HOW TO SEE ORANGE BOOK AND INTERPRET IT













TPYE ORANGE BOOK ON GOOGLE
Orange Book: Approved Drug Products with Therapeutic

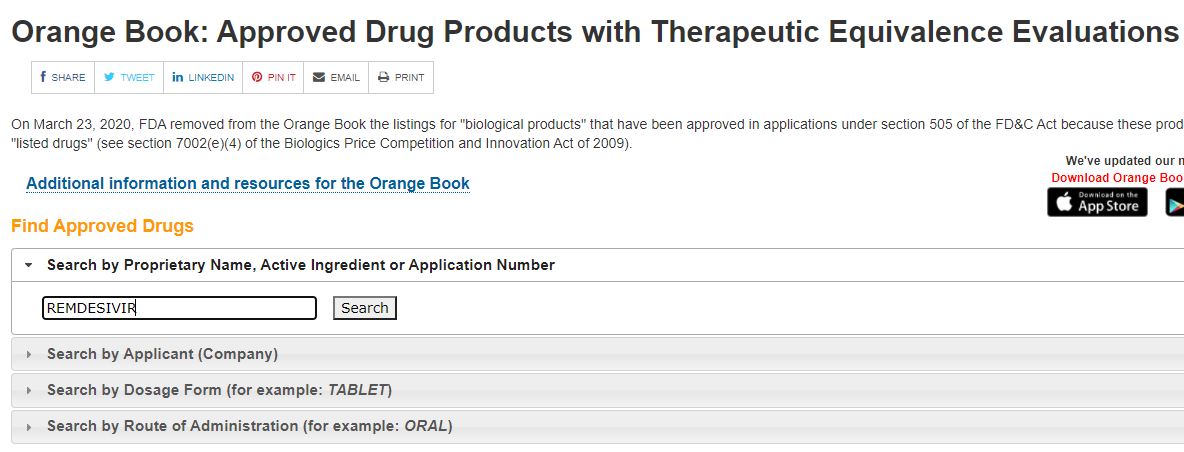
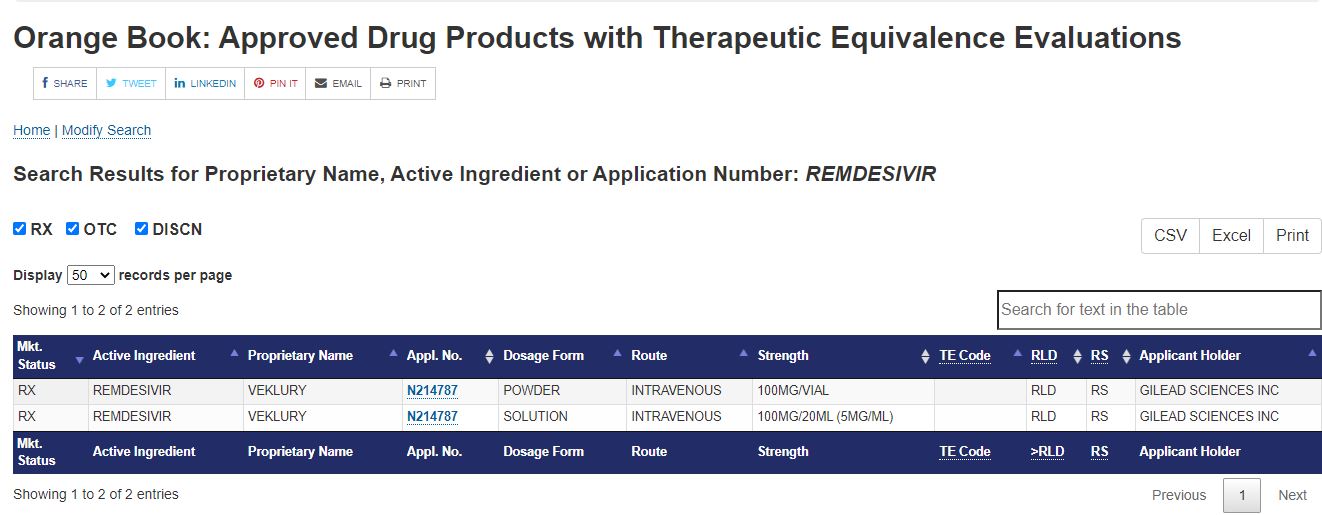
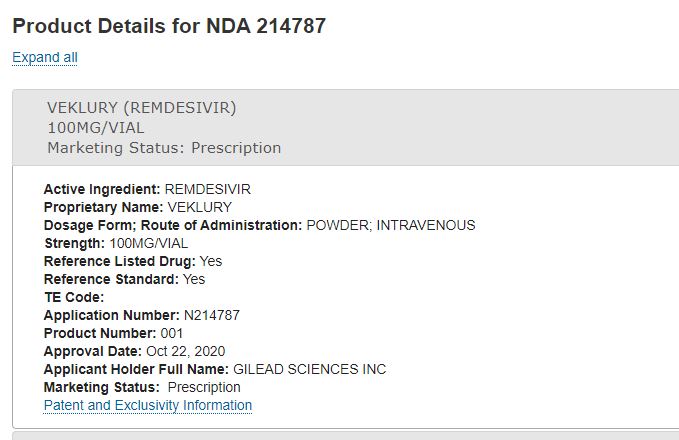

