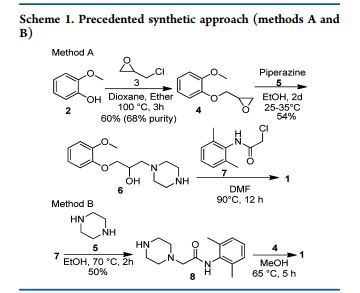
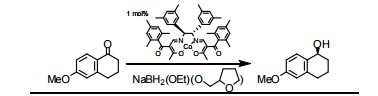
CSIR, Dr. D. Srinivasa Reddy
WO2016181414, PROCESS FOR THE SYNTHESIS OF IVACAFTOR AND RELATED COMPOUNDS
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016181414&recNum=1&maxRec=&office=&prevFilter=&sortOption=&queryString=&tab=PCTDescription
REDDY, Dumbala Srinivasa; (IN).
NATARAJAN, Vasudevan; (IN).
JACHAK, Gorakhnath Rajaram; (IN)
COUNCIL OF SCIENTIFIC & INDUSTRIAL RESEARCH [IN/IN]; Anusandhan Bhawan, Rafi Marg New Delhi 110001 (IN)
The present patent discloses a novel one pot two-step process for the synthesis of ivacaftor and related compounds of [Formula (I)], wherein R1, R2, R3, R4, R5, R6, R7 and Ar1are as described above; its tautomers or pharmaceutically acceptable salts thereof starting from indole acetic acid amides
See Eur J Org Chem, Nov 2015, for an article by the inventors, describing a process for preparing ivacaftor using 4-quinolone-3-carboxylic acid amides. The inventors appear to be based at National Chemical Laboratories of CSIR.
Ivacaftor, also known as N-(2,4-di-tert-butyl-5-hydroxyphenyl)-l,4-dihydro-4-oxoquinoline-3-carboxamide, having the following Formula (A):

Formula (A)
[003] Ivacaftor was approved by FDA and marketed by vertex pharma for the treatment of cystic fibrosis under the brand name KALYDECO® in the form of 150 mg oral tablets. Kalydeco® is indicated for the treatment of cystic fibrosis in patients age 6 years and older who have a G55ID mutation in the CFTR (cystic fibrosis transmembrane conductance regulator)gene.
[004] U.S. 20100267768 discloses a process for preparation of ivacaftor, which involves the coupling of 4-oxo-l,4-dihydro-3- quinoline carboxylic acid with hydroxyl protected phenol intermediate in the presence of propyl phosphonic anhydride (T3P®) followed by deprotection of hydroxyl protection group and optional crystallization with isopropyl acetate. The publication also discloses the use of highly expensive coupling reagent, propyl phosphonic anhydride; which in turn results to an increase in the manufacturing cost. The process disclosed is schematically represented as follows:

[005] Article titled “Discovery of N-(2,4-Di-te -butyl-5-hydroxyphenyl)-4-oxo-l,4-dihydroquinoline-3-carboxamide (VX-770, Ivacaftor), a Potent and Orally Bioavailable CFTR Potentiator” byHadida,S et. al in . Med. Chem., 2014, 57 (23), pp 9776-9795 reportsN-(2,4-di-teri-butyl-5-hydroxyphenyl)-4-oxo- 1 ,4-dihydroquinoline-3-carboxamide (VX-770, 48, ivacaftor), an investigational drug candidate approved by the FDA for the treatment of CF patients 6 years of age and older carrying the G551D mutation.

[006] WO 2014125506 A2 discloses a process for the preparation of ivacaftor in high yield and purity by using novel protected quinolone carboxylic acid compounds as intermediates.
[007] Article titled “Expeditious synthesis of ivacaftor” by Jingshan Shen et. al in Heterocycles, 2014, 89 (4), pp 1035 – 1040 reports an expeditious synthesis for ivacaftor featuring modified Leimgruber-Batcho procedure. The overall yield is 39% over six steps from commercially available 2-nitrobenzoyl chloride.
[008] U.S.2011/064811 discloses a process for preparation of ivacaftor, which involves condensation of 4-oxo-l,4-dihydro-3- quinolone carboxylic acid with 5- amino-2,4-di-(tert-butyl)phenol in the presence of HBTU followed by the formation of ethanol crystalate, which is then treated with diethyl ether to yield ivacaftor as a solid.

[010] U.S. 7,495,103 discloses modulators of ATP-binding cassette transporters such as ivacaftor and a process for the preparation of modulators of ATP-binding cassette transporters such as quinolone compounds. The process includes condensation of 4-oxo-l,4-dihydro-3 -quinolone carboxylic acid with aniline in presence of 2-(lH-7-azabenzotriazol-l-yl)-l,l,3,3-tetramethyluronium hexafluoro phosphate methanaminium (HATU) as shown:

[011] U.S. 2011/230519 discloses a process for preparation of 4-oxo-l,4-dihydro-3-quinoline carboxylic acid by reaction of aniline with diethylethoxymethylenemalonate at 100-110°C followed by cyclization in phenyl ether at temperature 228-232°C and then hydrolysis, as shown below:

[012] US 7,402,674 B2 discloses 7-Phenylamino-4-quinolone-3-carboxylic acid derivatives, process for their preparation and their use as medicaments.
[013] US 4,981,854 discloses l-aryl-4-quinolone-3 carboxylic acids, processes for their preparation and anti-bacterial agents and feed additives containing these compounds.
Article titled “Ozonolysis Applications in Drug Synthesis” by Van Ornum,S.G. ; Champeau,R.M.; Pariza,R. in Chem. Rev., 2006, 106 (7), pp 2990-3001 reports that ozonolysis for the synthesis of numerous interesting bioactive natural products and pharmaceutical agents.
[014] Article titled “Safe Execution of a Large-Scale Ozonolysis: Preparation of the Bisulfite Adduct of 2-Hydroxyindan-2-carbox-aldehyde and Its Utility in a Reductive Animation” by RaganJ.A. et. al. in Org. Proc. Res. Dev., 2003, 7 (2), pp 155-160 reports various routes to bisulfite adduct, the most efficient of which involved vinyl Grignard addition to 2-indanone followed by ozonolysis and workup with aqueous NaHS03 to effect reduction and bisulfite formation in a single pot. The utility of bisulfite adduct is as an aldehyde surrogate in a reductive amination reaction.

[015] The reported methods for the synthesis of ivacaftor suffered from several drawbacks such as harsh conditions, high temperature reactions and use of large excess of polyphosphoric acid and corrosive phosphoryl chloride etc. Furthermore, synthesis of ivacaftor requires use of high performance liquid chromatography (HPLC) techniques for the separation of ivacaftor and their analogues.
[016] Therefore, development of a simple and efficient synthetic route is in urgent need. Accordingly the present inventors developed environmentally benign, cost effective and short synthetic route for the synthesis of ivacaftor and their analogues.
Example 1:
Procedur A:

To a solution of indole acetic acid (500 mg, 2.85 mmol), aniline (2.85 mmol), HOBt (3.4 mmol) in acetonitrile (10 mL), EDC.HCl (3.4 mmol) followed by DIPEA (11.4 mmol) was added, and mixture was stirred for 16 h at ambient temperature. The
reaction mixture was evaporated to dryness, diluted with EtOAc (25 mL), washed with saturated aqueous NaHC03 solution (5 mL), H20 (5 mL), brine (5 mL), and dried over Na2S04. The crude material obtained after removal of solvent was purified by column chromatography (silica gel 230-400 mesh, ethyl acetate – pet ether) to afford corresponding amide as a colorless solid.
[040] Example 2:
2-(lH-indol-3-yl)-N-phenylacetamide (1) :
Yield: 570 mg; 80%; 1H NMR (200MHz, DMSO-d6) δ = 10.95 (brs, 1 H), 10.14 (s, 1 H), 7.64 (d, J = 7.8 Hz, 3 H), 7.47 – 7.24 (m, 4 H), 7.21 – 6.92 (m, 3 H), 3.76 (s, 2H); MS: 273 (M+Na)+.
[041] Example 3:
5-(2-(lH-indol-3-yl)acetamido)-2,4-di-tert-butylphenyl methyl carbonate (2): Yield: 800 mg; 64%; 1H NMR (200 MHz, DMSO-d6) δ = 11.51 (brs, 1 H), 9.41 (s, 1 H), 8.12 (d, J = 7.6 Hz, 1 H), 7.96 – 7.78 (m, 3 H), 7.71 – 7.42 (m, 3 H), 4.34 (s, 3 H), 4.30 (s, 2 H), 1.79 (s, 9 H), 1.64 (s, 9 H); MS: 459 (M+Na)+.
[042] Example 4:
(S)-2-(lH-indol-3-yl)-N-(l-phenylethyl)acetamide (3):
Yield: 620 mg; 78%; 1H NMR (400MHz ,DMSO-d6)5 = 10.88 (brs, 1 H), 8.48 (d, J = 8.1 Hz, 1 H), 7.59 (d, J = 7.8 Hz, 1 H), 7.39 – 7.26 (m, 5 H), 7.25 – 7.16 (m, 2 H), 7.08 (t, J = 7.3 Hz, 1 H), 7.02 – 6.95 (m, 1 H), 4.96 (t, J = 7.3 Hz, 1 H), 3.59 (s, 2H), 1.38 (d, J = 7.1 Hz, 3 H).
[043] Example 5:
N-(4-Fluorophenyl)-2-(lH-indol-3-yl)acetamide (4):
1H NMR (400 MHz, DMSO-d6) : δ 10.93 (brs, 1H), 10.17 (s, 1H), 7.68 – 7.61 (m, 3H), 7.36 (d, J= 8.1 Hz, 1H), 7.27 (d, J= 2.0 Hz, 1H), 7.15 – 7.13 (m, 3H), 7.11 – 6.99 (m, 1H), 3.73 (s, 2H); 13C NMR (100 MHz, DMSO-d6) : δ 170.1, 159.5, 157.1, 136.6, 136.3, 127.7, 124.4, 121.5, 121.3, 121.2, 119.1, 118.9, 115.8, 115.6, 111.8, 108.9, 34.2; MS: 269 (M+H)+
[044] Example 6:
N-(4-Chlorophenyl)-2-(lH-indol-3-yl)acetamide (5):
1H NMR (200 MHz, DMSO-d6): 510.93 (brs, 1H),10.24 (s, 1H), 7.67 – 7.59 (m, 3H), 7.36 – 7.27 (m, 4H), 7.12 – 6.98 (m, 2H), 3.74 (s, 2H); 13CNMR (100 MHz, DMSO-d6): 5170.4, 138.9, 136.7, 129.1, 127.8, 127.1, 124.5, 121.6, 121.2, 119.2, 119.0, 115.7, 111.9, 108.9, 34.3; MS: 285 (M+H)+.
[045] Example 7:
2-(lH-Indol-3-yl)-N-(p-tolyl)acetamide (6) :
1H NMR (400 MHz, DMSO-d6): 510.91 (brs, 1H), 10.01 (s, 1H), 7.62 (d, J= 7.8 Hz, 1H), 7.50 (d, J= 8.6 Hz, 2H), 7.37 (d, J= 8.1 Hz, 1H), 7.29 – 7.26 (m, 1H), 7.10 – 7.07 (m, 3H), 7.01 – 6.99 (m, 1H), 3.71 (s, 2H), 2.23 (s, 3H); 13C NMR (100 MHz, DMSO-de): 5170.0, 137.4, 136.6, 132.4, 129.5, 127.7, 124.3, 121.4, 119.6, 119.2, 118.8, 111.8, 109.1, 34.2, 20.9; MS: 265 (M+H)+.
[046] Example 8:
N-(4-Ethylphenyl)-2-(lH-indol-3-yl)acetamide (7):
XH NMR (400 MHz, DMSO-d6): 510.91 (brs, 1H), 10.01 (s, 1H), 7.61 (s, 1H), 7.52 (d, J= 8.3 Hz, 2H), 7.36 (d, J= 8.1 Hz, 1H), 7.26 (s, 1H), 7.15 – 7.04 (m, 3H), 6.99 (s, 1H), 2.55 (t, J= 7.5 Hz, 2H), 1.15 (t, J= 7.5 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): 5169.9, 138.9, 137.6, 136.6, 128.3, 127.7, 124.3, 121.4, 119.6, 119.2, 118.8, 111.8, 109.1, 40.6, 40.4, 40.2, 40.0, 39.8, 39.6, 39.4, 34.2, 28.0, 16.2; MS: 279 (M+H)+.
[047] Example 9:
2-(lH-Indol-3-yl)-N-(4-propylphenyl)acetamide (8):
1H NMR (400 MHz, DMSO-d6): 58.48 (brs, 1H), 7.64 (d, J = 8.1 Hz, 1H), 7.50 – 7.42 (m, 2H), 7.33 – 7.15 (m, 6H), 7.07 (d, J= 8.3 Hz, 2H), 3.92 (s, 2H), 2.52 (t, J= 7.6 Hz, 2H), 1.65 – 1.53 (m, 2H), 0.91 (t, J= 7.3 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): 5169.7, 138.9, 136.5, 135.2, 128.8, 126.9, 124.0, 122.8, 120.4, 120.1, 118.7, 111.6, 108.7, 37.4, 34.5, 24.6, 13.7; MS: 315 (M+Na)+.
[048] Example 10:
2-(lH-Indol-3-yl)-N-(4-isopropylphenyl)acetamide (9) :
yield 79% ; 1H NMR (400 MHz, DMSO-d6): δ 10.91 (brs, 1H), 10.01 (s, 1H), 7.62 (d, = 7.8 Hz, 1H), 7.55 – 7.49 (m, = 8.6 Hz, 2H), 7.37 (d, = 8.1 Hz, 1H), 7.26 (d, = 2.0 Hz, 1H), 7.18 – 7.11 (m, = 8.6 Hz, 2H), 7.11 – 7.05 (m, 1H), 7.02 – 6.95 (m, 1H), 2.95 – 2.71 (m, 1H), 1.17 (d, = 6.8 Hz, 6H); 13C NMR (100 MHz, DMSO-d6): δ 169.9, 143.5, 137.6, 136.6, 127.7, 126.8, 124.3, 121.4, 119.7, 119.2, 118.8, 111.8, 109.2, 24.4; MS: 315 (M+Na)+.
[049] Example 11:
2-(lH-indol-3-yl)-N-(4-(trifluoromethoxy)phenyl)acetamide (10):
Yield 85% ; 1H NMR (400 MHz, CDC13): δ 8.35 (brs., 1 H), 7.44 – 7.38 (m, 2 H), 7.27 – 7.21 (m, 3 H), 7.12 – 7.05 (m, 1H), 7.03 – 6.95 (m, 2H), 6.93 (d, = 8.6 Hz, 2H), 3.75 (s, 2H); 13C NMR (100 MHz, CDC13): δ 170.0, 145.3, 136.5, 136.2, 126.8, 124.1, 123.0, 121.6, 121.2, 120.5, 118.5, 111.7, 108.2, 34.4; MS: 335 (M+Na)+.
[050] Example 12:
N-(2-chloro-5-methoxyphenyl)-2-(lH-indol-3-yl)acetamide (11):
Yield 75% ; XH NMR (200 MHz, DMSO-d6): δ 10.98 (brs, 1H), 9.27 (s, 1H), 7.59 (d, = 7.8 Hz, 1H), 7.53 (d, = 2.9 Hz, 1H), 7.39 – 7.32 (m, 3H), 7.09 – 6.99 (m, 2H), 6.74 (dd, = 3.0, 8.8 Hz, 1H), 3.85 (s, 2H), 3.71 (s, 3H); 13C NMR (400 MHz, DMSO-d6): δ 170.4, 160.1, 141.1, 136.7, 130.0, 127.8, 124.4, 121.6, 119.2, 119.0, 111.9, 109.1, 105.4, 55.4, 34.4; MS: 315 (M+Na)+.
[051]Example 13:
N-(2-ethylphenyl)-2-(lH-indol-3-yl)acetamide (12):
Yield 78% ; 1H NMR (400 MHz, CDC13): δ 8.68 (brs, 1H), 7.95 (d, = 8.1 Hz, 1H), 7.67 (d, = 7.8 Hz, 1H), 7.48 – 7.44 (m, 2H), 7.29 – 7.23 (m, 1H), 7.22 – 7.20 (m, 3H), 7.05 (d, = 4.4 Hz, 2H), 2.00 (q, = 7.4 Hz, 2H), 0.67 (t, = 7.6 Hz, 3H); 13C NMR (100 MHz, CDC13): δ 169.9, 136.6, 135.0, 134.3, 128.7, 126.7, 125.1, 124.1, 123.0, 122.5, 120.4, 118.7, 111.6, 108.6, 34.4, 24.2, 13.6.
[052] Example 14:
N-(2-bromophenyl)-2-(lH-indol-3-yl)acetamide(13):
Yield 76%; 1H NMR (200 MHz, DMSO-d6): δ 11.00 (brs, 1H), 9.30 (s, 1H), 7.81 -7.77 (m, 1H), 7.63 – 7.56 (m, 2H), 7.41 – 7.35 (m, 3H), 7.11 – 7.05 (m, 3H), 3.85 (s, 2H);13C NMR (100 MHz, DMSO-d6): δ 169.9, 136.2, 132.5, 128.0, 127.2, 126.4, 125.5, 124.4, 121.2, 118.7, 118.5, 116.4, 111.4, 108.0, 33.2.
[053] Example 15:
N-benzyl-2-(lH-indol-3-yl)acetamide (14):
Yield 85%; 1H NMR (400 MHz, DMSO-d6): δ 10.89 (brs., 1H), 8.40 (t, = 5.7 Hz, 1H), 7.57 (d, = 7.8 Hz, 1H), 7.36 (d, = 8.1 Hz, 1H), 7.32 – 7.18 (m, 6H), 7.08 (t, = 7.5Hz, 1H), 7.03 – 6.90 (m, 1H), 4.28 (d, = 5.9Hz, 2H), 3.60 (s, 2H); 13C NMR (100 MHz, DMSO-de): δ 171.2, 140.1, 136.6, 128.7, 127.7, 127.2, 124.3, 121.4, 119.2, 118.7, 111.8, 109.3, 42.7, 33.2.
[054] Example 16:
2-(lH-indol-3-yl)-N-(4-methoxybenzyl)acetamide(15):
Yield 85% ; 1H NMR (400 MHz, DMSO-d6): δ 10.87 (brs, 1 H), 8.32 (t, = 5.6 Hz, 1 H), 7.55 (d, = 7.8 Hz, 1H), 7.35 (d, = 8.1 Hz, 1H), 7.22 – 7.13 (m, 3H), 7.11 – 7.05 (m, 1 H), 7.00 – 6.94 (m, 1H), 6.84 (d, = 8.6 Hz, 2H), 4.20 (d, = 6.1 Hz, 2H), 3.72 (s, 3H), 3.56 (s, 2H); 13C NMR (100 MHz, DMSO-d6): δ 171.1, 158.6, 136.6, 132.0, 129.0, 127.7, 124.2, 121.4, 119.2, 118.7, 114.1, 111.8, 109.4, 55.5, 42.1, 33.2.
[055] Example 17:
N,N-dibenzyl-2-(lH-indol-3-yl)acetamide (16):
Yield 70% ; 1H NMR (400 MHz, DMSO-d6): δ 10.91 (brs, 1H), 7.50 (d, = 7.8 Hz, 1H), 7.37 – 7.34 (m, 3H), 7.30 (d, = 6.6 Hz, 1H), 7.25 – 7.19 (m, 3H), 7.17 (t, = 6.6 Hz, 5H), 7.16 (d, = 7.8 Hz, 1H), 7.00 – 6.97 (m, 1H), 4.59 (s, 2H), 4.50 (s, 2H), 3.86 (s, 2H); 13C NMR (100 MHz, DMSO-d6): δ 171.7, 138.2, 136.6, 129.2, 128.8, 128.1, 127.8, 127.7, 127.5, 127.1, 124.2, 121.5, 119.2, 118.8, 111.8, 108.5, 50.7, 48.4, 31.2.
[056] Example 18:
2-(lH-indol-3-yl)-N-propylacetamide (17):
Yield 75% ; XH NMR (200 MHz, DMSO-d6): δ 10.86 (brs, 1H), 7.88 – 7.80 (m, 1H), 7.56 (d, = 7.6 Hz, 1H), 7.31 (d, = 7.8 Hz, 1H), 7.17 (d, = 2.3 Hz, 1H), 7.06 – 6.92 (m, 2H), 3.48 (s, 2H), 3.00 (q, J = 6.8 Hz, 2H), 1.39 (sxt, / = 7.2 Hz, 2H), 0.88 – 0.75 (t, = 7.2 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): δ 171.0, 136.6, 127.8, 124.2,
121.4, 119.2, 118.7, 111.8, 109.6, 39.4, 33.3, 22.9, 11.9.
[057] Example 19:
N-hexyl-2-(lH-indol-3-yl)acetamide (18) :
Yield 87% ; 1H NMR (400 MHz, DMSO-d6): δ 10.84 (brs, 1H), 7.83 (brs, 1H), 7.54 (d, = 7.8 Hz, 1H), 7.33 (d, = 8.1 Hz, 1H), 7.21 – 7.13 (m, 1H), 7.06 (t, = 7.6 Hz, 1H), 6.96 (t, J = 7.5 Hz, 1H), 3.47 (s, 2H), 3.03 (q, / = 6.8 Hz, 2H), 1.37 (t, = 6.5 Hz, 2H), 1.30 – 1.15 (m, 6H), 0.84 (t, = 6.7 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): δ 170.9, 136.6, 127.7, 124.2, 121.3, 119.1, 118.7, 111.7, 109.5, 39.06, 33.2, 31.5, 29.6, 26.5, 22.5, 14.4.
[058] Example 20:
Methyl (2-(lH-indol-3-yl)acetyl)-L-alaninate (19):
Yield 79% ; 1H NMR (400 MHz, CDC13): δ 8.53 (brs, 1H), 7.60 (d, = 7.8 Hz, 1H), 7.41 (d, = 8.1 Hz, 1H), 7.25 – 7.23 (m, 1H), 7.19 – 7.14 (m, 2H), 6.27 (d, = 7.3 Hz, 1H), 4.63 (t, = 7.3 Hz, 1H), 3.78 (s, 2H), 3.68 (s, 3H), 1.31 (d, = 7.3 Hz, 3H); 13C NMR (100 MHz, CDC13): δ 173.4, 171.2, 136.4, 127.0, 123.8, 122.5, 119.9, 118.7,
111.5, 108.5, 52.4, 48.0, 33.3, 18.2.
[059] Example 21:
-(6-chloro-lH-indol-3-yl)-N-phenylacetamide(20):

