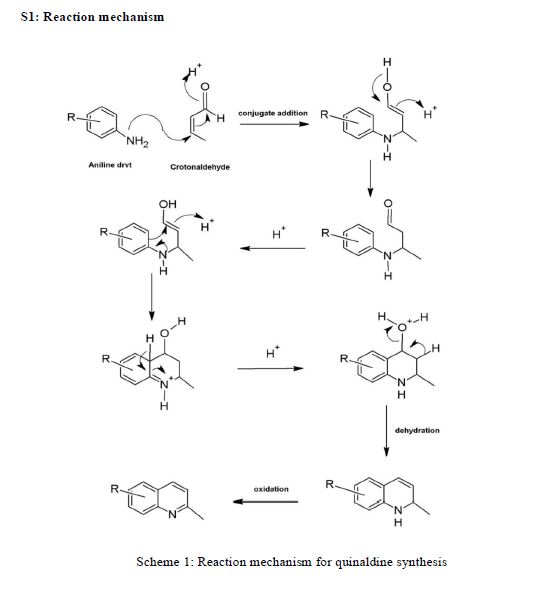
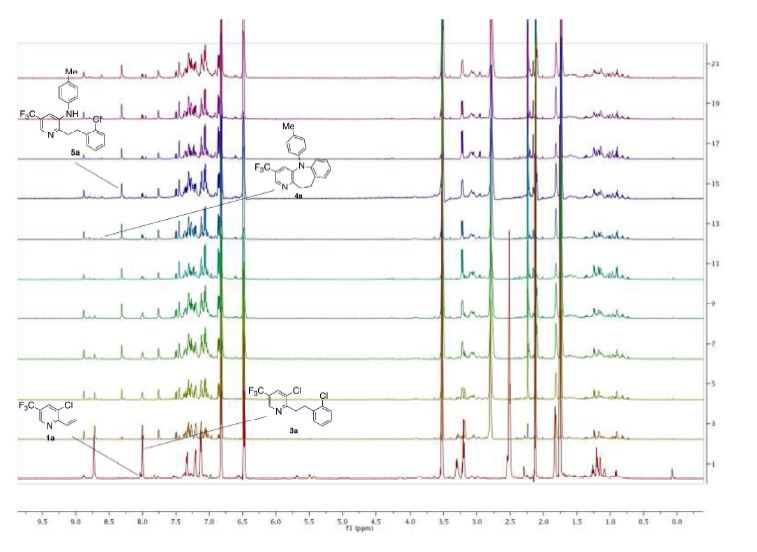
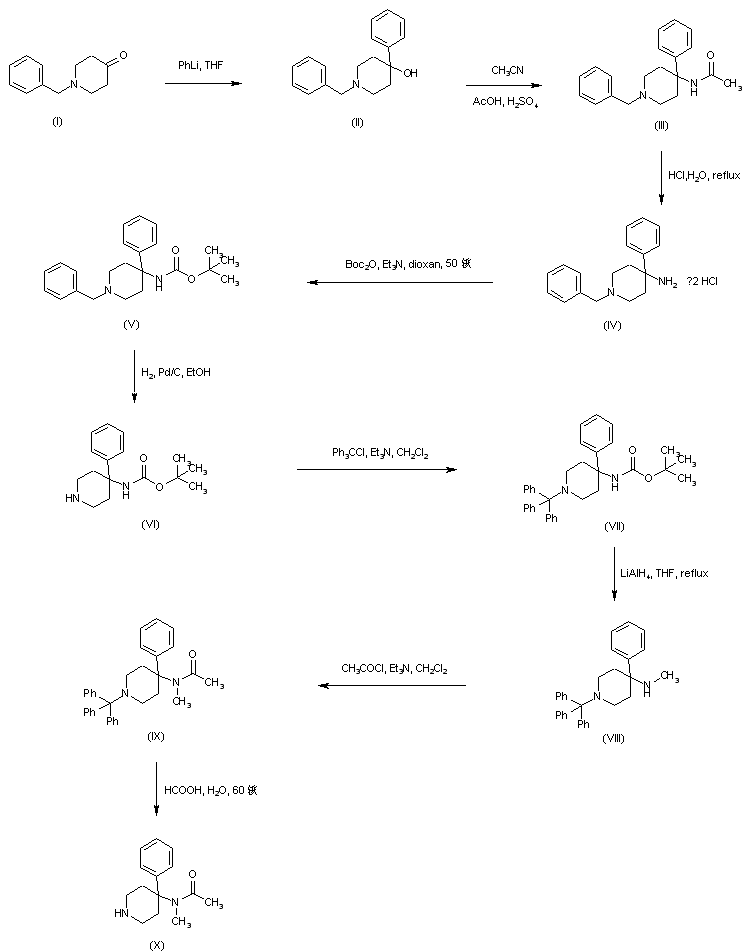
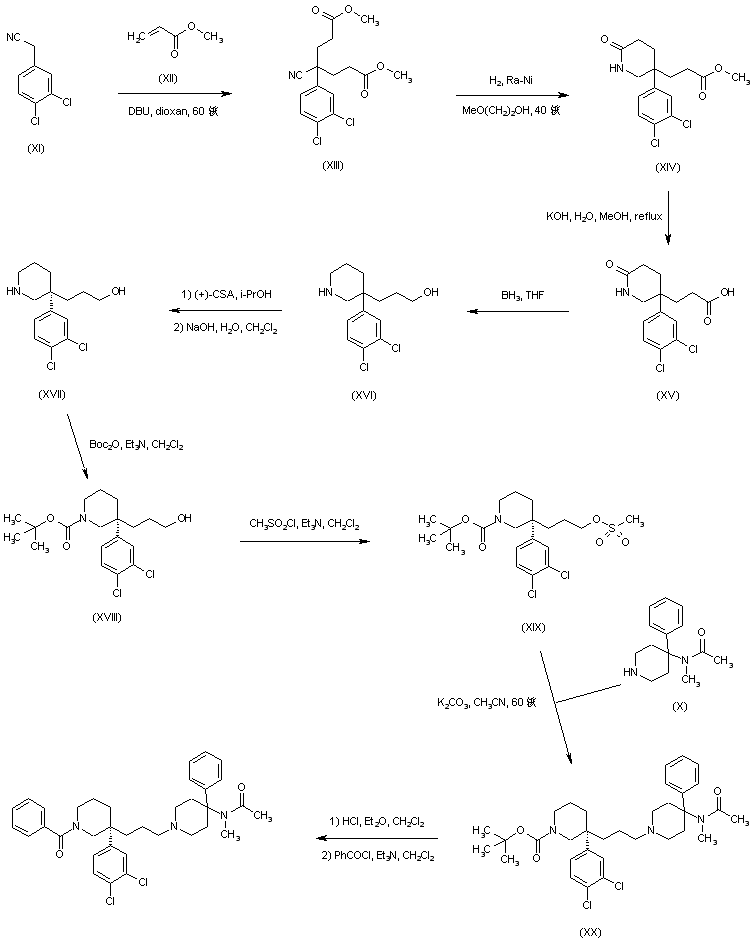
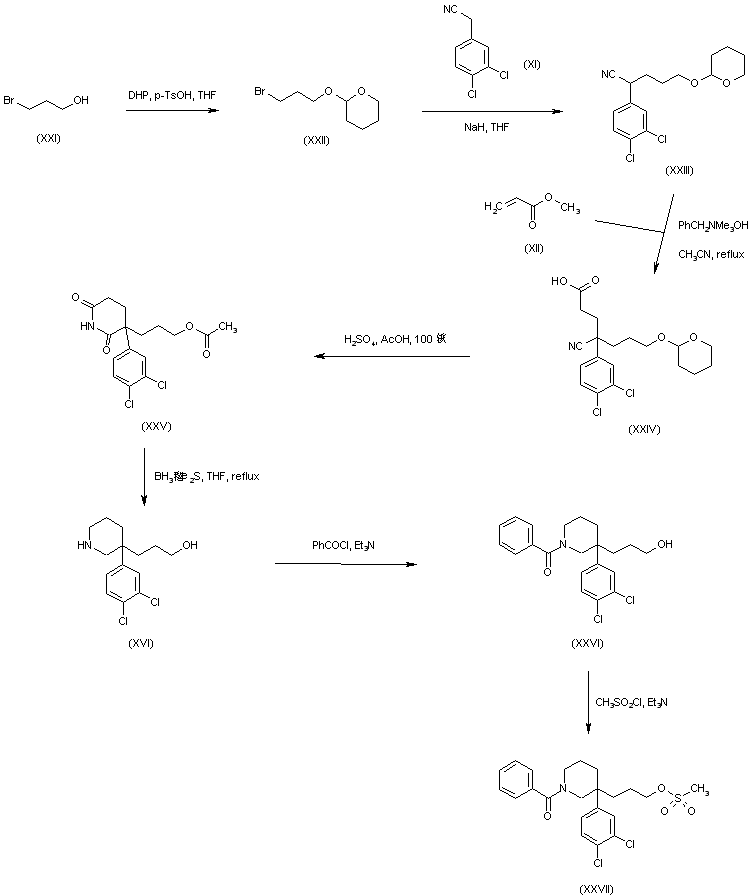
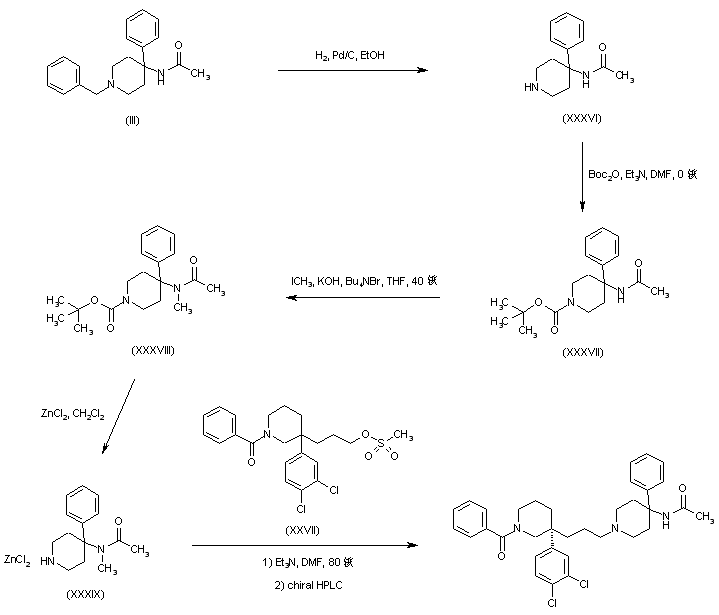
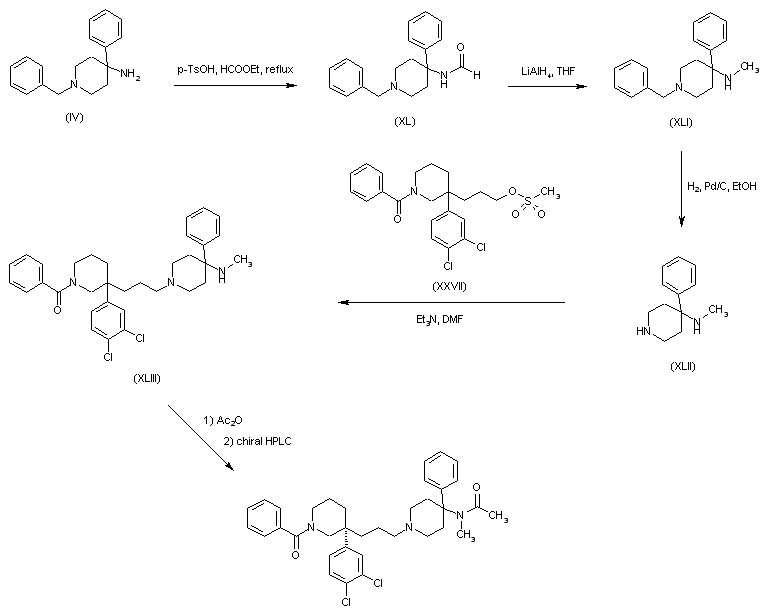
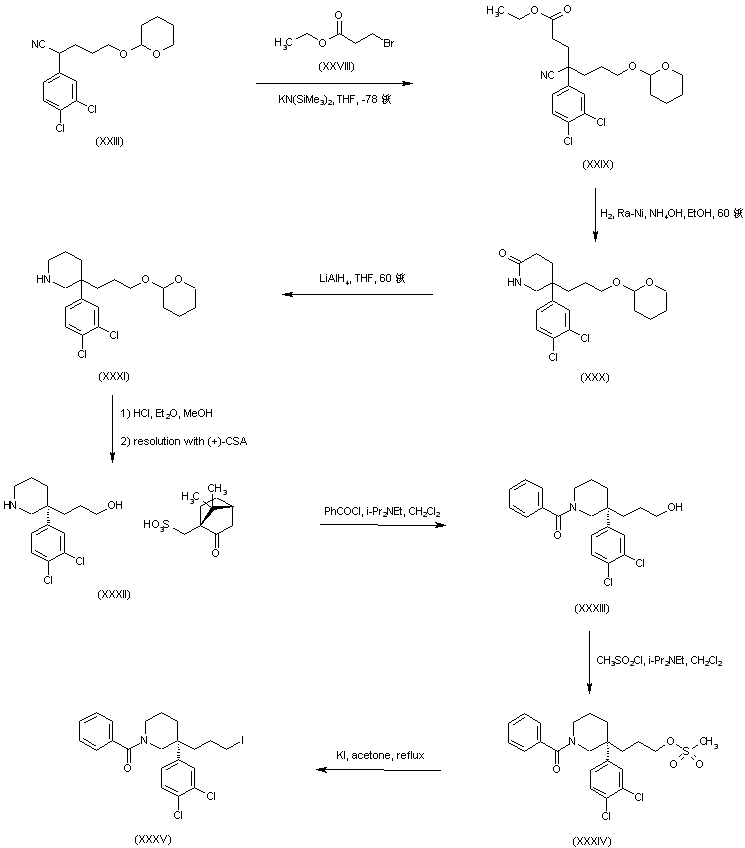
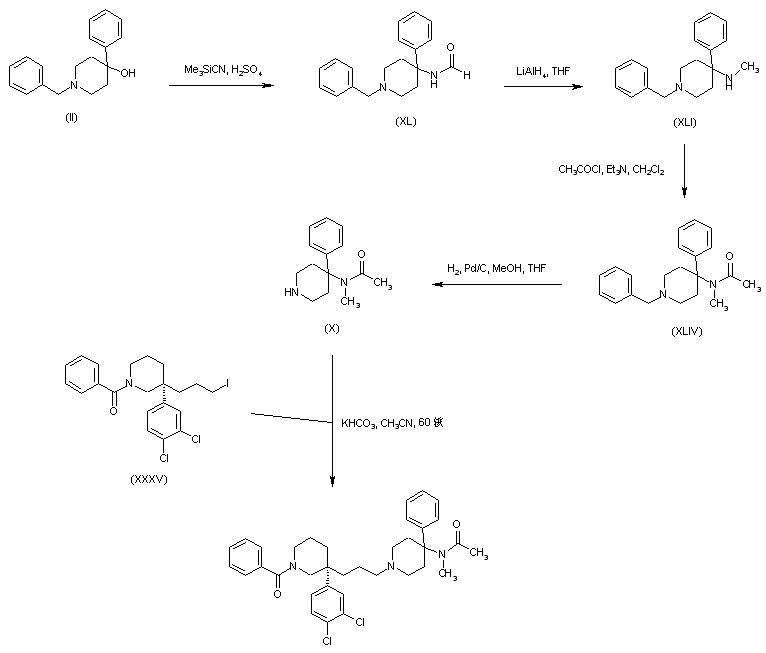
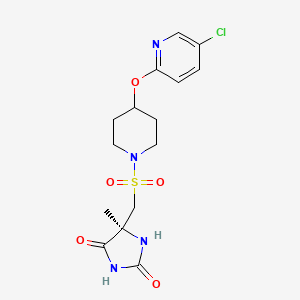
AZD1236
CAS 459814-89-2,
Mechanism of Action: Matrix metalloproteinase 9 & 12 (MMP9,12) inhibitor MMP9 MMP12i
Anders Eriksson, Matti Lepistö, Michael Lundkvist, af Rosenschöld Magnus Munck,Pavol Zlatoidsky,
Astrazeneca Ab INNOVATOR
- OriginatorAstraZeneca
- Class
- Mechanism of ActionMatrix metalloproteinase inhibitors
- Highest Development Phases
- DiscontinuedChronic obstructive pulmonary disease
Most Recent Events
- 29 Jul 2010Discontinued – Phase-II for Chronic obstructive pulmonary disease in Europe (PO)
- 29 Jul 2010Discontinued – Phase-I for Chronic obstructive pulmonary disease in Japan (PO)
