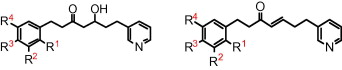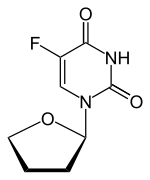
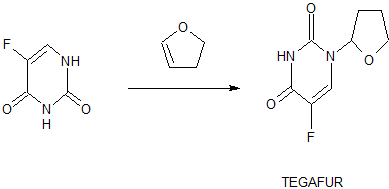
Tegafur
CAS 17902-23-7
2,4(1H,3H)-Pyrimidinedione, 5-fluoro-1-(tetrahydro-2-furanyl)-
- Molecular Weight,200.17, MF C8 H9 F N2 O3
-
Miyashita, Osamu; Chemical & Pharmaceutical Bulletin 1981, 29(11), PG 3181-90
Uracil, 5-fluoro-1-(tetrahydro-2-furyl)-
Utefos
Venoterpine
WY1559000
YR0450000
5-fluoro-1-tetrahydrofuran-2-ylpyrimidine-2,4(1H,3H)-dione
Carzonal
N1-(2′-Furanidyl)-5-fluorouracil
- Synonyms:Ftorafur
- ATC:L01BC03
- EINECS:241-846-2
- LD50:800 mg/kg (M, i.v.); 775 mg/kg (M, p.o.);
685 mg/kg (R, i.v.); 930 mg/kg (R, p.o.);
34 mg/kg (dog, p.o.)
Derivatives, monosodium salt
- Formula:C8H8FN2NaO3
- MW:222.15 g/mol
- CAS-RN:28721-46-2
Tegafur (INN, BAN, USAN) is a chemotherapeutic prodrug of 5-flourouracil (5-FU) used in the treatment of cancers. It is a component of the combination drug tegafur/uracil. When metabolised, it becomes 5-FU.[1]
Medical uses
As a prodrug to 5-FU it is used in the treatment of the following cancers:[2]
It is often given in combination with drugs that alter its bioavailability and toxicity such as gimeracil, oteracil or uracil.[2] These agents achieve this by inhibiting the enzyme dihydropyrimidine dehydrogenase (uracil/gimeracil) or orotate phosphoribosyltransferase (oteracil).[2]

Adverse effects
The major side effects of tegafur are similar to fluorouracil and include myelosuppression, central neurotoxicity and gastrointestinal toxicity (especially diarrhoea).[2] Gastrointestinal toxicity is the dose-limiting side effect of tegafur.[2] Central neurotoxicity is more common with tegafur than with fluorouracil.[2]

Pharmacogenetics
The dihydropyrimidine dehydrogenase (DPD) enzyme is responsible for the detoxifying metabolism of fluoropyrimidines, a class of drugs that includes 5-fluorouracil, capecitabine, and tegafur.[4] Genetic variations within the DPD gene (DPYD) can lead to reduced or absent DPD activity, and individuals who are heterozygous or homozygous for these variations may have partial or complete DPD deficiency; an estimated 0.2% of individuals have complete DPD deficiency.[4][5] Those with partial or complete DPD deficiency have a significantly increased risk of severe or even fatal drug toxicities when treated with fluoropyrimidines; examples of toxicities include myelosuppression, neurotoxicity and hand-foot syndrome.[4][5]
Mechanism of action
It is a prodrug to 5-FU, which is a thymidylate synthase inhibitor.[2]
Pharmacokinetics
It is metabolised to 5-FU by CYP2A6.[6][7]
Interactive pathway map
Click on genes, proteins and metabolites below to link to respective articles.[§ 1]
The interactive pathway map can be edited at WikiPathways: “FluoropyrimidineActivity_WP1601”.