To a solution of 6-Chloro indole 20a (300 mg, 1.98 mmol )in anhydrous THF, Oxalyl chloride (186 μΤ, 276 mg, 2.18 mmol) was added and the mixture stirred at room temperature. After 2 h, N,N-Diisopropylethylamine (758 μΤ, 562 mg, 4.35 mmol) was
introduced to the mixture, followed by the aniline (221.0 mg, 2.37 mmol). The temperature was raised to 45 °C, and heating continued for 18 h. The solvent was evaporated, and then mixture was diluted with EtOAC (15 mL), washed with brine and dried over anhydrous Na2S04. The crude material obtained after removal of solvent was purified by column chromatography (10 – 20% EtOAc : Petroleum ether) to afford 20b (295 mg, 51% yield) as a yellow coloured solid. IR Omax(film): 3346, 3307,2853, 1724, 1678 cm“1; 1H NMR (400 MHz, DMSO-d6): δ 12.40 (br. s., 1H), 10.68 (s, 1H), 8.79 (d, = 3.2 Hz, 1H), 8.25 (d, = 8.6 Hz, 1H), 7.85 (d, = 7.8 Hz, 2H), 7.62 (d, = 1.7 Hz, 1H), 7.41 – 7.30 (m, 3H), 7.19 – 7.13 (m, 1H); 13C NMR (100 MHz, DMSO-d6): δ 182.5, 162.5, 140.0, 138.4, 137.4, 129.2, 128.5, 125.4, 124.8, 123.4, 122.9, 120.8, 113.0, 112.3; HRMS (ESI) Calculated for Ci6HnN2OCl[M+H]+: 299.0582, found 299.0580;
A solution of 20b (300 mg, 0.99 mmol) dissolved in MeOH (40 mL) was added to NaBH4 (45 mg, 1.23 mmol). The reaction was stirred for 4h and then added to saturated solution of Na2S04. The reaction mixture was further stirred for lh and then filtered through Celite.The filtrate obtained was concentrated in vacuo, and then mixture was diluted with EtOAc (15 mL), washed with brine and dried over anhydrous Na2S04. The crude material obtained after removal of solvent was forwarded for next step without further purification.In an N2 atmosphere, TMSC1 (1.272 mL, 9.9 mmol) in CH3CN (40 mL) was added to sodium iodide (1.488 mg, 9.9 mmol) and stirred for 2h. The reaction mixture was cooled to 0 °C and a solution of above crude alcohol (0.99 mmol) in CH3CN (10 mL) was then added drop wise over 30 min, followed by stirring for 3h. The reaction mixture was poured into NaOH (7g in 40 mL of water) and then extracted with ethyl acetate (15×2). The organic layer was washed with aq.Na2S203, dried over Na2S04 and concentrated in vacuo. The residue was chromatographed on silica gel (EtOAc:Pet ether) to afford 20 as a off white solid (two steps 38 % ); IR Umax(film): 3273, 3084,2953, 2857, 1629, 1562 cm“1; 1H NMR (400 MHz, DMSO-d6): δ 11.06 (br. s., 1H), 10.13 (br. s., 1H), 7.62 – 7.57 (m, 3H), 7.40 (s, 1H), 7.30 – 7.25 (m, 3H), 7.04 – 6.99 (m, 2H), 3.71 (s, 2H); 13C NMR (100 MHz, DMSO-d6): δ 170.1,
139.7, 136.9, 129.2, 126.5, 126.3, 125.5, 123.7, 120.6, 119.6, 119.3, 111.5, 109.4, 34.0; HRMS (ESI):Calculated for Ci6Hi4N2OCl[M+H]+: 285.0789, found 285.0786.
[060] Example 22:
2-(5-chloro-lH-indol-3-yl)-N-phenylacetamide(21):

