16.83, 1 13.52, 76.18, 62.05, 50.39, 44.53, 29.21 , 15.28.
Compound 31 : 1H NMR (DMSO-d6): δ 7.67 (d, J = 7.4 Hz, 1 H), 6.28 (d, J = 7.4 Hz, 1 H), 5.29 (dd, J = 5.4, 3.8 Hz, 1 H), 4.82-4.75 (m, 1 H), 4.32 (dd, J = 13.6, 3.6 Hz, 1 H), 4.10 (dd, J = 13.5, 5.6 Hz, 1 H), 4.03-3.93 (m, 3H), 3.85 (ddd, J = 1 1 .6, 5.0, 2.2 Hz, 1 H), 1.97-1 .89 (m, 1 H), 1 .48 (dd, J = 13.8, 2.1 Hz, 1 H), 1.27 (d, J = 7.1 Hz, 3H), 1.26 (d, J = 7.0 Hz, 3H). 13C NMR (DMSO-d6): δ 174.38, 156.1 1 , 150.82, 139.48, 1 16.39, 1 13.52, 75.92, 67.31 , 61 .80, 51 .36, 44.22, 29.29, 15.76, 15.36.
Exa

The transformation of 6 to dolutegravir with sodium hydroxide in ethanol was monitored for the interconversion of intermediates. The suspension of 6 (0.44 g) and solid sodium hydroxide (0.20 g) in ethanol (3.33 ml.) was stirred at 22 °C. Samples of the reaction mixture were taken after 3, 8 and 24 h for UPLC analysis. After 24 h, the reaction mixture was quenched with 2 M H2S04 (5 ml_), and left stirring at room temperature. The reaction mixture was filtered through fritted funnel, the product rinsed with water (30 ml.) and dried in vacuo at 50 °C overnight to afford dolutegravir as a white solid (0.27 g, 64 %).
The results of reaction monitoring:
Time UPLC analysis (area%)
Entry
(h) compound 6 compound 29 dolutegravir
1 3 h 37.50 20.63 39.99
2 8 h 0.78 15.46 80.32
3 24h 0.31 8.56 88.21
Example 29:

The effect of added water and reaction temperature was evaluated by monitoring 4 reactions in parallel. To the suspensions of 27 (0.86 g) in MeOH were added solid sodium hydroxide (0.40 g) or aqueous solution of NaOH (5 M, 2 ml.) (see Table below). The reactions were stirred in parallel at 50 °C or 22 °C. Samples were taken in timely intervals for UPLC analysis.
The results of reaction monitoring demethylation of 27 in MeOH:

Example 30:

The effect of added water and reaction temperature was evaluated by monitoring 4 reactions in parallel. To the suspensions of 6 (0.88 g) in EtOH were added solid sodium hydroxide (0.40 g) or aqueous solution of NaOH (5 M, 2 mL) (see Table below). The reactions were stirred in parallel at 50 °C or 22 °C. Samples were taken in timely intervals for UPLC analysis.
The results of reaction monitoring of the transformations of 6 in ethanol with NaOH:

dol. = dolutegravir
Exa

The effect of added water and reaction temperature was evaluated by monitoring 4 reactions in parallel. To the suspensions of 27 (0.88 g) in EtOH were added solid sodium hydroxide (0.40 g) or aqueous solution of NaOH (5 M, 2ml_) (see Table below). The reactions were stirred in parallel at 50 °C or 22 °C. Samples were taken in timely intervals for UPLC analysis.
The results of reaction monitoring of the transformations of 27 in ethanol with NaOH:

dol. = dolutegravir
Example 32:

Compound 3 (30 g, 1 10 mmol; assay 99%) was suspended in acetonitrile (450 mL), acetic acid (73 mL) and methanesulfonic acid (25 mL) were added. The reaction mixture was stirred 4 h at 70 °C. The clear red solution was cooled to 25 °C. Triethylamine (77 mL) and (S)-2-aminopropanol (17 mL) were added and the mixture was stirred at reflux temperature for 20 h. The reaction mixture was cooled to 25 °C and the insoluble product filtered, washed with 1 M HCI(aq) (60 mL), water (3 * 60 mL) and dried to give 4c (19.49 g, 67%): mp = 313-315 °C; 1H NMR (DMSO-d6): δ 1.31 (d, J = 6.3 Hz, 3H), 3.65 (dd, J = 8.6, 6.8 Hz, 1 H), 4.13 (dd, J = 1 1.7, 10.3 Hz, 1 H), 4.28 (m, 1 H), 4.39 (dd, J = 8.6, 6.8 Hz, 1 H), 4.92 (dd, J = 12.3, 4.2 Hz, 1 H), 5.45 (dd, J = 10.2, 4.1 Hz, 1 H), 7.16 (s, 1 H), 8.84 (s, 1 H), 15.74 (s, 1 H); 13C NMR (DMSO-d6) 16.5, 51.6, 52.9, 72.4, 81.6, 1 15.8, 1 18.1 , 141.5, 147.6, 153.4, 165.3, 179.0.
PATENT
WO2016016279, NOVEL HYDRATES OF DOLUTEGRAVIR SODIUM
LEK PHARMACEUTICALS D.D. [SI/SI]; Verovskova 57 1526 Ljubljana (SI).
SANDOZ AG [CH/CH]; Lichtstrasse 35 CH-4056 Basel (CH)
HOTTER, Andreas; (AT).
THALER, Andrea; (AT).
LEBAR, Andrija; (SI).
JANKOVIC, Biljana; (SI).
NAVERSNIK, Klemen; (SI).
KLANCAR, Uros; (SI).
ABRAMOVIC, Zrinka; (SI)
The present invention relates to novel hydrates of sodium dolutegravir and their methods of preparation. In addition, the invention relates to a novel crystalline form of sodium dolutegravir, which is a useful intermediate for the preparation of one of the new hydrates. The invention also relates to the use of the new hydrates for the production of pharmaceutical compositions.
Finally, the invention relates to pharmaceutical compositions comprising an effective amount of the novel hydrates, oral dosage forms comprising these pharmaceutical compositions, a process for preparing said oral dosage forms, and the use of such pharmaceutical compositions or dosage forms in the treatment of retroviral infections such as HIV infections -1.
Dolutegravir, chemically designated (4f?, 12aS)-/V-(2,4-difluorobenzyl)-7-hydroxy-4-methyl-6,8-dioxo-3,4,6,8, 12, 12a-hexahydro-2H-pyrido[1 ‘,2’:4,5]pyrazino[2, 1- ?][1 ,3]oxazine-9-carboxamide, is a human immunodeficiency virus type 1 (HIV-1 ) integrase strand transfer inhibitor (INSTI) indicated in combination with other a nti retroviral agents for the treatment of HIV-1 infection. The marketed finished dosage form (TIVICAY™) contains dolutegravir as its sodium salt, chemically denominated sodium (4f?,12aS)-9-((2,4-difluorobenzyl)carbamoyl)-4-methyl-6,8-dioxo-3,4,6,8,12, 12a-hexahydro-2H-pyrido[1 ‘,2’:4,5]pyrazino[2, 1- ?][1 ,3]oxazin-7-olate, which is represented by the following general chemical formula (I):