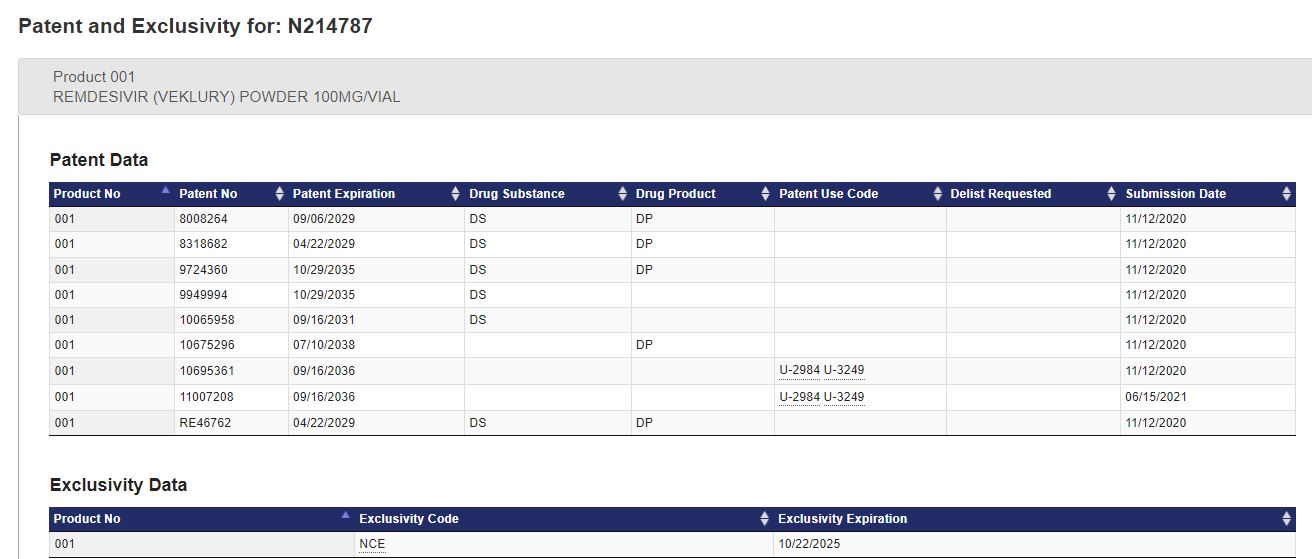
| 10/22/2025 |
NEXT
NCE-1 =9/22/2024
These NCE-1 dates indicate the first opportunity for generic drug companies to file Abbreviated New Drug Applications (ANDAs) for generic entry into branded drug markets. Generic launch is dependent on many factors, including FDA approval and patents.
.
.
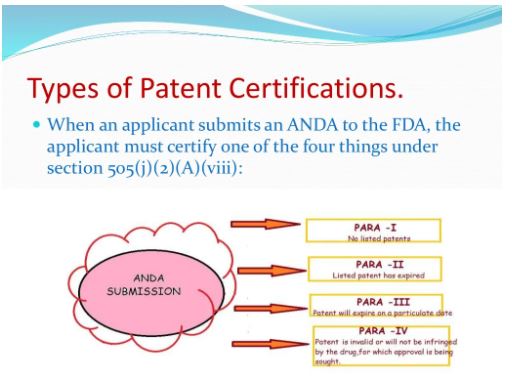
Patent Certifications and Suitability Petitions
FDA aspires to continually improve its pre-ANDA (abbreviated new drug application) interactions with applicants. To facilitate these interactions and keep stakeholders as informed as possible, the agency regularly publishes information on suitability petitions and Paragraph IV patent certifications.
- Paragraph IV Certifications List
- New Paragraph IV Certifications as of February 8, 2022
- Suitability Petitions
- Contact Us
Paragraph IV Patent Certifications
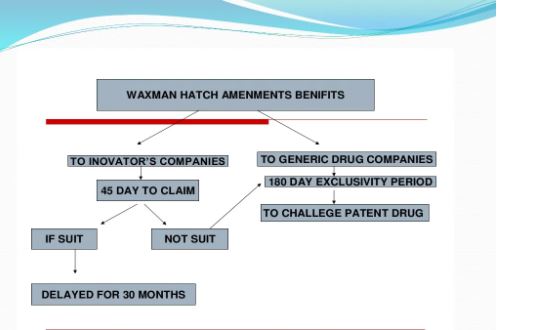
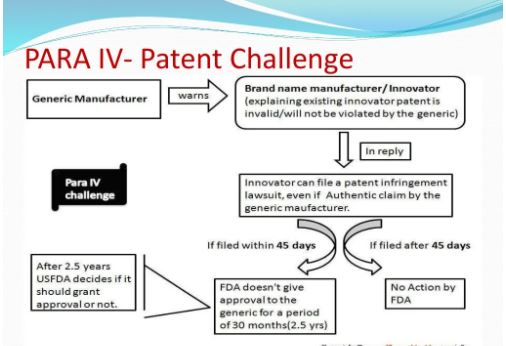
Under the Drug Price Competition and Patent Term Restoration Act of 1984, also known as the Hatch-Waxman Amendments, a company can seek FDA approval to market a generic drug before the expiration of patents related to the brand-name drug that the generic seeks to copy. To seek this approval, a generic applicant must provide in its application a “certification” that a patent submitted to FDA by the brand-name drug’s sponsor and listed in FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations (the Orange Book) is, in the generic applicant’s opinion and to the best of its knowledge, invalid, unenforceable, or will not be infringed by the generic product. This certification is called a “paragraph IV certification.” The first company or companies to submit an application that (1) is determined by the agency to be “substantially complete” upon submission and (2) contains a paragraph IV certification to at least one of the patents listed in the Orange Book is generally eligible for the exclusive right to market the generic drug for 180 days.
In order to challenge a patent in court, the generic applicant that submitted a paragraph IV certification must notify the brand product sponsor and any patent holder of the submission of the ANDA and patent challenge. If the brand product sponsor or patent holder files an infringement suit against the generic applicant within 45 days of the ANDA notification, FDA approval to market the generic drug is generally postponed for 30 months unless the patent expires or is judged to be invalid or not infringed before that time. This 30-month postponement, commonly referred to as the “30-month stay,” gives the brand product sponsor and patent holder a prescribed amount of time to assert patent rights in court before a generic competitor is approved and can market the drug.
Paragraph IV Certifications List
As part of its ongoing efforts to assist generic drug applicants in preparing their applications, the Food and Drug Administration (FDA or the Agency) regularly publishes information relevant to 180-day exclusivity for generic drug applicants provided under section 505(j) of the Federal Food, Drug, and Cosmetic Act (FD&C Act). Generally, this list describes drug products for which one or more substantially complete abbreviated new drug applications (ANDAs) containing a “paragraph IV” (PIV) patent certification have been submitted to FDA. (For more information on 180-day exclusivity, see FDA’s draft guidance for industry on 180-Day Exclusivity: Questions and Answers (Jan. 2017).)
Historically, this list has included the name of the drug product, dosage form, strength(s), reference listed drug (RLD)/new drug application (NDA) number, and, beginning on March 2, 2004, the date on which the first substantially complete ANDA(s) (or amendment or supplement to one) was submitted to the Agency that contained a paragraph IV certification.