21a 21b 21
To a solution of 5-Chloro indole 21a (300 mg, 1.98 mmol )in anhydrous THF(20 mL), Oxalyl chloride (186 ^L, 276 mg, 2.18 mmol) was added and the mixture stirred at room temperature. After 2 h, N,N-diisopropylethylamine (758 μΕ, 562 mg, 4.35 mmol) was introduced to the mixture, followed by the aniline (221.0 mg, 2.37 mmol). The tempera ture was raised to 45 °C, and heating continued for 18 h. The solvent was evaporated, and then mixture was diluted with EtOAC (15 mL), washed with brine and dried over anhydrous Na2S04. The crude material obtained after removal of solvent was purified by column chromatography (10 – 20% EtOAc : Petroleum ether) to afford (21b) (305 mg, 53% yield) as a yellow coloured solid. IR rjmax(film): 3346, 3307,2853, 1724, 1678 cm“1; 1H NMR (400 MHz, DMSO-d6): δ 12.40 (br. s., 1H), 10.68 (s, 1H), 8.79 (d, = 3.2 Hz, 1H), 8.25 (d, = 8.6 Hz, 1H), 7.85 (d, = 7.8 Hz, 2H), 7.62 (d, = 1.7 Hz, 1H), 7.42 – 7.30 (m, 3H), 7.20 – 7.14 (m, 1H); 13C NMR (100 MHz, DMSO-d6): δ 182.4, 162.4, 140.3, 138.4, 135.4, 129.2, 127.9, 124.8, 124.1, 120.8, 114.8, 112.0; HRMS (ESI) Calculated for Ci6HnN2OCl[M+H]+: 299.0582, found 299.0580; A solution of 21b (200 mg, 0.66 mmol) dissolved in MeOH (30 mL) was added to NaBH4 (30 mg, 0.82 mmol). The reaction was stirred for 4h and then added to saturated solution of Na2S04. The reaction mixture was further stirred for lh and then filtered through Celite. The filtrate obtained was concentrated in vacuo, and then mixture was diluted with EtOAc (15 mL), washed with brine and dried over anhydrous Na2S04. The crude material obtained after removal of solvent was forwarded for next step without further purification. In an N2 atmosphere, TMSC1 (848 mL, 6.6 mmol) in CH3CN (25 mL) was added to sodium iodide (992 mg, 6.6 mmol) and stirred for 2h. The reaction mixture was cooled to 0 °C and a solution of above crude alcohol(0.66 mmol) in CH3CN (5 mL) was then added dropwise over 30 min, followed by stirring for 3h. The reaction mixture was poured into NaOH (5g in 30 mL of water) and then extracted with ethyl acetate(15×2). The organic layer was washed with aq.Na2S203, dried over Na2S04 and concentrated in vacuo. The residue was chromatographed on silica gel (EtOAc:Pet ether) to afford 22 as a off white solid (two steps 42 % ); IR Umax(film): 3273, 3084,2955, 2857, 1629, 1562 cm“1; 1H NMR (400 MHz, DMSO-d6): δ 11.13 (br. s., 1H), 10.11 (s, 1H), 7.67 (s, 1H), 7.60 (d, = 7.8 Hz, 2H), 7.39 – 7.27 (m, 4H), 7.13 – 7.02 (m, 2H), 3.16 (s, 2H); 13C NMR (100 MHz, DMSO-d6): δ 169.9, 139.8, 135.0, 129.2, 128.9, 126.2, 123.6, 121.4, 119.6, 118.6, 113.4, 109.0, 34.0; HRMS (ESI) Calculated for Ci6H14N2OCl[M+H]+: 285.0789, found 285.0786.
[061] Example 23:
2-(l-benzyl-lH-indol-3-yl)-N-phenylacetamide (22):
Yield 79% ; 1H NMR (400 MHz, DMSO-d6): δ 7.67 (d, = 7.8 Hz, 1H), 7.54 (brs, 1H), 7.43 – 7.31 (m, 6H), 7.31 – 7.25 (m, 3H), 7.23 – 7.15 (m, 4H), 7.12 – 7.06 (m, 1H), 5.36 (s, 2H), 3.91 (s, 2H); 13C NMR (100 MHz, DMSO-d6): δ 169.7, 137.7, 137.2, 137.0, 128.9, 128.9, 127.9, 127.6, 126.9, 124.3, 122.7, 120.2, 119.9, 119.0, 110.2, 107.9, 77.4, 77.1, 76.8, 50.1, 34.5.
[062] Example 24:
Procedure B:

2-(lH-indol-3-yl)-N-phenylacetamidel(100 mg; 0.4 mmol) was dissolved in DCM:MeOH(50 mL; 5: 1), then a stream of 03 was passed through the solution until a blue color developed (10 min). The 03 stream was continued for 4 min. Then surplus O3 was removed by passing a stream of 02 through the solution for 10 min or until the blue colorcompletely vanished. Afterwards pyridine (0.1 mL;1.2mmol) was added to the cold (- 78 °C) mixture. The mixture was allowed to warm to room temperature (1 h) and then Et3N (0.35 mL; 2.4 mmol) were added. After stirring at room temperature overnight the reaction mass was concentrated under reduced pressure to dryness, diluted with EtOAc (30 mL), washed with H20 (5 mL), brine (5 mL), and dried over Na2S04. The crude material obtained after removal of solvent was purified by column chromatography (silica gel 230-400 mesh, MeOH – DCM) to give desired quinolone carboxamide as colorless solid.
[063] Example 25:
4-oxo-N-phenyl-l,4-dihydroquinoline-3-carboxamide (23):
Yield: 65 mg; 62%; XH NMR (200MHz ,DMSO-d6) δ = 12.97 (brs, 1 H), 12.49 (s, 1 H), 8.89 (s, 1 H), 8.33 (d, J = 8.2 Hz, 1 H), 7.91 – 7.69 (m, 4 H), 7.62 – 7.50 (m, 1 H), 7.37 (t, J = 7.8 Hz, 2 H), 7.18 – 7.01 (m, 1 H); MS: 287 (M+Na)+.
[064] Example 26:
2,4-di-tert-butyl-5-(4-oxo-l,4-dihydroquinoline-3-carboxamido)phenyl methyl carbonate (24):
Yield: 35 mg; 34%; 1H NMR (400MHz ,DMSO-d6) δ = 12.96 (brs, 1 H), 12.08 (s, 1 H), 8.94 – 8.82 (m, 1 H), 8.44 – 8.28 (m, 1 H), 7.86 – 7.79 (m, 1 H), 7.78 – 7.73 (m, 1 H), 7.59 (s, 1 H), 7.53 (t, J = 7.5 Hz, 1 H), 7.39 (s, 1 H), 3.86 (s, 3 H), 1.46 (s, 9 H), 1.32 (s, 9 H).
[065] Example 27:
(S)-4-oxo-N-(l-phenylethyl)-l,4-dihydroquinoline-3-carboxamide (25):
Yield: 56 mg; 53%; 1H NMR (500MHz ,DMSO-d6) δ = 12.75 (brs, 1H), 10.54 (d, J = 7.6 Hz, 1H), 8.73 (brs, 1H), 8.28 (d, J = 7.9 Hz, 1H), 7.78 (d, J = 7.9 Hz, 1H), 7.73 -7.68 (m, 1 H), 7.50 (t, J = 7.5 Hz, 1 H), 7.42 – 7.34 (m, 4 H), 7.29 – 7.23 (m, 1 H), 5.18 (t, J = 7.2 Hz, 1 H), 1.50 (d, J = 6.7 Hz, 3 H).
[066] Example 28:
Synthesis of ivacaftor (26):