(I)
WO 2010/068253 A1 discloses a monohydrate and an anhydrous form of dolutegravir sodium as well as a crystalline form of the free compound. Processes for the preparation of said forms are also provided in the application.
WO 2013/038407 A1 discloses amorphous dolutegravir sodium and processes for preparing the same.
Hydrates of pharmaceutical drug substances are of particular interest as they provide new opportunities for preparing novel pharmaceutical compositions with improved quality, activity and/or compliance. This is due to the fact that hydrates have different physicochemical properties compared to their anhydrous counterparts such as melting point, density, habitus, chemical and physical stability, hygroscopicity, dissolution rate, solubility, bioavailability etc., which influence the formulation process and also impact the final drug product.
If an anhydrous form is selected, phase changes during the formulation process induced by hydrate formation must be avoided. This can be particularly difficult if for example wet granulation is used with a substance that is able to form hydrates like dolutegravir sodium.
Hence, a stable hydrate of dolutegravir sodium would allow to easily formulate dolutegravir sodium in a controlled manner and subsequently also facilitate storage and packaging.
However, the so far known dolutegravir sodium monohydrate disclosed in WO 2010/068253 A1 shows excessive water uptake when exposed to moisture and on the other hand already dehydrates below 30% relative humidity.
Therefore, there is a need for hydrates of dolutegravir sodium with improved physicochemical properties, e.g. for hydrates which are stable over a broad humidity range, in particular for hydrates absorbing only low amounts of water at elevated humidity and on the other hand preserving their crystal structure also at dry conditions. In addition, there is a need for pharmaceutical compositions comprising these hydrates, and thus also for hydrates that allow for improved formulation of dolutegravir sodium in pharmaceutical compositions.
SUMMARY OF THE INVENTION
The present invention relates to novel hydrates of dolutegravir sodium and to processes for their preparation. Specifically, the present invention provides crystalline forms of dolutegravir sodium of formula (I) according to respective claims 1 , 5 and 6, with preferred embodiments being set forth in sub-claim 2. The present invention also provides processes for their preparation according to respective claims 3, 7 and 8, with preferred process embodiments being set forth in sub-claim 4. The present invention further provides the uses according to claims 9 and 16, and a pharmaceutical composition according to claim 10, and preferred embodiments thereof according to sub-claims 1 1 and 12. The present invention also provides a process for the preparation of the pharmaceutical composition according to claim
13, and preferred embodiments thereof according to sub-claim 14. The pharmaceutical composition for therapeutic use is set forth in claim 15.
The novel hydrates are physically and chemically stable over a broad humidity range, show only low water uptakes when exposed to moisture and are even stable at dry conditions. Therefore, the novel hydrates are especially suitable for the preparation of pharmaceutical compositions, e.g. in terms of time and costs.
In particular, it has been found that crystal Form HxA exhibits improved properties which allow for improved formulation of Form HxA in pharmaceutical compositions.
In addition, the present invention relates to a novel crystalline form of dolutegravir sodium, which, for the first time, allows the preparation of one of the novel hydrates and is therefore a valuable intermediate.
PATENT
1361/CHE/2013
Dolutegravir (I) is chemically known as (4/?,12aS)-N-[(2,4-difluorophenyl)methyl]-3,4,6,8,12,12a-hexahydro-7-hydroxy-4-methyl-6,8-dioxo-2//-pyrido[r,2′:4,5]pyrazino[2,l-b][l,3]oxazine-9-carboxamide. Dolutegravir is a human immunodeficiency virus type 1 (HIV-1) integrase strand transfer inhibitor (INSTI) indicated in combination with other antiretroviral agents for the treatment of HIV-1 infection. Dolutegravir is being marketed under the trade name Tivicay®. US 8,129,385 disclosed Dolutegravir or its pharmaceutically acceptable salts thereof. US ‘385 also discloses a process for the preparation of Dolutegravir (I). The process involves the condensation of 5-benzyloxy-4-hydroxy-6-hydroxymethyl nicotinic acid (II) with 2,4-difluorobenzylamine (III) to produce 5-benzyloxy-N-(2,4-difluorobenzyl)-4-hydroxy-6-hydroxymethyl nicotinic acid amide (IV), which is further under goes oxidation using manganese dioxide (Mn02) to produce 5-benzyloxy-N-(2,4-difluorobenzyl)-6-formyl-4-hydroxy-nicotinic acid amide (V). This amide compound (V) is reacted with sodium chlorite (NaClCh) to produce 3-benzyloxy-5-(2,4-difluorobenzylcarbamoyl)-4- hydroxy-pyridine-2-carboxylic acid (VI), which is further treated with methanol (MeOH) to produce 3-benzyloxy-5-(2,4-difluorobenzyl)-4-hydroxy-pyridine-2-carboxylic acid methyl ester (VII).