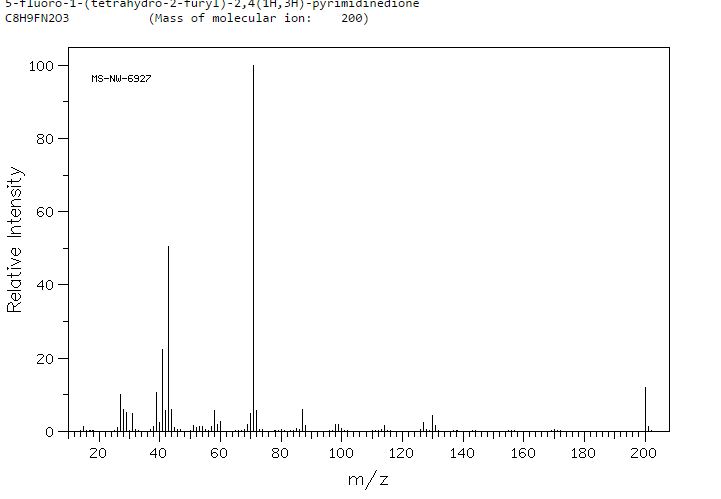
MASS SPECTRUM
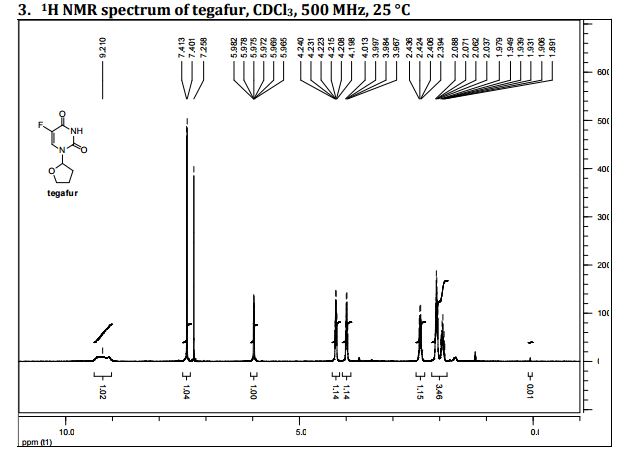
1H NMR
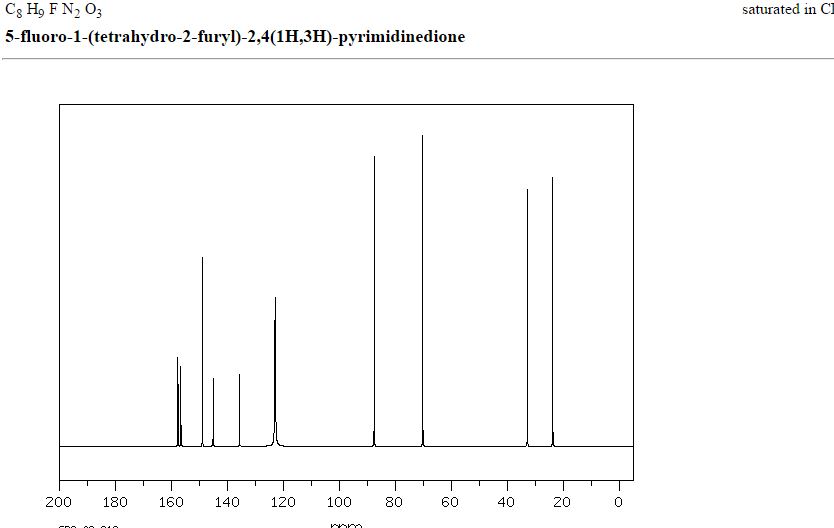
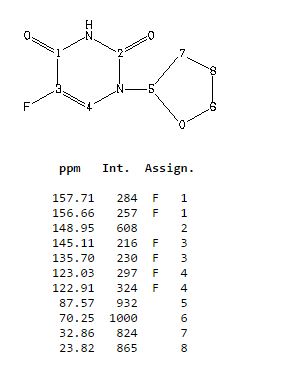
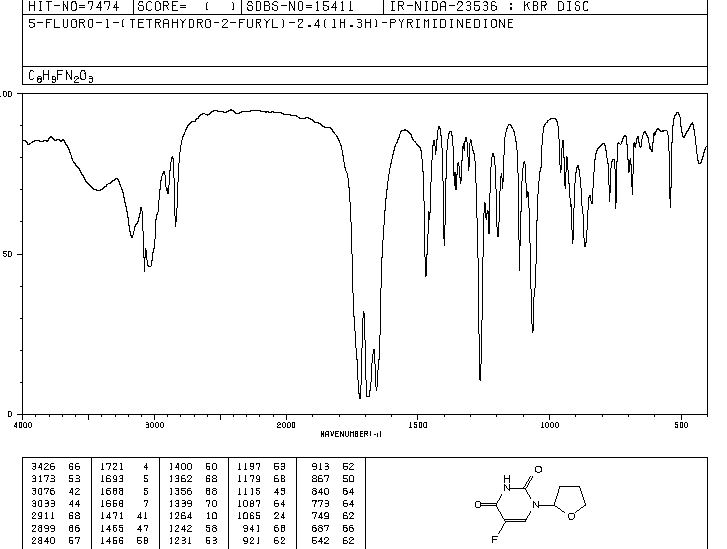
IR
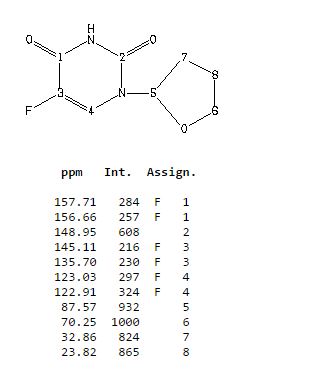
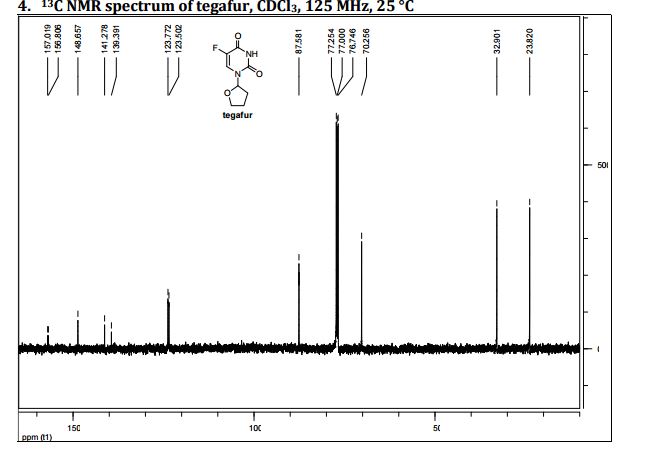
13C NMR
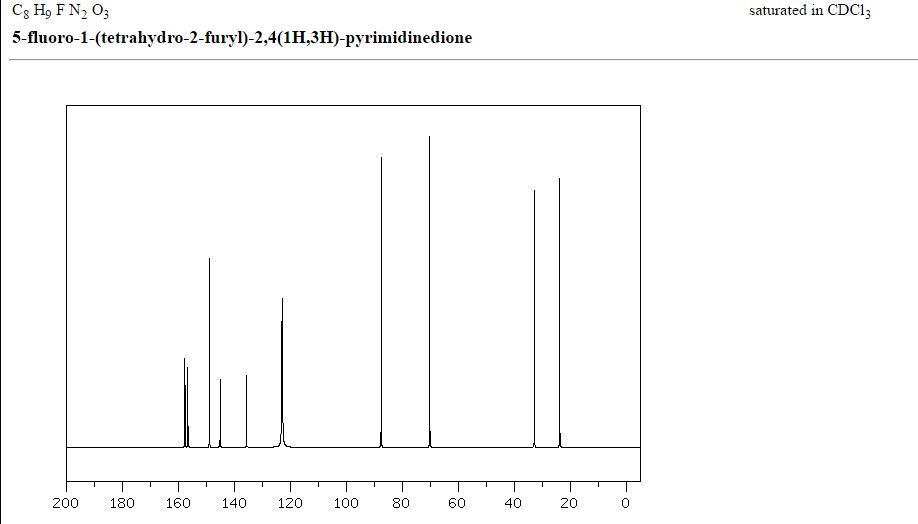
RAMAN
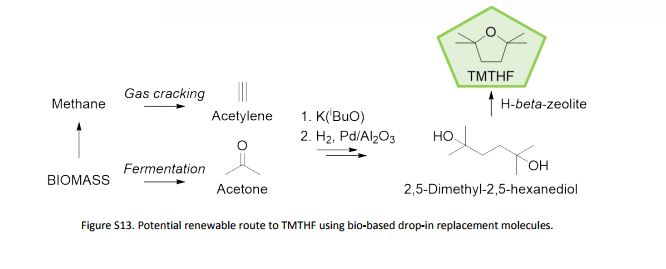
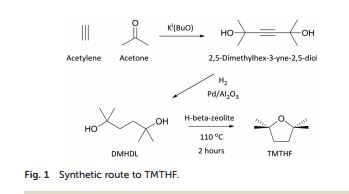
Synthesis

Substances Referenced in Synthesis Path
| CAS-RN |
Formula |
Chemical Name |
CAS Index Name |
| 58138-78-6 |
C10H19FN2O2Si2 |
1,3-bis(trimethylsilyl)fluorouracil |
2,4(1H,3H)-Pyrimidinedione, 5-fluoro-1,3-bis(trimethylsilyl)- |
| 13369-70-5 |
C4H7ClO |
2-chlorotetrahydrofuran |
Furan, 2-chlorotetrahydro- |
| 1191-99-7 |
C4H6O |
2,3-dihydrofuran |
Furan, 2,3-dihydro- |
| 51-21-8 |
C4H3FN2O2 |
5-fluorouracil |
2,4(1H,3H)-Pyrimidinedione, 5-fluoro- |



SYN1
CN 106397416
SYN 2

Advanced Synthesis & Catalysis, 356(16), 3325-3330; 2014
PATENTS
CN 106397416
CN 104513230
CN 103159746
PATENT
CN 102285972
tegafur is a derivative of 5-fluorouracil, and in 1967, Hiller of the former Soviet Union synthesized tegafur (SA Hiller, RA Zhuk, M. Yu. Lidak, et al. Substituted Uracil [ P, British Patent, 1168391 (1969)). In 1974, it was listed in Japan. China was successfully developed by Shandong Jinan Pharmaceutical Factory in 1979. Its present origin is Shanghai and Shandong provinces and cities. The anti-cancer effect of tegafur is similar to that of 5-fluorouracil and is activated in vivo by 5-fluorouracil through liver