| Active Pharmaceutical Ingredient (API) |
An active pharmaceutical ingredient (API) is a substance used in a finished pharmaceutical product, intended to furnish pharmacological activity or to otherwise have direct effect in the diagnosis, cure, mitigation, treatment or prevention of disease, or to have direct effect in restoring, correcting or modifying physiological functions in human beings.
|
| Annual Product Reviews (APR) |
The Annual Product Reviews (APR) include all data necessary for evaluation of the quality standards of each drug product to determine the need for changes in drug product specifications or manufacturing or control procedures. The APR is required by the U.S. Code of Federal Regulations. |
| ANVISA |
The Brazilian Health Surveillance Agency (in Portuguese, Agência Nacional de Vigilância Sanitária) is a governmental regulatory body in Brazil. Similar to the FDA in the United States, it oversees the approval of drugs and other health products and regulates cosmetics, food products, and other health-related industries. |
| Biologic License Application (BLA) |
The Biologics License Application (BLA) is a request for permission to introduce, or deliver for introduction, a biologic product into commerce in the U.S. |
| CFDA |
The China Food and Drug Administration is similar to the FDA in the United States and is responsible for regulating food and drug safety. |
| cGMP |
Current Good Manufacturing Practices govern the design, monitoring, and control of manufacturing facilities and processes and are enforced by the US FDA. Compliance with these regulations helps safeguard a drug’s identity, strength, quality, and purity. |
| COFEPRIS |
The Federal Commission for Protection against Sanitary Risks (in Spanish, Comisión Federal para la Protección contra Riesgos Sanitarios) is a government agency in Mexico. It regulates food safety, drugs, medical devices, organ transplants, and environmental protection. |
| Common Technical Document (CTD) |
The Common Technical Document (CTD) is the mandatory common format for new drug applications in the EU and Japan, and the U.S. The CTD assembles all the Quality, Safety and Efficacy information necessary for a drug application. |
| European Medicines Agency (EMA) |
The European Medicines Agency (EMA) is a decentralised agency of the European Union (EU), located in London. It began operating in 1995. The Agency is responsible for the scientific evaluation, supervision and safety monitoring of medicines developed by pharmaceutical companies for use in the EU. |
| Food and Drug Administration (FDA) |
The Food and Drug Administration (FDA) is an agency within the U.S. Department of Health and Human Services. The FDA is responsible for the approval of new pharmaceutical products for sale in the U.S. and performs audits at the companies participating in the manufacture of pharmaceuticals to ensure that they comply with regulations. |
| Human growth hormone |
A growth hormone (GH or HGH) is a peptide hormone produced by the pituitary gland that stimulates growth in children and adolescents. It is involved in several body processes, including cell reproduction and regeneration, regulation of body fluids, and metabolism. It can be produced by the body (ie, somatotropin) or genetically engineered (ie, somatropin). |
| In-Process Control (IPC) |
In-Process Controls (IPC) are checks performed during production in order to monitor and if necessary to adjust the process to ensure that the product conforms its specification. |
| Interferons (INFs) |
Interferons are proteins produced by the body as part of the immune response. They are classified as cytokines, proteins that signal other cells to trigger action. For example, a cell infected by a virus will release interferons to stimulate the defenses of nearby cells. |
| Interleukins |
Interleukins are proteins produced by cells as an inflammatory response. Most interleukins help leukocytes communicate with and direct the division and differentiation of other cells. |
| Investigational Medicinal Product Dossier (IMPD) |
The Investigational Medicinal Product Dossier (IMPD) is the basis for approval of clinical trials by the competent authorities in the EU. The IMPD includes summaries of information related to the quality, manufacture and control of the Investigational Medicinal Product, data from non-clinical studies and from its clinical use. |
| Investigational New Drug (IND) |
An Investigational New Drug application is provided to the FDA to obtain permission to test a new drug in humans in Phase I – III clinical studies. The IND is reviewed by the FDA to ensure that study participants will not be placed at unreasonable risk. |
| Marketing Authorization Application (MAA) |