The methyl ester compound (VII) is reacted with 3-bromopropene to produce l-allyl-3-benzyloxy-5-(2,4-difluorobenzyl)-4-oxo-l,4-dihydro-pyridine-2- carboxylic acid methyl ester (VIII), which is further reacted with potassium osmate dihydrate (K2OSO4.2H2O) to produce 3-benzyloxy-5-(2,4-difluorobenzylcarbamoyl)-4-oxo-l-(2-oxo-ethyl)-l,4-dihydropyridine-2-carboxylic acid methyl ester (IX). The compound (IX) is reacted with (R)-3-amino-l-butanol (X) to produce benzyloxy Dolutegravir (XI), which is deprotected by treating with TFA to produce Dolutegravir (I). The process is as shown in scheme-I below:
The major disadvantage with the above prior-art process is that it involves large no of steps and tedious work-up procedures to isolate the required product. This results a longer period of time cycle is required to produce Dolutegravir (I), which in turn renders the process more costly and less eco friendly. Further the above processes are low yielding and with less purity. US 8,217,034 discloses variant process for the preparation of Dolutegravir.
This process involves the reaction of methyl l-(2,2-dihydroxyethyl)-4-oxo-3-[(phenylmethyl)oxy]-l,4-dihydro-2-pyridine carboxylate (XII) with (R)-3-amino-l-butanol (X) to produce (4R, 12o5)-4-methyl-7-[(phenylmethyl)oxy]-3,4,12,12a-tetrahydro-2//-pyrido[ 1 \2′,4,5] pyrazino[2,l-b][l,3]oxazine-6,8-dione (XIII), which is further undergoes bromination using NBS to produce (4R,12aS)-9-bromo-4-methyl-7-[(phenylmethyl)oxy]-3,4,12,12a-tetrahydro-2H-pyrido[r,2′:4,5]pyrazino[2,l-b][l,3]oxazine-6,8-dione (XIV). The bromo Compound (XIV) is condensed with 2,4-difluorobenzylamine (III) in the presence of Tetrakis(triphenylphosphine)palladium (Pd(PPh3)4) to produce benzyloxy Dolutegravir (XI), which is hydrogenated in the presence of Pd/C to produce Dolutegravir (I). The process is as shown in Scheme-II below:

The major disadvantage with the above prior art process of preparing Dolutegravir is the use of expensive reagent tetrakis(triphenylphosphine)palladium (Pd(PPh3)4> in coupling step. Use of this reagent on industrial scale is not preferred, which makes the process more expensive. WO 2011/119566 discloses another variant process for the preparation of Dolutegravir.
This process involves the reaction of l-(2,2-dimethoxyethyl)-5-methoxy-6-(methoxycarbonyl)-4-oxo-l,4-dihydropyridine-3-carboxylic acid (XV) with acetic acid in presence of methane sulfonic acid to produce 5-methoxy-6-(methoxycarbonyl)-4-oxo-l-(2-oxoethyl)-l,4-dihydropyridine-3-carboxylic acid (XVI), which is further condensed with (R)-3-amino-l-butanol (X) to produce (4R,12aS)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2//-pyrido[ 1 ‘,2’:4,5]pyrazino[2,1 -b] [ 1,3]-oxazine-9-carboxylic acid (XVII). This acid Compound XVII is acylated with 2,4-difluorobenzylamine (III) in the presence of carbonyldiimidazole (CDI) to produce methoxy Dolutegravir (XVIII), which is demethylated in the presence of lithium bromide (LiBr) to produce Dolutegravir (I).
The process is as shown in Scheme-3 below:

The major disadvantage of the above prior art process of preparing Dolutegravir is the use of expensive and highly moisture sensitive reagent, 1,1-carbonyldiimidazole (CDI), during acylation. Use of this reagent on industrial scale is not preferred due to anhydrous conditions required in the process. However, there is always a need for alternative preparative routes, which for example, involve fewer steps, use reagents that are less expensive and/or easier to handle, consume smaller amounts of reagents, provide a higher yield of product, have smaller and/or more eco-friendly waste products, and/or provide a product of higher purity. Hence, there is a need to develop cost effective and commercially viable process for the preparation of Dolutegravir of formula (I). The present invention is related to a process for the preparation of pure Dolutegravir of formula (I), wherein optically active acid addition salt of (R)-3-amino-l-butanol (X) is directly condensed with 5-methoxy-6-(methoxycarbonyl)-4-oxo-l-(2-oxoethyl)-l,4-dihydropyridine-3-carboxylic acid (XVI) instead of condensing with free base of (R)-3-amino-1-butanol (X). The present invention is also related to a process for the preparation of pure Dolutegravir of formula (I), wherein, inexpensive and easily handling condensing reagents in the condensation of (4R, 12aS)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2//-pyrido[l’,2′:4,5]pyrazino [2,l-b][l,3]oxazine-9-carboxylic acid (XVII) with 2,4-difluorobenzylamine (III).
In another embodiment, 5-methoxy-6-(methoxycarbonyl)-4-oxo-l-(2-oxoethyl)-l,4- dihydropyridine-3-carboxylic acid (XVI) used in the present invention is prepared by reacting 4-methoxyacetoacetate (XIX) with N,N-dimethyl-l,l- bis(methyloxy)methanamine (DMF-DMA) (XX) to produce methyl-2- (dimethylaminomethylene)-4-methoxy-3-oxo-butanoate(methyl-3-(dimethylamino)-2 [(methyloxy)acetyl]-2-propenoate) (XXI), which is reacted with aminoacetaldehyde dimethyl acetal (XXII) to produce methyl-2-(2,2-dimethoxyethylaminomethylene)-4-methoxy-3-oxo-butanoate(methyl-3-{[2,2-bis(methyloxy)ethyl]amino}-2-[(methyloxy) acetyl]-2-propenoate) (XXIII).
The compound (XXIII) is contacted with dimethyl ethanedioate in presence of alkali metal alkoxide to produce dimethyl-1-(2,2-dimethoxyethyl)-3-methoxy-4-oxo-l ,4-dihydropyridine-2,5-dicarboxylate (XXIV), which is selectively hydrolyzed with a base to produce l-[2,2-bis(methyloxy)ethyl]-5-(methyloxy)-6-[(methyloxy)carbonyl]-4-oxo-l ,4-dihydro-3-pyridinecarboxylic acid (XV). The compound (XV) is treated with a catalytic amount of a strong protic acid in the presence of acetic acid in an organic solvent to produce a reaction mixture containing 5- methoxy-6-(methoxycarbonyl)-4-oxo-l-(2-oxoethyl)-l,4-dihydropyridine-3-carboxylic acid (XVI), The process is as shown in Scheme-IV below:
The following examples illustrate the nature of the invention and are provided for illustrative purposes only and should not be construed to limit the scope of the invention.
Example-1:
EXAMPLES: Example-1: Process for the preparation of Dolutegravir
Step-i: Preparation of (/?)-3-amino-l-butanol tartarate salt: D-(+) Tartaric acid (12.7 g, 0.085 mol) was added in to a solution of (i?,5)-3-amino-l-butnaol (7.5 g, 0.084 mol) in methanol (100 ml) at 40 °C. The reaction mixture was stirred for about 1 hour at 35-40 °C and the reaction mass was cooled to 0-5°C and maintained for 30-40 minutes. The obtained solid was filtered and washed with chilled methanol (10 ml) at 0-5 °C. The solid was dried to get (i?)-3-amino-l-butanol tartarate salt (8.0 g, 40%).