- 29 Jul 2010Discontinued – Phase-I for Chronic obstructive pulmonary disease in Japan (PO)
AZD1236 is a selective MMP-9 and MMP-12 inhibitor (IC50 4.5 and 6.1nM) from Astrazeneca that, since it failed biomarker endpoints for COPD is included in the AZ Open Innovation 2014 set for repurposing. Pending any published link the structure identification is tenatative but seems likely to be the structure crystalised in WO2007106022.
Matrix metallopeptidase 9 and 12 (MMP9|MMP12) inhibitor http://www.ncbi.nlm.nih.gov/gene/4318; http://www.ncbi.nlm.nih.gov/gene/4321 Preclinical Pharmacology AZD1236 is a potent and reversible inhibitor of human MMP9 and MMP12 (IC50’s = 4.5 and 6.1nM, respectively), with 10 – 15-fold selectivity to MMP2 and MMP13 and >350-fold selectivity to other members of the enzyme family. Its activity is approximately 20- to 50-fold lower at the rat, mouse, and guinea pig orthologues. In acute models of lung injury, AZD1236 inhibited the hemorrhage and inflammation induced by instillation of human MMP12 into rat lungs by ~80% at 0.81 mg/kg, and also abolished macrophage infiltration into BAL fluid induced by tobacco smoke inhalation in the mouse. Safety and Tolerability In healthy human volunteers, AZD1236 was well tolerated in single doses from 2 to 1500 mg and in multiple doses of 15, 75 and 500 mg for periods of up to 13 days. AZD1236 was also well tolerated in COPD patients with moderate to severe disease when given at 75 mg BID for 6 weeks. Pre-clinical toxicology studies of up to 12 month duration have been performed. Toxicologically important findings mainly relate to chronic treatment and included: diffuse eye lens opacities after 6 months administration to rats and fibrodysplasia in the subcutis after 12 months to dogs. Clinical Pharmacology Target coverage data to date have been mixed. In healthy subjects, single dose of 30 or 75 mg inhibited ex vivo zymosanstimmulated whole blood MMP activity (the 75 mg dose yielding plasma compound levels at Cmax steady state of ~120 x IC50). In contrast, 75 mg BID for 6 wks in COPD patients compared to placebo did not identify any significant change in whole blood MMP activity.

PATENT
WO 2002074750
WO 02/074767 further discloses a specific metalloproteinase inhibitor compound identified therein as (5S)-5-[4-(5-chloro-pyridin-2-yloxy)-piperidine-l-sulfonylmethyl]-5- methyl-imidazolidine-2,4-dione (page 65, lines 15 to 27; and page 120, lines 23 to 29). This compound is designated herein as compound (I).
(I)
WO 02/074767 further discloses processes for the preparation of compound (I). Thus, in one embodiment, compound (I) is prepared by a route analogous to that shown in the following Scheme (WO 02/074767; pages 87, 113 and 120) but substituting the appropriate amine in step (d):
Scheme 1
Reagents and conditions for Scheme 1: a) KCN, (NHLj)2CO3, EtOHTH2O, +900C, 3h;. b) Chiral separation, CHIRALPAK AD, methanol as eluent;. c) Cl2 (g), AcOH/H2O, <+15 0C, 25min; d) Diisopropylethylamine, THF. -20 0C, 30 min.