The Marketing Authorization Application (MAA) is a common document used as the basis for a marketing application across all European markets, plus Australia, New Zealand, South Africa, and Israel. This application is based on a full review of all quality, safety, and efficacy data, including clinical study reports. |
| Master batch records |
These general manufacturing instructions, which are required by cGMP, are the bases for a precise, detailed description of a pharmaceutical manufacturing process. They ensure that all proper ingredients are included, each process step is completed, and the process is controlled. |
| Medicines and Healthcare Products Regulatory Agency (MHRA) |
The Medicines and Healthcare products Regulatory Agency (MHRA) regulates medicines, medical devices and blood components for transfusion in the UK. MHRA is an executive agency, sponsored by the Department of Health. |
| MFDS |
The Ministry of Food and Drug Safety (formerly the Korean Food & Drug Administration) is a government agency that oversees the safety and efficacy of drugs and medical devices in South Korea. |
| Monoclonal antibodies |
Monoclonal antibodies are antibodies made in a laboratory from identical immune cells that are clones of a single cell. They are distinct from polyclonal antibodies, which are made from different immune cells. |
| NDA |
A New Drug Application (NDA) is the vehicle submitted to the FDA by drug companies in order to gain approval to market a new product. Safety and efficacy data, proposed package labeling, and the drug’s manufacturing methods are typically included in an NDA. |
| New Drug Application (NDA) |
The New Drug Application (NDA) is the vehicle through which drug sponsors formally propose that the FDA approve a new chemical pharmaceutical for sale and marketing in the U.S.
|
| Oligonucleotides |
These short nucleic acid chains (made up of DNA or RNA molecules) are used in genetic testing, research, and forensics. |
| Parenteral |
Parenteral medicine is taken or administered in a manner other than through the digestive tract. Intravenous and intramuscular injections are two examples. |
| Peptide hormones |
Peptide hormones are proteins secreted by organs such as the pituitary gland, thyroid, and adrenal glands. Examples include follicle-stimulating hormone (FSH) and luteinizing hormone. Similar to other proteins, peptide hormones are synthesized in cells from amino acids. |
| PMDA |
The Pharmaceuticals Medical Devices Agency is an independent administrative agency that works with the Ministry of Health, Labour and Welfare to oversee the safety and quality of drugs and medical devices in Japan. |
| Process Analytical Technology (PAT) |
These analytical tools help monitor and control the manufacturing process, including accommodating for variability in material and equipment, in order to ensure consistent quality. |
| Product Quality Reviews (PQR) |
The Product Quality Reviews (PQR) of all authorized medicinal products, is conducted with the objective of verifying the consistency of the existing process, the appropriateness of current specifications for both starting materials and finished product, to highlight any trends and to identify product and process improvements. The PQR is required by the EU GMP Guideline. |
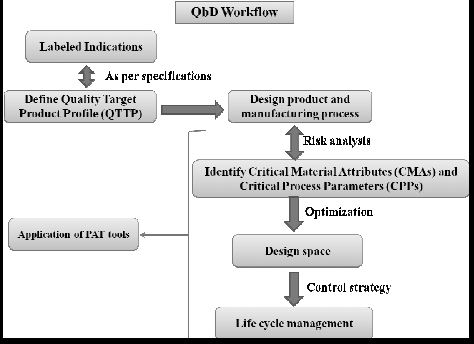
| Quality by Design (QbD) |
This concept involves a holistic, proactive, science- and risk-based approach to the development and manufacturing of drugs. At the heart of QbD is the idea that quality is achieved through in-depth understanding of the product and the process by which it is developed and manufactured. |
| Restricted Access Barrier System (RABS) |
This advanced aseptic processing system provides an enclosed environment that reduces the risk of contamination to the product, containers, closures, and product contact surfaces. As a result, it can be used in many applications in a fill-finish area. |
| Scale-up |
Scale-up involves taking a small-scale manufacturing system developed in the laboratory to a commercially viable, robust production process. |
| Six Sigma |
Six Sigma is a set of quality management methods, techniques, and tools used to improve manufacturing, transactional, and other business processes. The goal is to enhance quality (as well as employee morale and profits) by identifying and eliminating the cause of errors and process variations. |
| Target Product Profile (TPP) |
This key strategic document summarizes the features of an intended drug product. Characteristics may include the dosage form, route of administration, dosage strength, pharmacokinetics, and drug product quality criteria. |
| TFDA |
The Taiwan Food & Drug Administration is a governmental body devoted to enhancing food safety and drug quality in that country. |