Step-ii: Preparation of (4rt,12a£)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[l’,2′;4,5]pyrazino[2,l-b][l,3]oxazine-9-carboxylic acid (XVII): l-[2,2-Bis(methyloxy)ethyl]-5-(methyloxy)-6-[(methyloxy)carbonyl]-4-oxo-l,4-dihydro-3-pyridinecarboxylic acid (XV) (lOOg; 0.3175 moles) was suspended in acetonitrile (800 ml) and heated to 80-82°C. A mixture of acetic acid (95.25 g), methanesulfonic acid (9.14 g; 0.09525 moles) and acetonitrile (200 ml) were added to the slurry at 80-82°C. The reaction mass was continued at 80-82°C to complete the reaction. After completion of the reaction, anhydrous sodium acetate (65 g) and (/?)-3-amino-l-butanol tartrate salt (79.68g; 0.3334 moles) were added at 20-25°C and stirred at 60-65°C to complete the reaction. The reaction mass was concentrated and acidified with IN aqueous hydrochloric acid (750 ml) and extracted with methylene chloride (1500 ml) at ice cold temperature. The organic layer was separated, concentrated, treated with hot methanol (350 ml) for 2 h, filtered, washed with methanol and dried to yield (4R,12aS)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[ 1′ ,2′ :4,5]pyrazino[2,1 -b] [ 1,3]oxazine-9-carboxylic acid (XVII) (72 g; HPLC purity: 99.07%).
Step-iii: Process for the preparation of Dolutegravir (I). Method A: Triethylamine (3.61 g; 0.0357 moles) was added to the suspension of (4R,12aS)-7- methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[ 1′ ,2′ :4,5]pyrazino[2,1 – b][l,3]oxazine-9-carboxylic acid (XVII) (10 g; 0.0325 moles) in methylene chloride (50 ml), and cooled to 10-15°C. Pivaloyl chloride (4.3 g; 0.0357 moles) was added to the reaction mass, and stirred at 10-15°C for 1 h. Thereafter, 2,4-difiuorobenzylamine (5.58 g; 0.0389 moles) was added at 10-15°C and then warmed to 20-25°C to complete the reaction. After completion of the reaction, IN aqueous hydrochloric acid (20 ml) was added, organic layer was separated, washed with 5% w/w aqueous sodium bicarbonate solution (10 ml) followed by 15% w/w aqueous sodium chloride solution (10 ml) and concentrated. To the concentrated mass, acetonitrile (100 ml) and Lithium bromide (5.08 g; 0.0584 moles) were added and heated to 65-70°C for 3 h to complete the reaction. After completion of the reaction, the reaction mass was acidified with 5N aqueous hydrochloric acid (40 ml), concentrated to about 50 ml and DM water was added to crystallize the product at 20-25°C. The slurry was stirred for 2 h, filtered, washed with DM water and dried to yield (4R,12aS)-N-(2,4-difluorobenzyl)-7-hydroxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a,-hexahydro-2H-pyrido[ 1′ ,2′ :4,5]pyrazino[2,1 -b] [ 1,3]oxazine-9-carboxamide (I) (11.5 g, HPLC purity: 99.63%).
Method B: Isobutyl chloroformate (4.65 gm, 0.03404 moles) in methylene chloride (10 ml) was added to the solution of N-methylmorpholine (3.45 gm, 0.03410 moles) and (4R,12aS)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[ 1′ ,2′ :4,5]pyrazino-[2,1 -b][l,3]oxazine-9-carboxy!ic acid (XVII) (10.0 gm, 0.03245 moles) in methylene chloride (60 ml) at -10 to 0°C in about 1 h. 2,4-Difloro benzyl amine (4.88 gm, 0.03409 moles) in methylene chloride (10 ml) was added to the cold reaction mass, and stirred at 20-30°C for completion of reaction. After completion of reaction, the reaction mass was washed with 5%w/w aqueous sodium bicarbonate solution (20 ml), IN hydrochloric acid (20 ml), DM water (20 ml) and concentrated. Acetonitrile (120 ml) and lithium bromide (4.8 gm, 0.05516 moles) were added to the concentrated mass, and stirred at 70-80°C for 3 h to complete the reaction. After completion of reaction, the reaction mass was acidified with 5N aqueous hydrochloric acid (40 ml) and concentrated to about 50 ml. DM Water (100 ml) was added to the concentrated reaction mass and stirred for 2 h at 25-30°C to crystallize the product. The product was filtered, washed with DM Water (50 ml) and dried to yield Dolutegravir (I) (10.7 gm, HPLC purity: 99.60%).
Example-2: Process for the preparation of Dolutegravir (I) (4R, 12aS)-N-(2,4-difluorobenzyl)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a,-hexahydro-2H-pyrido[r,2′:4,5]pyrazino[2,l-b][l,3]oxazine-9-carboxamide (XVIII) (2 g, 0.0046 moles) was suspended in isopropyl alcohol (20 ml) and lithium bromide (0.8 g, 0.00924 moles) was added and stirred at 70-80°C for 15 h to complete the reaction. After completion of reaction the reaction mass was acidified with 5N aqueous hydrochloric acid (5 ml) and concentrated. DM Water (20 ml) was added to the concentrated mass and stirred at 25-30°C to crystallize the product. The product was filtered, washed with DM Water and dried to yield Dolutegravir (I) (1.5 g, HPLC purity: 97.93%).

