Catalytic C-H amination at its limits: challenges and solutions
DOI: 10.1039/C7QO00547D, Review Article
Pushing C-H amination to its limits fosters innovative synthetic solutions and offers a deeper understanding of the reaction mechanism and scope.
Catalytic C–H amination at its limits: challenges and solutions
Abstract
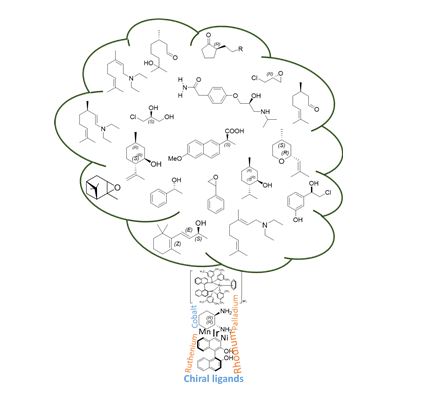
Catalytic C–H amination reactions enable direct functionalization of non-activated C(sp3)–H bonds with high levels of regio-, chemo- and stereoselectivity. As a powerful tool to unlock the potential of inert C–H bonds, C–H amination chemistry has been applied to the preparation of synthetically challenging targets since major simplification of synthetic sequences are expected from this approach. Pushing C–H amination to its limits has led to a deeper understanding of the reaction mechanism and scope. In this review, we present a description of the specific challenges facing catalytic C–H amination in the synthesis of natural products and related compounds, as well as innovative tactics created to overcome them. By identifying and discussing the major insights gained and strategies designed, we hope that this review will stimulate further progress in C–H amination chemistry and beyond.
Conclusion Since the seminal works of Du Bois in the early 2000s, the pace of discovery in the field of metal-catalysed C–H amination has been breath-taking. Not surprisingly, this synthetic tool has been applied to the total synthesis of many compounds of interest, given the high prevalence of the amino group in natural products and synthetic pharmaceuticals.67 Chemist’s confidence in the high potential of the C–H amination methodology to unlock inert C–H bonds has been demonstrated by its application to more and more challenging substrates. This has been a powerful drive for progress in the field. New valuable insights have been gained allowing, for example, a better regiochemical control via stereoelectronic and/or conformational effects. Innovative strategies have been discovered to direct the insertion event in substrates bearing a large degree of attendant functionality. Solutions have spanned from the elegant exploitation of kinetic isotope effects to the tactical use of protecting groups with different sizes or electronic characteristics. Systematic exploration of different catalytic systems has been also performed leading to the opening of new possibilities in C–H amination technology. Manganese-based catalysts have thus given rise to nitrenoids that overcome the low reactivity of primary aliphatic C–H bonds without interfering with weaker secondary/tertiary C–H bonds. Despite these impressive achievements, much remains to be done. Counterintuitive selectivity and unexplained reactivity should serve as a reminder that further studies are highly needed to understand in depth catalytic C–H amination chemistry. Many challenges remain on the way, from basic to applied research. A clear mechanistic view based on definitive evidence concerning the details of the C–N bond forming process would undoubtedly facilitate the rational design of efficient catalytic systems leading to higher regio-, chemio- and stereoselectivity. In particular, the quest for site-selective C–H amination through catalyst control has to be pursued.10d,e In this context, the development of efficient intermolecular C–H amination process still represents a major challenge and upcoming advancements are expected to increase the impact of this technology in organic synthesis. Future progress made in the field of catalytic C–H amination chemistry might also lead to industrial-scale applications in the next decade. It is likely that total synthesis of synthetically challenging targets related to natural products will continue to be a powerful driving force towards this goal.