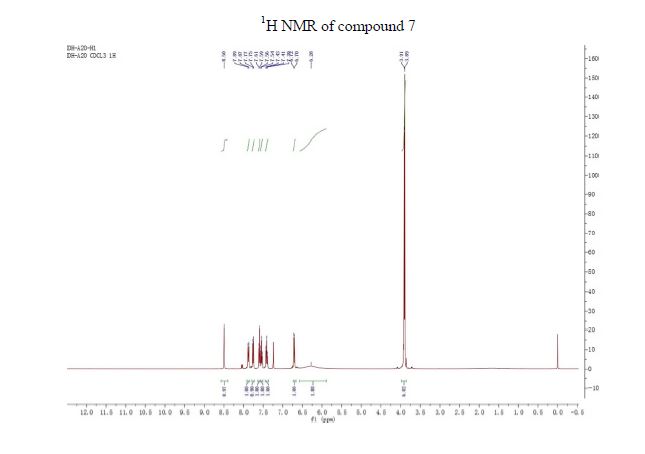
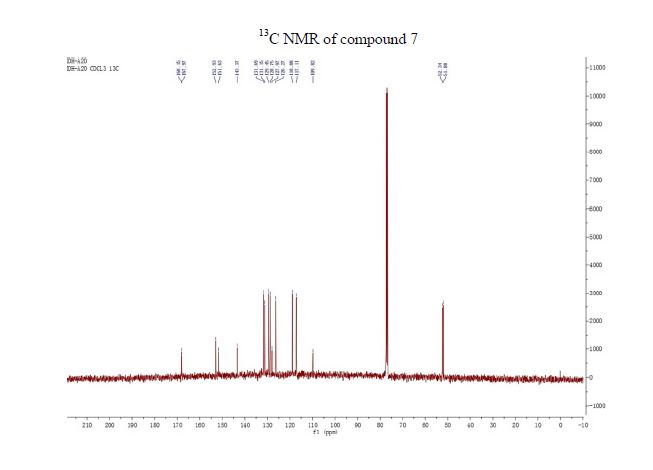
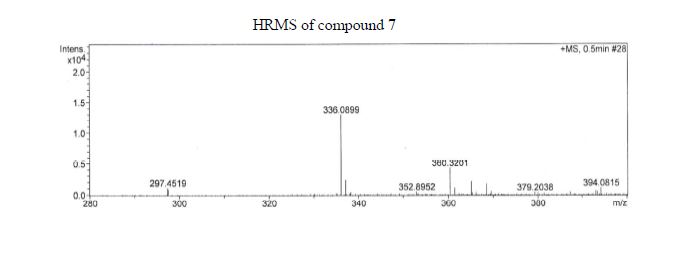
To a solution of 2,4-di-tert-butyl-5-(4-oxo-l,4-dihydroquinoline-3-carboxamido)phenyl methyl carbonate 5 (30 mg, 0.06mmol) in MeOH (2 mL) was added NaOH (5.3 mg, 0.13mmol) dissolved in H20 (2 mL), and the reaction mixture was stirred at room temperature for 5h. Reaction mass was evaporated to one third of its volume (temperature not exceeding 40°C) and acidified with aq.2N HC1 to pH 2-3. The resulting precipitate was collected by suction filtration give desired compound 7 (19 mg, 76%) as off white solid H NMR (400MHz ,DMSO-d6) δ = 12.88 (d, J = 6.6 Hz, 1 H), 11.81 (s, 1 H), 9.20 (s, 1 H), 8.86 (d, J = 6.6 Hz, 1 H), 8.32 (d, J = 7.8 Hz, 1 H), 7.88 – 7.65 (m, 2 H), 7.51 (t, J = 7.5 Hz, 1 H), 7.16 (s, 1 H), 7.10 (s, 1 H), 1.38 (s,9H), 1.36 (s, 9H).
[067] Example 29:
N-(4-fluorophenyl)-4-oxo-l,4-dihydroquinoline-3-carboxamide (27):
Yield 56% ; 1H NMR (400 MHz, DMSO-d6): δ 12.96 (br. s., 1H), 12.50 (s, 1H), 8.88 (s, 1H), 8.33 (d, = 7.3 Hz, 1H), 7.86 – 7.72 (m, 4H), 7.54 (t, = 7.3 Hz, 1H), 7.20 (t, = 8.8 Hz, 2H); 13C NMR (400 MHz, DMSO-d6): δ 176.8, 163.2, 159.7, 157.3, 144.6, 139.6, 135.7, 133.5, 126.4, 125.9, 125.8, 121.8, 119.7, 116.1, 115.9, 110.9.
[068] Example 30:
N-(4-chlorophenyl)-4-oxo-l,4-dihydroquinoline-3-carboxamide (28):
Yield 51% ; 1H NMR (400 MHz, DMSO-d6): δ 13.00 (brs., 1H), 12.59 (br. s., 1H), 8.89 (s, 1H), 8.34 (d, = 7.6 Hz, 1H), 7.83 – 7.76 (m, 4H), 7.56 (s, 1H), 7.42 (d, = 7.9 Hz, 2H); 13C NMR (400 MHz, DMSO-d6): δ 176.8, 163.4, 144.7, 139.6, 138.2, 133.5, 129.4, 127.4, 126.4, 125.9, 125.8, 121.6, 119.7, 110.8.
[069] Example 31:
4-oxo-N-(p-tolyl)-l,4-dihydroquinoline-3-carboxamide (29):
Yield 57% ; 1H NMR (400 MHz, DMSO-d6): δ 12.94 (brs., 1H), 12.40 (s, 1H), 8.88 (s, 1H), 8.33 (d, = 7.8Hz, 1H), 7.82 – 7.80 (m, 1H), 7.76 – 7.7 (m, 1H), 7.63 (d, = 8.3 Hz, 2H), 7.53 (t, = 7.3 Hz, 1H), 7.17 (d, = 8.1 Hz, 2H), 2.29 (s, 3H); 13C NMR (100 MHz, DMSO-de): δ 176.8, 163.1, 144.5, 139.6, 136.8, 133.4, 132.8, 129.9, 126.4, 125.9, 125.7, 120.0, 119.6, 111.1, 20.9; HRMS (ESI):Calculated for Ci7H1502N2[M+H]+: 279.1128, found 279.1127.
[070] Example 32:
N-(4-ethylphenyl)-4-oxo-l,4-dihydroquinoline-3-carboxamide (30):
Yield 51% ; 1H NMR (400 MHz, DMSO-d6): δ 12.95 (br. s., 1H), 12.40 (d, = 7.8 Hz, 1H), 8.87 (d, = 6.1 Hz, 1H), 8.33 (d, = 8.1 Hz, 1H), 7.81 – 7.76 (m, 2H), 7.66 – 7.62 (m, = 8.3 Hz, 2H), 7.53 (t, 7 = 7.5 Hz, 1H), 7.22 – 7.17 (m, = 8.3 Hz, 2H), 2.58 (q, = 7.6 Hz, 2H), 1.18 (t, = 7.6 Hz, 3H); 13C NMR (400 MHz, DMSO-d6): δ 181.5, 167.8, 149.3, 144.3, 144.0, 141.7, 138.2, 133.4, 131.1, 130.7, 130.5, 124.8, 124.4, 115.9, 32.8, 20.9.
[071] Example 33:
4-Oxo-N-(4-propylphenyl)-l,4-dihydroquinoline-3-carboxamide (31):
Yield 51%; 1H NMR (500 MHz, DMSO-d6): δ12.93 (brs, 1H), 12.40 (s, 1H), 8.87 (s, 1H), 8.36 – 8.29 (m, 1H), 7.86 – 7.78 (m, 1H), 7.75 (d, J= 7.9 Hz, 1H), 7.68 – 7.61 (m, J= 8.2 Hz, 2H), 7.54 (t, J= 7.6 Hz, 1H), 7.22 – 7.14 (m, J= 8.2 Hz, 2H), 2.55 – 2.51 (m, 2H), 1.64 – 1.53 (m, 2H), 0.90 (t, J= 7.3 Hz, 3H); 13C NMR (500 MHz, DMSO-d6): 176.8, 163.1, 144.5, 139.6, 137.6, 137.0, 133.5, 129.3, 126.4, 125.9, 125.7, 120.0, 119.7, 111.1, 37.2, 24.6, 14.1.
[072] Example 34:
N-(4-isopropylphenyl)-4-oxo-l,4-dihydroquinoline-3-carboxamide (32):
Yield 46% ; 1H NMR (500 MHz, DMSO-d6): δ 12.93 (br. s., 1H), 12.40 (br. s., 1H), 8.89 – 8.86 (m, 1H), 8.33(d, = 7.6 Hz, 1H), 7.81 – 7.50 (m, 5H), 7.25 – 7.21 (m, 2H), 2.90-2.83 (m, 1H), 1.22-1. l l(m, 6H); 13C NMR (100 MHz, DMSO-d6): δ 176.8, 163.1, 144.5, 143.9, 139.6, 137.1, 133.4, 127.2, 126.4, 125.9, 125.7, 120.1, 119.6, 111.1, 33.4, 24.4.
[073] Example 35:
4-oxo-N-(4-(trifluoromethoxy)phenyl)-l,4-dihydroquinoline-3-carboxamide(33):
Yield 57% ; 1H NMR (400 MHz, DMSO-d6): δ 12.98 (br. s., 1H), 12.63 (s, 1H), 8.88 (d, = 4.9 Hz, 1H), 8.32 (d, = 7.8 Hz, 1H), 7.89 – 7.83 (m, = 8.8 Hz, 2H), 7.79 (d, = 7.6 Hz, 1H), 7.77 – 7.73 (m, 1H), 7.53 (t, J = 7.5 Hz, 1H), 7.40 – 7.34 (m, = 8.6 Hz, 2H); 13C NMR (100 MHz, DMSO-d6): δ 176.8, 163.5, 144.7, 144.0, 139.5, 138.5, 133.5, 126.3, 125.9, 125.8, 122.3, 121.4, 119.7, 110.7.
[074] Example 36:
N-(2-chloro-5-methoxyphenyl)-4-oxo-l,4-dihydroquinoline-3-carboxamide(34):
Yield 54% ; XH NMR (400 MHz, DMSO-d6): δ 12.98 (br. s., 1H), 12.49 (s, 1H), 8.88 (s, 1H), 8.33 (d, = 7.8 Hz, 1H), 7.83 – 7.75 (m, 1H), 7.56-7.48 (m, 3H), 7.27 – 7.21 (m, 1H), 6.67 (d, = 7.8 Hz, 1H), 3.77 (s, 3H); 13C NMR (400 MHz, DMSO-d6): δ 176.8, 163.4, 160.2, 144.7, 140.4, 139.6, 133.5, 130.3, 126.4, 125.9, 125.8, 119.7, 112.3, 111.0, 109.5, 105.7, 55.5.
[075] Example 37:
N-(2-ethylphenyl)-4-oxo-l,4-dihydroquinoline-3-carboxamide(35):