The obtained compound (I) is then purified either by precipitation and washing with ethanol/water or by preparative HPLC. In a second embodiment, the racemate of compound (I), (5RS)-5-[4-(5-chloro-pyridin-2- yloxy)-piperidine-l-sulfonylmethyl]-5-methyl-imidazolidine-2,4-dione, was prepared by reacting l-[4-(5-chloro-pyridin-2-yloxy)-piperidine-l-sulfonyl]-propan-2-one with an excess of potassium cyanide and ammonium carbonate in ethanol, and isolating the product by precipitation. Compound (I), the (5S)-enantiomer, was then obtained by chiral HPLC (WO 02/074767; pages 55 and 65).
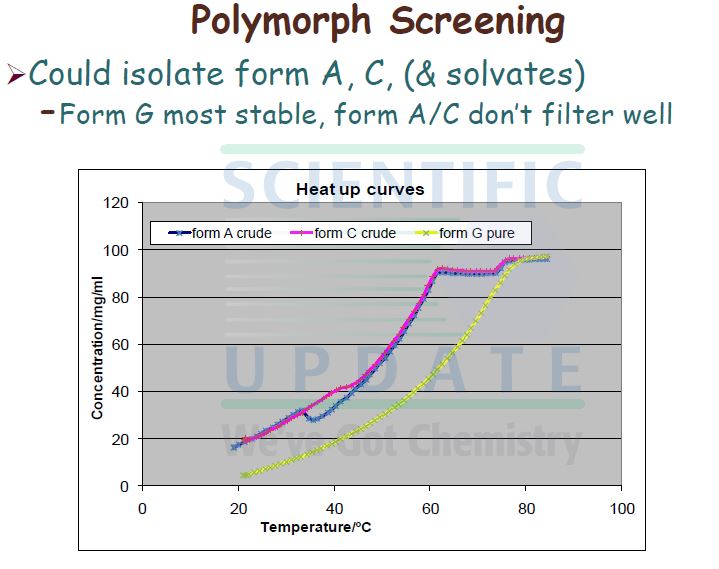
No crystalline forms of compound (I) are disclosed in WO 02/074767.
Compound (I) is a potent metalloproteinase inhibitor, particularly a potent inhibitor of
MMP 12, and as such is useful in therapy. However, when made according to the processes described in WO 02/074767, compound (I) exhibits unpredictable solid state properties with respect to thermodynamic stability. To prepare pharmaceutical formulations containing compound (I) for administration to humans in accordance with the requirements of U.S. and other international health registration authorities, there is a need to produce compound (I) in a stable form, such as a stable crystalline form, having constant physical properties.


PATENT
WO 2007106022
WO 02/074767 further discloses a specific metalloproteinase inhibitor compound identified therein as (5S)-5-[4-(5-chloro-pyridin-2-yloxy)-piperidine-l-sulfonylmethyl]-5- methyl-imidazolidine-2,4-dione (page 65, lines 15 to 27; and page 120, lines 23 to 29). This compound is designated herein as compound (I).
(I)
WO 02/074767 further discloses processes for the preparation of compound (I). Thus, in one embodiment, compound (I) is prepared by a route analogous to that shown in the following Scheme (WO 02/074767; pages 87, 113 and 120) but substituting the appropriate amine in step (d):
Scheme 1
Reagents and conditions for Scheme 1: a) KCN, (NHLj)2CO3, EtOHTH2O, +900C, 3h;. b) Chiral separation, CHIRALPAK AD, methanol as eluent;. c) Cl2 (g), AcOH/H2O, <+15 0C, 25min; d) Diisopropylethylamine, THF. -20 0C, 30 min.
The obtained compound (I) is then purified either by precipitation and washing with ethanol/water or by preparative HPLC. In a second embodiment, the racemate of compound (I), (5RS)-5-[4-(5-chloro-pyridin-2- yloxy)-piperidine-l-sulfonylmethyl]-5-methyl-imidazolidine-2,4-dione, was prepared by reacting l-[4-(5-chloro-pyridin-2-yloxy)-piperidine-l-sulfonyl]-propan-2-one with an excess of potassium cyanide and ammonium carbonate in ethanol, and isolating the product by precipitation. Compound (I), the (5S)-enantiomer, was then obtained by chiral HPLC (WO 02/074767; pages 55 and 65).
No crystalline forms of compound (I) are disclosed in WO 02/074767.
Compound (I) is a potent metalloproteinase inhibitor, particularly a potent inhibitor of
MMP 12, and as such is useful in therapy. However, when made according to the processes described in WO 02/074767, compound (I) exhibits unpredictable solid state properties with respect to thermodynamic stability. To prepare pharmaceutical formulations containing compound (I) for administration to humans in accordance with the requirements of U.S. and other international health registration authorities, there is a need to produce compound (I) in a stable form, such as a stable crystalline form, having constant physical properties.
A preferred process for the synthesis of compound (I) is shown in Scheme 2.