activation. Unlike 5-fluorouracil, tegafur is fat-soluble, has good oral absorption, maintains high concentrations in the blood for a long time and easily passes through the blood-brain barrier. Clinical and animal experiments show that tegafur on gastrointestinal cancer, breast cancer is better, the role of rectal cancer than 5-fluorouracil good, less toxic than 5-fluorouracil. Teflon has a chemotherapy index of 2-fold for 5-fluorouracil and only 1 / 4-1 / 7 of toxicity. So the addition of fluoride is widely used in cancer patients with chemotherapy.
[0003] The first synthesis of tegafur is Hiller ([SA Hiller, RA Zhuk, Μ. Yu. Lidak, et al. Substituted Uracil [P], British Patent, 1168391 (1969)]. 5-fluorouracil or 2,4-bis (trimethylsilyl) -5-fluorouracil (Me3Si-Fu, 1) and 2-chlorotetrahydrofuran (Thf-Cl), and it is reported that this synthesis must be carried out at low temperature (- 20 to -40 ° C), because Thf-Cl is unstable, and excess Thf-Cl results in a decomposition reaction, thereby reducing the yield of Thf-Fu.
[0004] Earl and Townsend also prepared 1_ (tetrahydro-2-furyl) uracil using Thf-Cl and 2,4-bis (trimethylsilyl) uracil, and then using trifluoromethyl fluorite to product Fluorination. Mitsugi Yasurnoto reacts with the Friedel-Crafts catalyst in the presence of 2,4-bis (trimethylsilyl) -5-fluorouracil (Me3Si-U, 1) 2-acetoxytetrahydrofuran (Thf-OAc, 2) (Kazu Kigasawa et al., 2-tert-Butoxy), & lt; RTI ID = 0.0 & gt;, & lt; / RTI & gt; (K. Kigasawa, M. Hiiragi, K. ffakisaka, et al. J. Heterocyclic Chem. 1977, 14: 473-475) was reacted with 5-Fu at 155-160 ° C. Reported in the literature for the fluoride production route there are the following questions: 1, high energy consumption. In the traditional synthesis method, in order to obtain the product, the second step of the reaction needs to continue heating at 160 ° C for 5-6 hours, high energy consumption; 2, difficult to produce, low yield: 5-fluorouracil as a solid powder The reaction needs to be carried out at a high temperature (160 ° C), which requires the use of a high boiling solvent N, N-dimethylformamide (DMF). But it is difficult to completely remove the fluoride from the addition of fluoride, because DMF can form hydrogen bonds with the fluoride molecules, difficult to separate from each other; 3, in order to unreacted 5-fluorouracil and tegafur separation and recycling , The use of carcinogenic solvent chloroform as a extractant in the conventional method to separate 5-fluorouracil and tegafur. However, the main role of chloroform on the central nervous system, with anesthesia, the heart, liver, kidney damage; the environment is also harmful to the water can cause pollution. Therefore, the use of volatile solvent chloroform, even if the necessary measures to reduce its volatilization, will still cause harm to human health and the environment; 4, low yield. Since both NI and N-3 in the 5-fluorouracil molecule react with 2-tert-butoxytetrahydrofuran, the addition of tegafur is also the addition of 1,3-bis (tetrahydro-2-furyl) -5 – Fluorouracil. Therefore, the improvement of the traditional production process of tegafur is a significant and imminent task.