/////////////
“ALL FOR DRUGS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This is a compilation for educational purposes only. P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent




 Googleplus
Googleplus











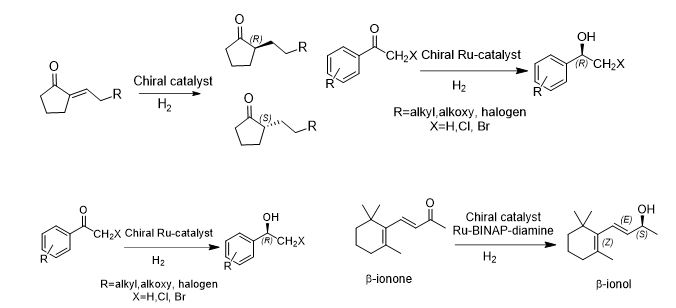
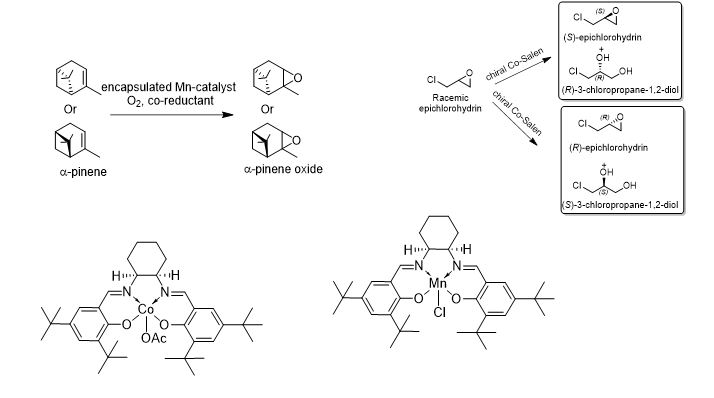
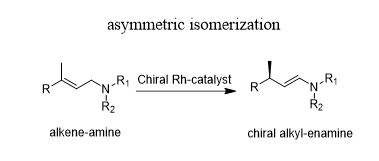
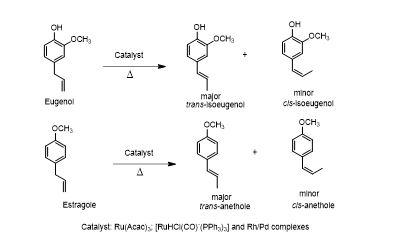
















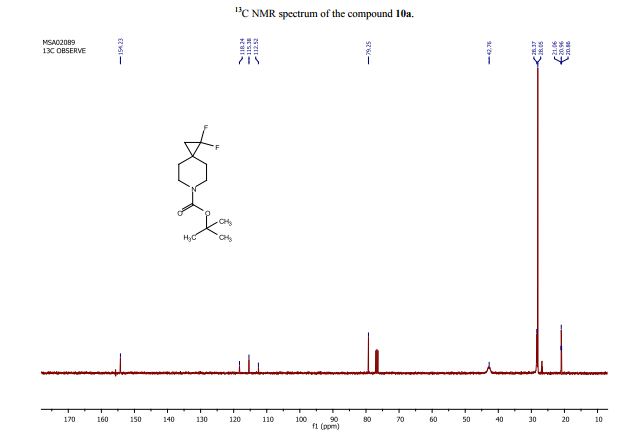
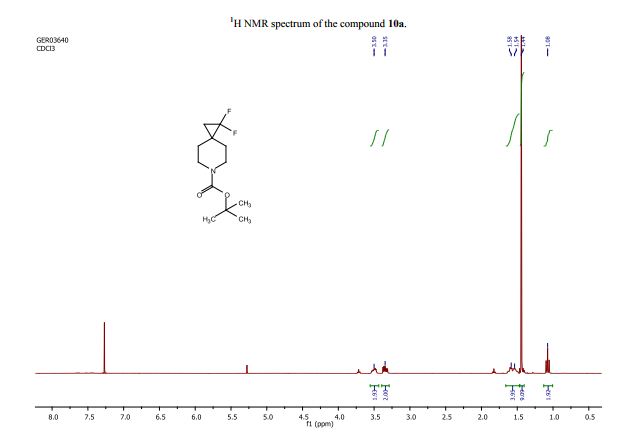







 学 系
学 系