Under the FDA’s Drug Competition Action Plan (DCAP), FDA committed to enhancing efficiency of the development and approval of ANDAs, with the ultimate goal of more approvals to help increase access to high-quality, lower cost generic drugs. FDA believes that additional information regarding 180-day exclusivity for specific drug products may support this DCAP goal by providing greater clarity to ANDA applicants regarding the earliest date when they may be able to obtain final approval. Accordingly, on a prospective basis beginning June 18, 2019, the Agency also will include the additional information described in detail below.
The newly added columns to the Paragraph IV Certification List will be updated retrospectively as practicable. Thus, entries in the newly added columns for drug products that reflect a date of first paragraph IV certification prior to June 18, 2019 may be blank because we have not updated that drug product’s information. By contrast, if entries for the newly added columns for drug products that reflect a date of paragraph IV certification after June 18, 2019 are blank, it means, in general, that FDA has not yet made a determination regarding or an event has not yet occurred for that column.
The Agency aims to provide accurate information in this list. However, FDA will make regulatory decisions based on the data and information in any relevant applications, and not the information on this list. Any discrepancies or disparities should be discussed with FDA’s Patent and Exclusivity Team (PET) by emailing CDER-OGDPET@fda.hhs.gov.
Beginning with the June 18, 2019 update, the Agency plans to include the following information for individual drug products on the Paragraph IV Certification List:
- Active Ingredient Name
This column reflects the established (nonproprietary) name of the active ingredient(s) in the drug product. - Dosage Form
- Strength
- RLD Name and NDA Number
This column reflects the proprietary name, if any, of the RLD and its NDA number. - Date of First PIV Submission
This column reflects the first date that a substantially complete application containing a paragraph IV certification(s) to at least one patent listed for the RLD in the FDA’s Approved Drug Products With Therapeutic Equivalence Evaluations (the Orange Book) was submitted. The date of first PIV submission may correspond to the date of submission of an original ANDA, an amendment to a pending ANDA, or a supplement to an approved ANDA.
Drug products on the list that have “pre-MMA” in the “Date of First PIV Submission” column are subject to the provisions of the FD&C Act in effect prior to the amendments made by the Medicare Prescription Drug, Improvement, and Modernization Act of 2003 (MMA). Due to the “patent-by-patent” analysis needed to determine eligibility for 180-day exclusivity under the pre-MMA statutory scheme, listing individual dates of relevant submissions is not practical. (For more information on the applicability of the “pre-MMA” provisions of the FD&C Act related to 180-day exclusivity, see FDA’s draft guidance for industry on 180-Day Exclusivity: Questions and Answers, at Q1.). - Number of Potential First Applicant ANDAs Submitted
This column indicates the number of substantially complete ANDAs that contained a paragraph IV certification to at least one patent listed for the RLD in the Orange Book on the first date such a certification was submitted. FDA may adjust this number over time for a drug product, for example, if the agency grants a request for reconsideration of FDA’s refusal to receive a relevant ANDA. - 180-Day Decision Status