Example 1 (for example, the best reaction conditions):
Weigh 3.5 g (50 mmol) of 2,3-dihydrofuran, 1.9 g (50 mmol) of ethanol was added to a one-necked flask. To this was added 15 ml of tetrahydrofuran (THF). And then weighed 10. 0 mg CuCl2, microwave irradiation 250W at 25 ° C reaction 0. 6h. Cool to room temperature, add 1.95 g (15 mmol) of 5-fluorouracil (5-Fu), and microwave irradiation at 400 ° C for 100 ° C. After distilling off the low boiling solvent, the oil was obtained. Rinsed with ether to give a white solid which was recrystallized from anhydrous ethanol to give 1.34349 g of product. Melting point: 160-165 ° C. The yield was 75%.
[0011] Example 2
Weigh 3,5 g (50 mmol) of 2,3-dihydrofuran and 3.8 g (100 mmol) of ethanol were added to a single-necked flask. To this was added 15 ml of tetrahydrofuran (THF). And then weighed 5mg CuCl2, microwave irradiation 250W at 25 ° C for 0.6h. Cool to room temperature, add 1.95 g (15 mmol) of 5-fluorouracil (5-Fu), microwave irradiation 400W, reaction temperature 60 ° C under the reaction pool. The low boiling solvent was distilled off to give an oil. Rinsed with ether to give a white solid which was recrystallized from absolute ethanol to give the product 0. 46 g. Melting point: 160-165 ° C. The yield was 15%.
[0012] Example 3
Weigh 3.5 g (50 mmol) of 2,3-dihydrofuran, 1.9 g (50 mmol) of ethanol was added to a one-necked flask. To this was added 15 ml of tetrahydrofuran (THF). And then weighed 20mg CuCl2, microwave irradiation 250W at 25 ° C for 0.6h. Cooled to room temperature, add 1.95 g (15 to 01) 5-fluorouracil (5 call 11), microwave irradiation 2001, reaction temperature 1301: reaction lh. The low boiling solvent was distilled off to give an oil. Rinsed with ether to give a white solid which was recrystallized from anhydrous ethanol to give the product 1.81 g. Melting point: 160-165 ° C. The yield was 61%.
[0013] Example 4
Weigh 3.5 g (50 mmol) of 2,3-dihydrofuran and 19 g (500 mmol) of ethanol were added to a single-necked flask. To this was added 20 ml of tetrahydrofuran (THF). And then weighed IOmg CuCl2, microwave irradiation 250W at 25 ° C for 0.6h. Cooled to room temperature, add 1.95 g (15 to 01) 5-fluorouracil (5 call 11), microwave irradiation 2001, reaction temperature 1101: reaction lh. The low boiling solvent was distilled off to give an oil. Rinsed with ether to give a white solid which was recrystallized from absolute ethanol to give product U6g. Melting point: 160-165 ° C. The yield was 43%.
[0014] Example 5
Weigh 3,5 g (50 mmol) of 2,3-dihydrofuran and 9.5 g (250 mmol) of ethanol were added to a single-necked flask. To this was added 30 ml of tetrahydrofuran (THF). And then weighed IOmg CuCl2, microwave irradiation 250W at 25 ° C for 0.6h. Cooled to room temperature, add 1.95 g (15 to 01) 5-fluorouracil (5 call 11), microwave irradiation 6001, reaction temperature 1001: reaction lh. The low boiling solvent was distilled off to give an oil. Rinsed with ether to give a white solid which was recrystallized from absolute ethanol to give 1.15 g of product. Melting point: 160-165 ° C. The yield was 38%.
[0015] Example 6
Weigh 3.5 g (50 mmol) of 2,3-dihydrofuran, 1.9 g (50 mmol) of ethanol was added to a one-necked flask. To this was added 25 ml of tetrahydrofuran (THF). And then weighed 15mg CuCl2, microwave irradiation 250W at 25 ° C for 0.6h. Cooled to room temperature, add 1.95 g (15 to 01) 5-fluorouracil (5 call 11), microwave irradiation 5001, reaction temperature 1101: reaction lh. The low boiling solvent was distilled off to give an oil. Rinsed with ether to give a white solid which was recrystallized from anhydrous ethanol to give product 2.10 g. Melting point: 160-165 ° C. The yield was 70%.
Paper
A novel protocol for preparation of tegafur (a prodrug of 5-fluorouracil) is reported. The process involves the 1,8-diazabicycloundec-7-ene-mediated alkylation of 5-fluorouracil with 2-acetoxytetrahydrofuran at 90 °C, followed by treatment of the prepurified mixture of the alkylation products with aqueous ethanol at 70 °C. The yield of the two-step process is 72%.