Dolutegravir
Experimental:
1H NMR (CDCl3) δ 12.45 (s, 1H), 10.38 (br s, 1H), 8.30 (s, 1H), 7.40-7.30 (m, 1H), 6.85-6.75 (m, 2H), 5.26 (d, J = 5.8, 4.1 Hz, 2H), 5.05-4.95 (m, 1H), 4.64 (d, J = 5.9 Hz, 2H), 4.27 (dd, J = 13.4, 4.2 Hz, 1H), 4.12 (dd, J = 13.6, 6.0 Hz, 1H), 4.05 (t, J = 2.3 Hz, 1H), 4.02 (d, J = 2.2 Hz, 1H), 2.30-2.19 (m, 1H), 1.56 (dd, J = 14.0, 2.0 Hz, 1H), 1.42 (d, J = 7.0 Hz, 3H).////////////LINK


















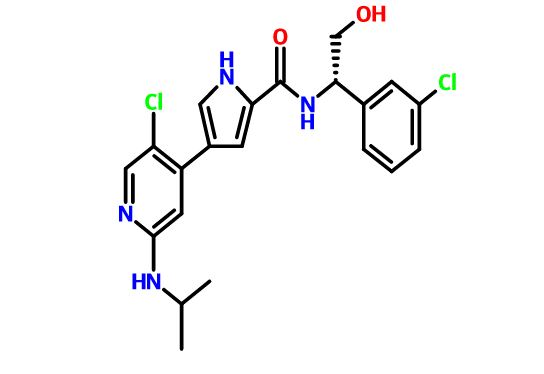
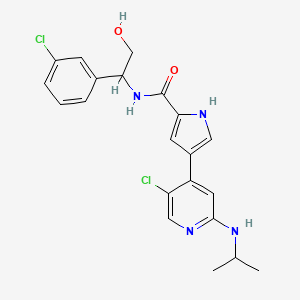




















 .
.
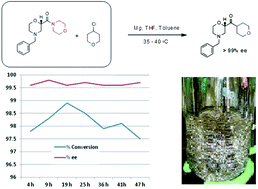

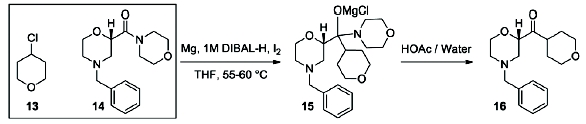
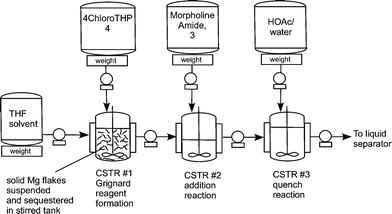
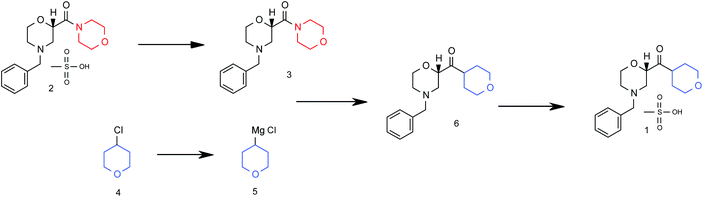
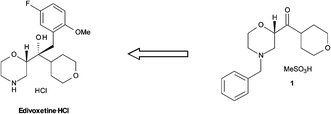
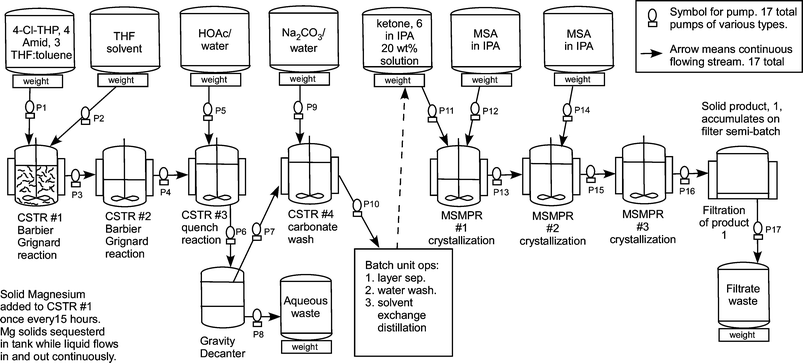 Flow diagram for the whole continuous process from amide 3 to product 1.
Flow diagram for the whole continuous process from amide 3 to product 1.