This column indicates if FDA has made (or has deferred) a decision regarding eligibility for 180-day exclusivity for a drug product. Specifically, this column indicates whether: (1) FDA approved at least one first applicant’s ANDA and considered that first applicant’s drug product eligible for 180-day exclusivity at the time of approval (“eligible”); (2) FDA approved a first applicant but did not make a determination regarding eligibility for exclusivity at the time of approval (“deferred”); (3) FDA tentatively approved a subsequent applicant solely on the basis of a first applicant’s eligibility for 180-day exclusivity at a time that none of the grounds for forfeiture were found to apply (“non-forfeiture”); or (4) FDA determined that 180-day exclusivity has been extinguished, for example, if all first applicants have forfeited or voluntary relinquished eligibility for 180-day exclusivity (“extinguished”).
There may be instances in which the 180-Day Decision Status reflects that FDA made a non-forfeiture decision at one point in time, e.g., non-forfeiture under the statutory provision related to obtaining approval or tentative approval within 30 months of submission (failure-to-obtain-TA in 30 months provision), and then subsequently determined based on new events that the first applicant forfeited its eligibility for exclusivity, e.g., forfeiture under the “failure to market” statutory provision. If FDA has not made any of the above determinations, this column will be blank. This column will be cumulative, with the most recent decision reflected first.
For certain 180-day exclusivity decisions that require analysis by FDA, including those under the failure-to-obtain-TA in 30 months and failure-to-market provisions, it is FDA’s practice to make these decisions in the context of specific ANDAs that are otherwise eligible for approval (i.e., when a first applicant’s ANDA or a subsequent applicant’s ANDA is ready for approval). Many factors may influence eligibility for exclusivity up to the time an application is ready for approval (e.g., patent expiration, failure to obtain a tentative or final approval within 30 months, withdrawal of ANDA) and thus could render a premature eligibility determination incorrect.
180-day exclusivity decisions under the withdrawal-of-application, amendment-of-certification, and expiration-of-all-patents forfeiture provisions generally are made once all first applicants have forfeited eligibility for 180-day exclusivity under these forfeiture provisions and the Agency confirms the forfeiture(s), unless there are complicating factors that would require analysis. In such cases, FDA generally would make a decision in the context of specific ANDAs that are otherwise eligible for approval, and any updates to this column would be posted after the Agency’s decision had been made. - 180-Day Decision Posting Date
This column reflects the month and year that FDA updated the PIV Certification List to reflect the corresponding 180-day decision status. This column will be cumulative, with the most recent decision date reflected first. - Date of First Approval of “First Applicant” ANDA
This column indicates the first date on which a first applicant’s ANDA received final approval. If there are multiple first applicants, only the date of the first approval is posted. - Date of First Commercial Marketing
This column reflects the date of first commercial marketing by any first applicant. This column, in conjunction with the Date of First Approval of “First Applicant” ANDA, provides the time frame between the dates a first applicant’s ANDA was approved and the date when a first applicant began commercial marketing. To note, if an ANDA is determined to be eligible for 180-day exclusivity, the Orange Book posts a Patent Challenge (PC) code in the “Exclusivity Data” section of the Orange Book to only the approved ANDA(s) eligible for 180-day exclusivity upon any first applicant’s commercial marketing, indicating the date the 180-day exclusivity will expire. If the agency deferred in making a decision as to whether or not an approved ANDA(s) is eligible for 180-day exclusivity, a PC code will not be entered for this ANDA(s) in the Orange Book. The approval letters for first applicant ANDA(s) are posted on the Drugs@FDA website. - Expiration Date of Last Qualifying Patent