Scheme 2
KCN, (NH4)2CO3
(H) 2-propanol
Chromatography KOBu’
Cl2
AcOH AcOH, H2O
Compound (I)
Recrystallisation EtOH, H2O
Compound (I) Form G
Example 5
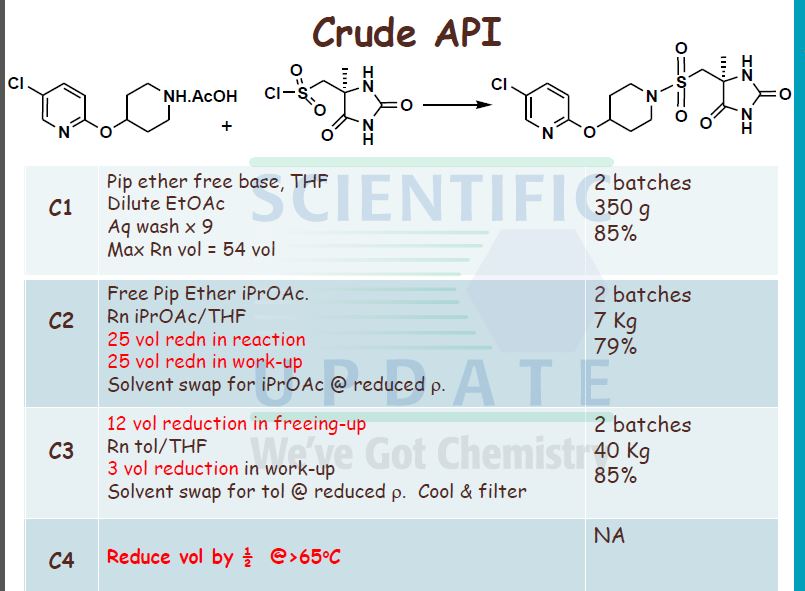
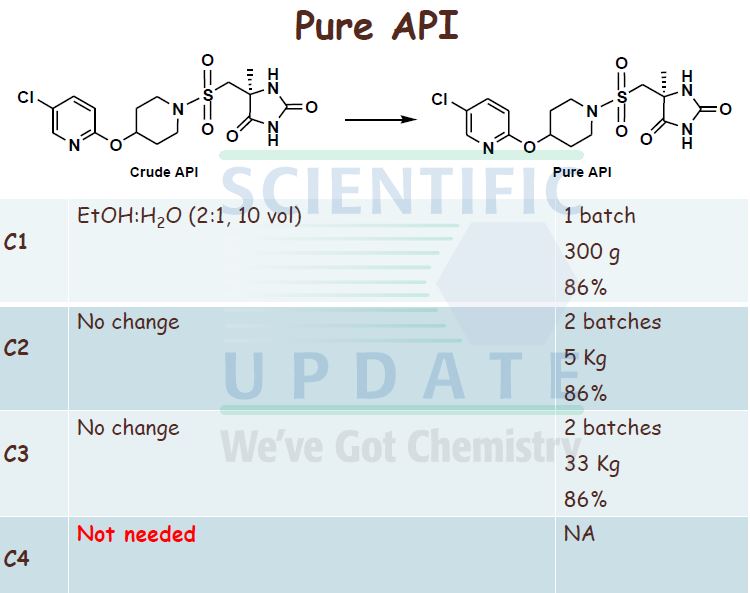
(S)-5-[4-(5-ChIoro-pyridin-2-yloxy)-piperidine-l-suIfonylmethyl]-5-methyl- imidazoIidine-2,4-dione Process 1
I5 a) 5-Chloro-2-(piperidin-4-yloxy)-pyridine
5-Chloro-2-(piperidin-4-yloxy)-pyridine acetate (40 g, 0.146 mol) was slurried in iso- PrOAc (664 mL) at 300C. To this slurry was added Na2CO3 (1.5 mol per litre; 196 mL, 2 mol eq.). The slurry was then rapidly stirred at 30 0C for 15 minutes. The biphasic mixture was allowed to settle, and the bottom aqueous phase was separated and discarded.
20 The above base washing procedure was repeated twice more. The organic phase was then washed once with water (200 mL). The resulting iso-VxOAc solution was reduced in volume to approximately 300 mL by distillation under reduced pressure. The solution was then diluted with zsø-PrOAc (400 mL) and again distilled down to approximately 300 mL. This procedure was repeated once more. A sample was removed for analysis of 5-chloro-
25 2-(piperidm-4-yloxy)-pyridine content and water content. The weight or the volume of the solution was measured in order to calculate the concentration of 5-chloro-2-(piperidin-4- yloxy)-pyridme in the Z-PrOAc solution.
fr) rSV5-r4-(5-Chloro-pyridin-2-yloxyVpiperidine-l-sulfonylmethvn-5-methyl- 30 imidazolidine-2 ,4-dione Diisopropylethylamine (24.3 mL, 0.139 mol, 1 mol eq.) was added to the iso-PrOAc solution prepared in part (a) [ca. 300 mL; equivalent to 31.2 g, 0.146 mol, 1.05 mol eq. of 5-chloro-2-(piperidin-4-yloxy)-pyridine] in one portion at RT. The solution was then cooled to -15 °C.