Synthesis of Tegafur by the Alkylation of 5-Fluorouracil under the Lewis Acid and Metal Salt-Free Conditions
Aleksandra Zasada, Ewa Mironiuk-Puchalska, and Mariola Koszytkowska-Stawińska*
Faculty of Chemistry, Warsaw University of Technology, ul. Noakowskiego 3, 00-664 Warszawa, Poland
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.7b00103
*E-mail: mkoszyt@ch.pw.edu.pl.
http://pubs.acs.org/doi/abs/10.1021/acs.oprd.7b00103
http://pubs.acs.org/doi/suppl/10.1021/acs.oprd.7b00103/suppl_file/op7b00103_si_001.pdf
Tegafur, a prodrug of 5-fluorouracil (5-FUra), was discovered in 1967. The compound features high lipophilicity and water solubility compared to 5-FUra. Tegafur is used as a racemate since no significant difference in antitumor activity of enantiomers was observed.
The prodrug is gradually converted to 5-FUra by metabolism in the liver. Hence, a rapid breakdown of the released 5-FUra in the gastrointestinal tract is avoided.(6) In injectable form, tegafur provoked serious side effects, such as nausea, vomiting, or central nervous system disturbances.
The first generation of oral formulation of tegafur , UFT) is a combination of tegafur and uracil in a fixed molar ratio of 1:4, respectively. The uracil slows the metabolism of 5-FUra and reduces production of 2-fluoro-α-alanine as the toxic metabolite. UFT was approved in 50 countries worldwide excluding the USA.
S-1 is the next generation of oral formulation of tegafur.(7) It is a combination of tegafur, gimeracil, and oteracil in a fixed molar ratio of 1:0.4:1, respectively.
Gimeracil inhibits the enzyme responsible for the degradation of 5-FUra. Oteracil prevents the activation of 5-FUra in the gastrointestinal tract, thus minimizing the gastrointestinal toxicity of 5-FUra. S-1 is well-tolerated, but its safety can be influenced by schedule and dose, similar to any other cytotoxic agent. Since common side effects of S-1 can be managed with antidiarrheal and antiemetic medications, the drug can be administered in outpatient settings. S-1 was approved in Japan, China, Taiwan, Korea, and Singapore for the treatment of patients with gastric cancer.
In 2010, the Committee for Medicinal Products for Human Use (CHMP), a division of the European Medicines Agency (EMA), recommended the use of S-1 for the treatment of adults with advanced gastric cancer when given in a combination with cisplatin. Currently, S-1 has not been approved by the FDA in the United States.
There is a great interest in further examination of S-1 as an anticancer chemotherapeutic. Currently, 23 clinical trials with S-1 has been registered in National Institutes of Health (NIH). Combinations of S-1 and other anticancer agents have been employed in a majority of these trials.
5-Fluoro-1-(tetrahydrofuran-2-yl)pyrimidine-2,4(1H,3H)-dione (Tegafur)
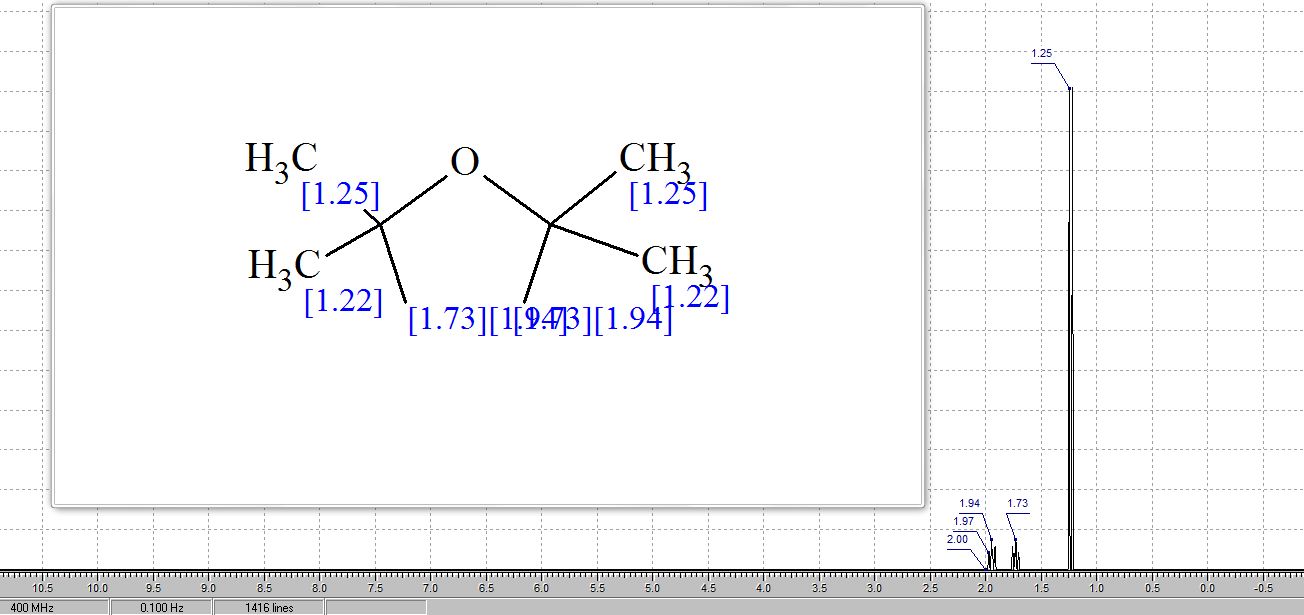
δH 1.89–2.10 (m, 3H), 2.38–2.45 (m, 1H), 3.97–4.01 (q-like m, 1H), 4.20–4.24 (dq-like m), 5.97–5.98 (m, 1H), 7.41 (d, 3JHF 6.1), 9.21 (bs, 1H, NH).