This column lists the last expiration date of any patent(s) that qualifies any first applicant for 180-day exclusivity for a given drug product. This column indicates when 180-day exclusivity will no longer be available due to patent expiry. If there are multiple applications submitted on the first day, the patents that have at least one PIV certification amongst all the submissions will be posted. For example, if applicant 1 has PIV certifications to patents A, B, C and PIII certifications to patents D, E and F, but applicant 2 has PIV certifications to patents A, B, D and E, and PIII certifications to patents C and F, then the expiration date that is last-in-time for patents A, B, C, D and E would be posted on the PIV Certification List since there is a PIV certification for each of the patents; only one expiration date will be posted. The expiration date for patent F would not be considered because both applicants have a PIII certification to this patent. This column does not reflect any pediatric exclusivity for the RLD associated with the last qualifying patent.
Paragraph IV Certifications
| Drug Name | Dosage Form | Strength | RLD/NDA | Date of Submission |
|---|---|---|---|---|
| Angiotensin II Acetate | Injection | 2.5 mg/mL | Giapreza 209360 |
12/21/2021 |
| Cyclosporine | Ophthalmic Emulsion | 0.05% | Restasis 50790 |
1/13/2014 |
| Cysteamine Bitartrate | Delayed-release Granules | 75 mg/Packet and 300 mg/Packet |
Procysbi 213491 |
12/16/2021 |
| Ertugliflozin | Tablets | 5 mg and 15 mg | Steglatro 209803 |
12/20/2021 |
| Latanoprost and Netarsudil Dimesylate | Ophthalmic Solution | 0.005%/0.02% | Rocklatan 208259 |
12/20/2021 |
Suitability Petitions
Certain differences between a reference listed drug (RLD) and a proposed generic drug product may be permitted in an abbreviated new drug application (ANDA) if these differences are the subject of an approved suitability petition submitted under section 505(j)(2)(C) of the Federal Food, Drug, and Cosmetic Act, and pursuant to 21 CFR 314.93.
An applicant may submit a suitability petition to the FDA requesting permission to submit an ANDA for a generic drug product that differs from an RLD in its:
- route of administration,
- dosage form,
- strength, or
- if it has one different active ingredient in a fixed-combination drug product.
An ANDA citing a suitability petition that has not been approved will not be received for review because the application lacks a legal basis for the submission.
A generic applicant cannot submit an ANDA for such a product until FDA has approved the related petition. The grounds for FDA approval of such a petition are set out in 21 CFR 314.93(e). The determination that an ANDA will be approved is not made until the ANDA itself is submitted and is reviewed by the Agency.
Accessing and Tracking Suitability Petitions
You may view copies of submitted suitability petitions and responses from FDA at Regulations.gov, which is searchable by docket number or drug product name. You may also submit comments to pending suitability petitions through this site.
The Office of Generic Drugs (OGD) has previously posted suitability petition tracking reports; however, these lists are not regularly updated as they are burdensome to maintain. For the most up-to-date information, OGD recommends searching for suitability petitions on regulations.gov.
- OGD Suitability Tracking Report Sorted by Drug Name (PDF – 1 MB) (updated August 2015)
- OGD Suitability Tracking Report Sorted by Petition Number (PDF – 1 MB) (updated August 2015)
Contact Us
Questions about suitability petitions can be directed to
FDA’s Division of Dockets Management
5630 Fishers Lane
Room 1061, HFA-305
Rockville, MD 20852
Resources For You
.
.
.