((S)-4-Methyl-2,5-dioxo-imidazolidin-4-yl)-methanesulfonyl chloride (31.65 g, 0.139 mol, 1 mol eq.) was dissolved in dry THF (285 mL) at RT with stirring. The resulting solution was then added to the iso-PrOAc solution of 5-chloro-2-(piperidin-4-yloxy)- pyridine dropwise at -15 0C over about 1.5 h. A precipitate was seen on addition of the ((S)-4-methyl-2,5-dioxo-imidazolidin-4-yl)-methanesulfonyl chloride. At the end of the addition, dry THF (32 mL) was added to the reaction mixture to wash the line and the mixture was stirred for 1 h at — 15 0C. It was then warmed to 20 °C over 1 h and stirred at 20 °C for 1 h further. The reaction was quenched with 10 wt% NaHSO4 (157 mL) with rapid stirring. After about 15 minutes, the biphasic mixture was allowed to settle, and the bottom aqueous phase was separated and discarded. This acid wash procedure was repeated once more. The organic phase was then washed with water (157 mL) using rapid stirring and allowing complete phase separation before partitioning. The reaction solution was then warmed to 40 °C and washed again with water (157 mL). THF (95 mL) was added to the organic layer that was then warmed to 40 0C and filtered at 400C to remove any particulate matter. The solvent volume was then reduced to about 157 mL by reduced pressure distillation with the jacket temperature at 55 °C. zso-PrOAc (317 mL) was then added and the volume was again reduced to about 157 mL. Two more put-and-takes of zsø-PrOAc (317 mL) were carried out. Solids began to precipitate out during the distillations and a suspension resulted. The volume was reduced to about 157 mL each time and after the final distillation a small sample of solvent was then removed from the reaction mixture for residual THF analysis. The 1H NMR showed no THF peaks. The contents of the reaction were then cooled to 0 °C and the product was collected by filtration. The reaction vessel was washed with zsø-PrOAc (63 mL) and this rinse was used to wash the product on the filter. The product was dried overnight in a vacuum oven at 40 °C. The required (S)-5-[4- (5-chloro-pyridin-2-yloxy)-piperidine-l-sulfonyhnethyl]-5-methyl-imidazolidine-2,4-dione was isolated as a white solid in 71% yield (41.8 g).
1H NMR (300MHz, d6-DMSO) δ 10.74 (IH, s), 8.20 (IH, d), 8.01 (IH, s), 7.81 (IH, dd), 6.87 (IH, d), 5.09 (IH, m), 3.52-3.35 (4H, m), 3.13 (2H, m), 2.02 (2H, m), 1.72 (2H, m), 1.33 (3H, s).
Example 6
(S)-5-[4-(5-Chloro-pyridin-2-yloxy)-piperidine-l-sulfonylmethyl]-5-methyl- imidazolidine-2,4-dione Process 2 a) 5-Chloro-2-(piperidm-4-yloxy)-pyridine
5-Chloro-2-(piperidm-4-yloxy)-pyridine acetate (70 g, 257 mmol) was slurried in toluene
(560 mL) at RT. IM NaOH (420 mL) was added and the slurry was then rapidly stirred at RT for 15 min. The biphasic mixture was allowed to settle, and the bottom aqueous phase was separated and discarded. The organic phase was then washed with water (2 x 420 mL). A sample was removed from the organic phase and assayed for 5-chloro-2-(piperidin-
4-yloxy)-pyridine.
The resulting toluene solution was then reduced in volume by distillation at reduced pressure, down to approximately 168 mL (2.4 vol eq. with respect to 5-chloro-2-(piperidin-
4-yloxy)-pyridine acetate charge). The solution was then diluted with toluene (420 mL) and again distilled down to approx 168 mL (2.4 vol eq.). A sample was removed for analysis of water content.
b*) (S)-5-r4-r5-Chloro-pyridm-2-yloxy)-piperidine-l-sulfonylmethvH-5-methyl- imidazolidine-2 ,4-dione
Diisopropylethylamine (38.4 mL, 220 mmol) was added to the toluene solution of 5-chloro-2-(piperidin-4-yloxy)-pyridine obtained in step (a) (containing 236 mmol) in one portion followed by dry THF (151 mL) as a line wash. ((S)-4-Methyl-2,5-dioxo- imidazolidin-4-yl)-methanesulfonyl chloride (48.7 g, 215 mmol) was dissolved in dry THF (352 mL) at RT with stirring. The resulting solution of the sulfonyl chloride was then added dropwise to the toluene/THF solution of 5-chloro-2-(piperidin-4-yloxy)-pyridine and diisopropylethylamine at RT over 1 to 2 h. A precipitate was seen on addition of the sulfonyl chloride. At the end of the addition, dry THF (50 mL) was added to the reaction 5 mixture as a line wash. After the addition was complete, the reaction was stirred for about 30 min at RT.
The reaction was quenched with 10 wt% NaHSO4 (251 mL) with rapid stirring for approx 15 min. The biphasic mixture was allowed to settle, when the bottom aqueous phase was io separated and discarded. This acid wash procedure was repeated once more. The solvent volume was then reduced to about 220 mL by reduced pressure distillation. Toluene (300 mL) was then added and the volume was reduced to about 245 mL Solids begin to precipitate during the distillations and a suspension resulted. After the final distillation, a small sample of solvent was then removed from the reaction mixture for residual THF i5 analysis.