δC 23.82, 32.90, 70.26, 87.58, 123.63 (d, 2JCF 33.89), 140.33 (d, 1JCF 237.20) 148.66, 156.9 (d, 2JCF 26.81).
HRMS m/z calcd for C8H10N2O3F [M – H]+ 201.0670, found 201.0669.
Elemental analysis. Found C%, 46.42; H%, 4.45; N%, 13.35. Calcd for 3(C8H9N2O3F)·H2O: C%, 46.61; H%, 4.73; N%, 13.59.
| Cited Patent |
Filing date |
Publication date |
Applicant |
Title |
| CN85108855A * |
Nov 6, 1985 |
Sep 24, 1986 |
Central Chemical Research Institute |
Preparation of 1- (2-tetrahydrofuryl) -5-fluorouracil |
| GB1168391A * |
|
|
|
Title not available |
| JPS5452085A * |
|
|
|
Title not available |
| JPS5455581A * |
|
|
|
Title not available |
| JPS5459288A * |
|
|
|
Title not available |
| JPS52118479A * |
|
|
|
Title not available |
| JPS54103880A * |
|
|
|
Title not available |
| US4256885 * |
Dec 10, 1976 |
Mar 17, 1981 |
Mitsui Toatsu Kagaku Kabushiki Kaisha |
Process for the preparation of 1- (2-tetrahydrofuryl) -5-fluorouracil |
| US5075446 * |
Oct 12, 1990 |
Dec 24, 1991 |
Korea Advanced Institute Of Science & Technology |
Synthesis of tetrahydro-2-furylated pyrimidine derivatives |
References
1
- El Sayed, YM; Sadée, W (1983). “Metabolic activation of R,S-1-(tetrahydro-2-furanyl)-5-fluorouracil (ftorafur) to 5-fluorouracil by soluble enzymes”. Cancer Research. 43 (9): 4039–44. PMID 6409396.
- 2
- Sweetman, S, ed. (14 November 2011). “Martindale: The Complete Drug Reference”. Pharmaceutical Press. Retrieved 12 February 2014.
- 3
- Ishikawa, T (14 May 2008). “Chemotherapy with enteric-coated tegafur/uracil for advanced hepatocellular carcinoma.” (PDF). World Journal of Gastroenterology : WJG. 14 (18): 2797–2801. doi:10.3748/wjg.14.2797. PMC 2710718
 . PMID 18473401.
. PMID 18473401.
- 4
- Caudle, KE; Thorn, CF; Klein, TE; Swen, JJ; McLeod, HL; Diasio, RB; Schwab, M (December 2013). “Clinical Pharmacogenetics Implementation Consortium guidelines for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing.”. Clinical pharmacology and therapeutics. 94 (6): 640–5. doi:10.1038/clpt.2013.172. PMC 3831181 . PMID 23988873.
- 5
- Amstutz, U; Froehlich, TK; Largiadèr, CR (September 2011). “Dihydropyrimidine dehydrogenase gene as a major predictor of severe 5-fluorouracil toxicity.”. Pharmacogenomics. 12 (9): 1321–36. doi:10.2217/pgs.11.72. PMID 21919607.