Yield 58% ; 1H NMR (400 MHz, DMSO-d6): δ 12.94 (br. s., 1H), 12.37 (s, 1H), 8.90 (s, 1H), 8.36 (dd, = 8.1, 1.4 Hz, 2H), 8.32 (dd, = 8.1, 1.4 Hz, 2H), 7.82 – 7.74 (m, 1H), 7.53- 7.19 (m, 3H), 7.15 – 7.06(m, 1H), 2.79 (q, = 7.3 Hz, 2H), 1.26 (t, = 7.5 Hz, 3H); 293 (M+H)+.
[076] Example 38:
N-(2-bromophenyl)-4-oxo-l,4-dihydroquinoline-3-carboxamide(36):
Yield 47% ; 1H NMR (200 MHz, DMSO-d6): δ 12.98 (br. s., 1H), 12.69 (s, 1H), 8.90 (d, = 5.9 Hz, 1H), 8.54 (dd, 7 = 1.4, 8.3 Hz, 1H), 8.34 (d, = 7.6 Hz, 1H), 7.86 – 7.67 (m, 3H), 7.57 – 7.49 (m, 1H), 7.40 (t, = 7.2 Hz, 1H), 7.10 – 7.05 (m, 1H); 13C NMR (100 MHz, DMSO-de): δ 176.7, 163.7, 145.0, 139.5, 137.7, 133.5, 133.1, 128.6, 126.4, 126.0, 125.8, 125.3, 122.9, 119.7, 113.4, 110.8.
[077] Example 39:
N-benzyl-4-oxo-l,4-dihydroquinoline-3-carboxamide(37):
Yield 58% ; 1H NMR (400 MHz, CD3OD-d6): δ 8.82 (s, 1 H), 8.35 (d, = 8.1 Hz, 1 H), 7.79 – 7.77 (m, 1 H), 7.65 (d, = 8.3 Hz, 1 H), 7.52 (t, = 7.6 Hz, 1 H), 7.42 – 7.34 (m, 4 H), 7.31 – 7.26 (m, 1 H), 4.67 (s, 2 H); 13C NMR (400 MHz, DMSO-d6): δ 176.6, 165.0, 144.2, 140.0, 139.5, 133.2, 128.9, 128.7, 127.8, 127.3, 126.6, 125.9, 125.4, 119.5, 111.2, 42.6.
[078] ] Example 40:
N-(4-methoxybenzyl)-4-oxo-l,4-dihydroquinoline-3-carboxamide(38):
Yield 56% ; 1H NMR (400 MHz, DMSO-d6): δ 12.73 (br. s., 1H), 10.35 (t, = 5.3 Hz, 1H), 8.78 (d, = 6.1 Hz, 1H), 8.24 (d, = 8.1 Hz, 1H), 7.76 (d, = 7.1 Hz, 1H), 7.73 -7.68 (m, 1H), 7.48 (t, = 7.5 Hz, 1H), 7.28 (d, = 8.3 Hz, 2H), 6.91 (d, = 8.1 Hz, 2H), 4.49 (d, = 5.6 Hz, 2H), 3.74 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 176.6, 164.8, 158.8, 144.1, 139.5, 133.1, 131.9, 129.2, 126.6, 125.8, 125.4, 119.5, 114.3, 111.3, 55.5, 42.0.
[079] Example 41:
N,N-dibenzyl-4-oxo-l,4-dihydroquinoline-3-carboxamide(39):
Yield 43% ; 1H NMR (400 MHz, DMSO-d6): δ 12.21 (br. s., 1H), 8.27 (d, = 4.9 Hz, 1H), 8.21 (d, = 7.6 Hz, 1H), 7.49 – 7.41 (m, 2H), 7.41 – 7.35 (m, 3H), 7.33 – 7.20 (m, 5H), 7.20 – 7.11 (m, 7 = 7.1 Hz, 2H), 4.59 (br. s., 2H), 4.42 (s, 2H).
[080] Example 42:
4-oxo-N-propyl-l,4-dihydroquinoline-3-carboxamide(40):
Yield 47% ;1H NMR (400 MHz, DMSO-d6): δ 12.7 (br.s., 1H)10.05 (t, = 5.5 Hz, 1H), 8.74 (s, 1H), 8.26 (d, = 8.1 Hz, 1H), 7.83 – 7.66 (m, 2H), 7.52 – 7.44 (m, 1H), 3.33 – 3.22 (m, 2H), 1.61 – 1.49 (m, 2H), 0.93 (t, = 7.5 Hz, 3H); 13C NMR (100 MHz, DMSO-de): δ 176.6, 164.8, 143.9, 139.5, 133.1, 126.6, 125.9, 125.3, 119.4, 111.4, 39.3, 23.1, 12.0
[081] Example 43:
N-hexyl-4-oxo-l,4-dihydroquinoline-3-carboxamide(41):
Yield 51% ;1H NMR (400 MHz, DMSO-d6): δ 12.68 (m, 1H), 10.02 (t, = 5.5 Hz, 1H), 8.73 (d, = 6.1 Hz, 1H), 8.27 – 8.25 (m, 1H), 7.77 – 7.67 (m, 2H), 7.47 (t, = 7.5 Hz, 1H), 3.33 – 3.29 (m, 2H), 1.56 – 1.45 (m, 2H), 1.34 – 1.25 (m, 6H), 0.88 – 0.82 (m, 3H); 13C NMR (100 MHz, DMSO-d6): δ 176.6, 164.8, 143.9, 139.5, 133.1, 126.6, 125.9, 125.3, 119.4, 111.4, 38.7, 31.5, 29.8, 26.7, 22.5, 14.4.
[082] Example 44:
Methyl (4-oxo-l,4-dihydroquinoline-3-carbonyl)-L-alaninate(42):
Yield 38% ; 1H NMR (400 MHz, CD3OD): δ 8.74 (s, 1H), 8.47 – 8.29 (m, 1H), 7.86 -7.76 (m, 1H), 7.64 (d, = 8.3 Hz, 1H), 7.58 – 7.44 (m, 1H), 4.69 (d, = 7.3 Hz, 1H), 3.79 (s, 3H), 1.55 (d, = 7.3 Hz, 3H); 13C NMR (100 MHz, CD3OD): δ 177.3, 173.3, 165.5, 143.6, 139.2, 132.9, 126.3, 125.4, 125.2, 118.5, 110.3, 51.5, 47.0, 17.0.
[083] Example 45:
7-chloro-4-oxo-N-phenyl-l,4-dihydroquinoline-3-carboxamide(43):
Yield 48% ; IR Omax(film): 2920, 2868, 1661, 1601 cm” 1; 1H NMR (400 MHz, DMSO-de): δ 12.91 (br. s., 1H), 12.30 (s, 1H), 8.90 (s, 1H), 8.29 (d, = 8.8 Hz, 1H), 7.80 -7.67 (m, 3H), 7.58 – 7.51 (m, 1H), 7.36 (t, = 7.7 Hz, 2H), 7.09 (t, = 7.3 Hz, 1H); 13C NMR (100 MHz, DMSO-d6): δ 176.3, 162.9, 145.4, 140.3, 139.2, 138.0, 129.5, 128.2, 126.1, 125.1, 123.9, 120.1, 118.8, 111.6.
[084] Example 46:
6-chloro-4-oxo-N-phenyl-l,4-dihydroquinoline-3-carboxamide(44):
Yield 52% ; 1H NMR (400 MHz, DMSO-d6): δ 13.05 (brs, 1H), 12.27 (s, 1H), 8.88 (s, 1H), 8.21 (d, = 2.2 Hz, 1H), 7.86 – 7.67 (m, 4H), 7.36 (t, = 7.8 Hz, 2H), 7.16 – 7.04 (m, 1H); 13C NMR (100 MHz, DMSO-d6): δ 175.6, 162.9, 144.9, 139.1, 138.2, 133.5, 130.4, 129.5, 127.5, 124.9, 123.9, 122.0, 120.1, 111.4.
[085] Example 47:
l-benzyl-4-oxo-N-phenyl-l,4-dihydroquinoline-3-carboxamide(45)
Yield 55% ; 1H NMR (400 MHz, DMSO-d6): δ 12.30 (s, 1H), 9.05 (s, 1H), 8.60 (dd, = 1.7, 8.1 Hz, 1H), 7.82 (d, = 7.8 Hz, 2H), 7.69 – 7.62 (m, 1H), 7.55 – 7.45 (m, 2H), 7.43 – 7.34 (m, 5H), 7.24 – 7.18 (m, 2H), 7.17 – 7.10 (m, 1H), 5.53 (s, 2H); 13C NMR (100 MHz, DMSO-d6): δ 176.9, 162.9, 148.7, 139.3, 138.7, 134.1, 133.1, 129.4, 128.9, 128.7, 128.0, 127.4, 126.2, 125.5, 123.9, 120.5, 116.9, 112.3, 57.9; HRMS (ESI): Calculated for C23H1802N2Na [M+Na]+: 377.1260, found 377.1259; MS: 355 (M+H)+.
[086] Advantages of invention:
1. Cost-effective process for synthesis.
2. Carried out at environmentally benign conditions.
3. Short synthetic route.
4. Useful for making several related compounds of medicinal