The contents of the reaction mixture were then cooled to 0 °C, stirred for about 30 minutes at this temperature and the product was collected by filtration. The reaction vessel was washed with toluene (100 mL) and this rinse was used to wash the product on the filter. 20 The product was dried in a vacuum oven at 40 0C to constant weight. (S)-5-[4-(5-Chloro- pyridin-2-yloxy)-piperidine-l-sulfonylmethyl]-5-methyl-imidazolidine-2,4-dione was isolated as a white solid in typically 85 to 88% yield over the two steps.

Aerial view of Mölndal
Patent
WO 2003106689

Paul Hudson, President, AstraZeneca U.S. and Executive Vice President, North America, joined by AstraZeneca volunteers to celebrate the AstraZeneca Hope Lodge’s fifth birthday.
CLIPS

Astra boss Pascal Soriot

Massachusetts Economic Development Secretary Jay Ash (left) congratulates Kumar Srinivasan, Head of AstraZeneca R&D Boston (right), at a ceremony to launch AstraZeneca’s Gatehouse Park BioHub.


| References |
1. AstraZeneca.
AZD1236.
Accessed on 31/10/2014. Modified on 31/10/2014. Open Innovation, http://openinnovation.astrazeneca.com/what-we-offer/compound/azd1236/ |
2. Dahl R, Titlestad I, Lindqvist A, Wielders P, Wray H, Wang M, Samuelsson V, Mo J, Holt A. (2012)
Effects of an oral MMP-9 and -12 inhibitor, AZD1236, on biomarkers in moderate/severe COPD: a randomised controlled trial.
Pulm Pharmacol Ther, 25 (2): 169-77. [PMID:22306193] |
https://ncats.nih.gov/files/AZD1236.pdf
AZD1236
| WO1992001062A1 * |
Jul 4, 1991 |
Jan 23, 1992 |
Novo Nordisk A/S |
Process for producing enantiomers of 2-aryl-alkanoic acids |
| WO1996021640A1 * |
Jan 16, 1996 |
Jul 18, 1996 |
Teva Pharmaceutical Industries, Ltd. |
Optically active aminoindane derivatives and preparation thereof |
| WO2002074767A1 * |
Mar 13, 2002 |
Sep 26, 2002 |
Astrazeneca Ab |
Metalloproteinase inhibitors |
| WO2003093260A1 * |
Apr 29, 2003 |
Nov 13, 2003 |
Biogal Gyogyszergyar Rt. |
Novel crystal forms of ondansetron, processes for their preparation, pharmaceutical compositions containing the novel forms and methods for treating nausea using them |
| WO2003094919A2 * |
May 12, 2003 |
Nov 20, 2003 |
Teva Pharmaceutical Industries Ltd. |
Novel crystalline forms of gatifloxacin |
| EP0175312A2 * |
Sep 14, 1985 |
Mar 26, 1986 |
Kanegafuchi Kagaku Kogyo Kabushiki Kaisha |
Process for preparing optically active hydantoins |
| EP0255390A2 * |
Jul 30, 1987 |
Feb 3, 1988 |
MediSense, Inc. |
Rhodococcus bacterium for the production of aryl acylamidase |
| EP0442584A1 * |
Feb 14, 1991 |
Aug 21, 1991 |
Dsm N.V. |
Process for the preparation of an optically active amino acid amide |
| EP0580210A1 * |
Jul 6, 1993 |
Jan 26, 1994 |
Dsm N.V. |
Process for the preparation of optically active methionine amide |
| EP0909754A1 * |
Oct 13, 1998 |
Apr 21, 1999 |
Eli Lilly And Company |
Process to make chiral compounds |
| EP1550725A1 * |
Jun 5, 2003 |
Jul 6, 2005 |
Kaneka Corporation |
PROCESS FOR PRODUCING OPTICALLY ACTIVE alpha-METHYLCYSTEINE DERIVATIVE |
| US4983771 * |
Sep 18, 1989 |
Jan 8, 1991 |
Hexcel Corporation |
Method for resolution of D,L-alpha-phenethylamine with D(-)mandelic acid |
| US20040044215 * |
Aug 28, 2003 |
Mar 4, 2004 |
Alain Alcade |
Crystalline forms of osanetant |
| US20040266832 * |
Jun 24, 2004 |
Dec 30, 2004 |
Li Zheng J. |
Crystal forms of 2-(3-difluoromethyl-5-phenyl-pyrazol-1-yl)-5-methanesulfonyl pyridine |
| Citing Patent |
Filing date |
Publication date |
Applicant |
Title |
| US7625934 |
|
Dec 1, 2009 |
Astrazeneca Ab |
Metalloproteinase inhibitors |
| US7772403 |
Mar 15, 2007 |
Aug 10, 2010 |
Astrazeneca Ab |
Process to prepare sulfonyl chloride derivatives |
///////AZD1236, AZD-1236, AZD 1236,
O=S(=O)(C[C@@]1(C)NC(=O)NC1=O)N3CCC(Oc2ccc(Cl)cn2)CC3