- 6
- Nakayama, T; Noguchi, S (January 2010). “Therapeutic usefulness of postoperative adjuvant chemotherapy with Tegafur-Uracil (UFT) in patients with breast cancer: focus on the results of clinical studies in Japan.” (PDF). The Oncologist. 15 (1): 26–36. doi:10.1634/theoncologist.2009-0255. PMC 3227888 . PMID 20080863.
- 7
Matt P, van Zwieten-Boot B, Calvo Rojas G, Ter Hofstede H, Garcia-Carbonero R, Camarero J, Abadie E, Pignatti F (October 2011). “The European Medicines Agency review of Tegafur/Gimeracil/Oteracil (Teysuno™) for the treatment of advanced gastric cancer when given in combination with cisplatin: summary of the Scientific Assessment of the Committee for medicinal products for human use (CHMP).” (PDF). The Oncologist. 16 (10): 1451–1457. doi:10.1634/theoncologist.2011-0224. PMC 3228070 . PMID 21963999.
- (1) Hirose, Takashi; Oncology Reports 2010, V24(2), P529-536
- (2) Fujita, Ken-ichi; Cancer Science 2008, V99(5), P1049-1054
- (3) Tahara, Makoto; Cancer Science 2011, V102(2), P419-424
- (4) Chu, Quincy Siu-Chung; Clinical Cancer Research 2004, V10(15), P4913-4921
- (5) Tominaga, Kazunari; Oncology 2004, V66(5), P358-364
- (6) Peters, Godefridus J.; Clinical Cancer Research 2004, V10(12, Pt. 1), P4072-4076
- (7) Kim, Woo Young; Cancer Science 2007, V98(10), P1604-1608
- Hillers, Solomon; Puti Sinteza i Izyskaniya Protivoopukholevykh Preparatov 1970, VNo. 3, P109-12
- Grishko, V. A.; Trudy Kazakhskogo Nauchno-Issledovatel’skogo Instituta Onkologii i Radiologii 1977, V12, P110-14
- Ootsu, Koichiro; Takeda Kenkyushoho 1978, V37(3-4), P267-77
- “Drugs – Synonyms and Properties” data were obtained from Ashgate Publishing Co. (US)
- Yabuuchi, Youichi; Oyo Yakuri 1971, V5(4), P569-84
- Germane, S.; Eksperimental’naya i Klinicheskaya Farmakoterapiya 1970, (1), P85-92
- JP 56046814 A 1981
MORE
- AIST: Integrated Spectral Database System of Organic Compounds. (Data were obtained from the National Institute of Advanced Industrial Science and Technology (Japan))
- ACD-A: Sigma-Aldrich (Spectral data were obtained from Advanced Chemistry Development, Inc.)
- Nomura, Hiroaki; Chemical & Pharmaceutical Bulletin 1979, V27(4), P899-906
- Sakurai, Kuniyoshi; Chemical & Pharmaceutical Bulletin 1978, V26(11), P3565-6
- Miyashita, Osamu; Chemical & Pharmaceutical Bulletin 1981, V29(11), P3181-90
- Lukevics, E.; Zhurnal Obshchei Khimii 1981, V51(4), P827-34
- Needham, F.; Powder Diffraction 2006, V21(3), P245-247
-
- Nomura, Hiroaki; Chemical & Pharmaceutical Bulletin 1979, V27(4), P899-906
- Sakurai, Kuniyoshi; Chemical & Pharmaceutical Bulletin 1978, V26(11), P3565-6
- “Drugs – Synonyms and Properties” data were obtained from Ashgate Publishing Co. (US)
- Miyashita, Osamu; Chemical & Pharmaceutical Bulletin 1981, V29(11), P3181-90
- “PhysProp” data were obtained from Syracuse Research Corporation of Syracuse, New York (US)
- Lukevics, E.; Zhurnal Obshchei Khimii 1981, V51(4), P827-34
- Lukevics, E.; Latvijas PSR Zinatnu Akademijas Vestis, Kimijas Serija 1982, (3), P317-20
- Kruse, C. G.; Recueil des Travaux Chimiques des Pays-Bas 1979, V98(6), P371-80
- Lukevics, E.; Latvijas PSR Zinatnu Akademijas Vestis, Kimijas Serija 1981, (4), P492-3
- Kametani, Tetsuji; Heterocycles 1977, V6(5), P529-33
- Kametani, Tetsuji; Journal of Heterocyclic Chemistry 1977, V14(3), P473-5
- Hillers, S.; GB 1168391 1969
///////////TEGAFUR
FC1=CN(C2CCCO2)C(=O)NC1=O































 The presentation will load below
The presentation will load below