DR SRINIVASA REDDY recieving NASI – Reliance Industries Platinum Jubilee Award (2015) for Application Oriented Innovations in Physical Sciences.

MYSELF WITH HIM

From left to right: Dr. D. Srinivasa Reddy, Shri Y. S. Chowdary, Dr. Harsh Vardhan, Dr. Girish Sahni
- Dr D. Srinivasa Reddy receiving the prestigious “SHANTI SWARUP BHATNAGAR” award at the occasion of the 75th Foundation day of CSIR.
.JPG)
Shanti Swarup Bhatnagar awardees with the honorable Prime Minister of India

NCL PUNE

DSR Group
//////////WO-2016181414, WO 2016181414, IVACAFTOR, new patent, COUNCIL OF SCIENTIFIC & INDUSTRIAL RESEARCH, Anusandhan Bhawan, Rafi Marg New Delhi, INDIA, CSIR, Dr. D. Srinivasa Reddy














 Know what kind of patent you have. In the US, 2 main types of patents are given: utility patents or design patents. Utility patents cover the function of an invention and design patents protect the way an invention looks. Utility patents generally last 20 years, while design patents last 14 years or 15 years for those filed on or after May 13, 2015. There are also 20 year long plant patents for inventors who asexually reproduce a newly discovered or invented variety of plant.[2]
Know what kind of patent you have. In the US, 2 main types of patents are given: utility patents or design patents. Utility patents cover the function of an invention and design patents protect the way an invention looks. Utility patents generally last 20 years, while design patents last 14 years or 15 years for those filed on or after May 13, 2015. There are also 20 year long plant patents for inventors who asexually reproduce a newly discovered or invented variety of plant.[2] Find out if you qualify. Patent extensions are sometimes granted if there are government regulatory delays or if newer laws extend the length of a patent. Sometimes, with very strong justification, you can try to get Congress to pass a bill to extend your patent. If you fall into one of these categories, then you may be able to get your patent extended.
Find out if you qualify. Patent extensions are sometimes granted if there are government regulatory delays or if newer laws extend the length of a patent. Sometimes, with very strong justification, you can try to get Congress to pass a bill to extend your patent. If you fall into one of these categories, then you may be able to get your patent extended. Be aware that extensions may not be an option. For most inventions, the given term for your patent will stand. Recognize that you may not be able to extend your patent for this particular invention. Focus, instead, on developing a new invention that you can then get a new patent for.
Be aware that extensions may not be an option. For most inventions, the given term for your patent will stand. Recognize that you may not be able to extend your patent for this particular invention. Focus, instead, on developing a new invention that you can then get a new patent for.


 Get a term adjustment. If you filed your patent after May 29, 2000 and your patent was delayed because the USPTO was taking longer than normal to process the paperwork, you may be eligible to file for an extension. The extension will cover the time lost from your patent term for the delay. The length of the extension you are approved for will depend on the delay time frame, but will not be longer than 5 years.
Get a term adjustment. If you filed your patent after May 29, 2000 and your patent was delayed because the USPTO was taking longer than normal to process the paperwork, you may be eligible to file for an extension. The extension will cover the time lost from your patent term for the delay. The length of the extension you are approved for will depend on the delay time frame, but will not be longer than 5 years. Increase your original patent term. If you were initially granted less time than later legislation allows, you may be eligible to request an extension on your patent for the newer patent term. Under the Uruguay Rounds Agreements Act, utility patents granted before June of 1995 may be given an extension to 20 years instead of the original 17 years. This does not apply to design patents.
Increase your original patent term. If you were initially granted less time than later legislation allows, you may be eligible to request an extension on your patent for the newer patent term. Under the Uruguay Rounds Agreements Act, utility patents granted before June of 1995 may be given an extension to 20 years instead of the original 17 years. This does not apply to design patents. Get an extension under the Hatch-Waxman Act. A patent term restoration under the Hatch-Waxman Act is sometimes given to those who qualify.This applies to those whose products or processes, such as medications, medical devices, food and color additives, require testing and approval by the Food and Drug Administration’s (FDA) before they can be marketed. The period of time that you were unable to sell your product because you were awaiting FDA approval may be restored as an extension to the original patent. [3]
Get an extension under the Hatch-Waxman Act. A patent term restoration under the Hatch-Waxman Act is sometimes given to those who qualify.This applies to those whose products or processes, such as medications, medical devices, food and color additives, require testing and approval by the Food and Drug Administration’s (FDA) before they can be marketed. The period of time that you were unable to sell your product because you were awaiting FDA approval may be restored as an extension to the original patent. [3]
 Wait to hear back from the USPTO. It can take up to several months for the USPTO to process your request. As with any government process, patience is best. If you are eligible and have a good reason for an extension, then there’s a chance you could be approved so waiting is worth it.
Wait to hear back from the USPTO. It can take up to several months for the USPTO to process your request. As with any government process, patience is best. If you are eligible and have a good reason for an extension, then there’s a chance you could be approved so waiting is worth it.
 Meet with an intellectual property lawyer. It could be very beneficial to consult with an attorney to review your options, especially if your request is denied. Your lawyer may be able to offer suggestions and ways to supplement your application. Filing for a patent extension can be complex and your lawyer can ensure that it is done correctly.
Meet with an intellectual property lawyer. It could be very beneficial to consult with an attorney to review your options, especially if your request is denied. Your lawyer may be able to offer suggestions and ways to supplement your application. Filing for a patent extension can be complex and your lawyer can ensure that it is done correctly.







 Find a representative. Do some research on representatives in your area or someone you think would want to sponsor you in extending your patent. You will need to convince him or her that there is a very important reason you must extend your patent. It is best if they have a record of supporting the type of invention you have or are connected in some way to that field.
Find a representative. Do some research on representatives in your area or someone you think would want to sponsor you in extending your patent. You will need to convince him or her that there is a very important reason you must extend your patent. It is best if they have a record of supporting the type of invention you have or are connected in some way to that field. Draft a bill. It’s a good idea to do as much of the legwork as possible before approaching your representative. Your bill should be written in legal language and go over the reasons your patent should be granted an extension. You can check existing bills on the Congress website to get an idea what a bill looks like. It might be helpful to consult with a patent attorney when you’re writing this as legalese can be difficult to master.[5]
Draft a bill. It’s a good idea to do as much of the legwork as possible before approaching your representative. Your bill should be written in legal language and go over the reasons your patent should be granted an extension. You can check existing bills on the Congress website to get an idea what a bill looks like. It might be helpful to consult with a patent attorney when you’re writing this as legalese can be difficult to master.[5] Submit your bill to your potential sponsor. Contact your representative by phone or email. Many have websites where you can fill out a form to submit your case. Be sure to ask what the process is like and when you can expect to hear back.
Submit your bill to your potential sponsor. Contact your representative by phone or email. Many have websites where you can fill out a form to submit your case. Be sure to ask what the process is like and when you can expect to hear back. Get a lobbyist to represent you. If your patent is important to certain groups or not extending it could cause harm, then look for someone with contacts to represent you. Lobbyist groups can put pressure on Congress to extend your patent. In order to do this, you need to have a good cause with far-reaching effects if your patent is not extended.
Get a lobbyist to represent you. If your patent is important to certain groups or not extending it could cause harm, then look for someone with contacts to represent you. Lobbyist groups can put pressure on Congress to extend your patent. In order to do this, you need to have a good cause with far-reaching effects if your patent is not extended. Be patient. The legislative process can take time. It must go through multiple committees before the house will vote on it. After that, it must be signed in. The length of time this will take varies and is something you should discuss with your sponsor.
Be patient. The legislative process can take time. It must go through multiple committees before the house will vote on it. After that, it must be signed in. The length of time this will take varies and is something you should discuss with your sponsor.













 The presentation will load below
The presentation will load below