.
.
NEXT
.
.
What is the difference between patent and exclusivity?
How long does an exclusivity period last?
It depends on what type of exclusivity is at issue.
- Orphan Drug Exclusivity (ODE) – 7 years
- New Chemical Entity Exclusivity (NCE) – 5 years
- Generating Antibiotic Incentives Now (GAIN) Exclusivity– 5 years added to certain exclusivities
- New Clinical Investigation Exclusivity – 3 years
- Pediatric Exclusivity (PED) – 6 months added to existing Patents/Exclusivity
- Patent Challenge (PC) – 180 days (this exclusivity is for ANDAs only)
- Competitive Generic Therapy (CGT) – 180 days (this exclusivity is for ANDAs only)
See 21 C.F.R. 314.108, 316.31, 316.34 and sections 505A, 505E, 505(j)(5)(B)(iv), and Section 505(j)(5)(B)(v) of the FD&C Act.
This certification is called a “paragraph IV certification.” The first company or companies to submit an application that (1) is determined by the agency to be “substantially complete” upon submission and (2) contains a paragraph IV certification to at least one of the patents listed in the Orange Book
A paragraph IV filing is a subset of an ANDA application, where the generic applicant is claiming that the patent they are targeting is unenforceable either due to i) invalid or, ii) not infringed or iii) both invalid and non infringed by their product
To obtain approval of a generic drug, an ANDA applicant is not required to provide independent 64 evidence of the safety and effectiveness of the proposed generic drug. Instead, the applicant relies on FDA’s previous finding that the reference listed drug (RLD)8 65 relied upon by the ANDA 66 applicant is safe and effective. The ANDA applicant must identify the RLD on which it seeks to 67 rely and, among other things, demonstrate, with limited exceptions, that the proposed generic 68 drug has the same active ingredient(s), route of administration, dosage form, and strength as the RLD.9 69 A generic drug also must have the same conditions of use and the same labeling as the RLD (except for certain permissible labeling differences), 10 70 and the applicant must demonstrate that its proposed generic drug is bioequivalent to the RLD.11
A Reference Listed Drug (RLD) is an approved drug product to which new generic versions are compared to show that they are bioequivalent. A drug company seeking approval to market a generic equivalent must refer to the Reference Listed Drug in its Abbreviated New Drug Application (ANDA).
With respect to each patent listed in the Orange Book for the RLD, the applicant’s patent 87 certification must state one of the following: 88 89 2. That such patent has expired (a paragraph II certification) 90 3. The date on which such patent will expire (a paragraph III certification) 91 4. That such patent is invalid, unenforceable, or will not be infringed by the manufacture, 92 use, or sale of the new drug for which the application is submitted (a paragraph IV certification).

.
.

.
.

.
.

..
.

.
.


.

.
.
.
.
.
.
.
.

.
.

.
How does an ANDA applicant qualify as a “first applicant”?
A first applicant is an ANDA “applicant that, on the first day on which a substantially complete
application containing a [paragraph IV] certification … is submitted for approval of a drug, 196 submits a substantially complete application that contains and lawfully maintains a [paragraph IV] certification … for the drug.”23
An applicant that previously submitted a substantially complete ANDA that did not contain a
paragraph IV certification may become eligible for 180-day exclusivity by amending its ANDA
to contain a paragraph IV certification to a listed patent for the RLD on the first day that an ANDA or amendment containing a paragraph IV certification is submitted.
The listed patent or patents to which an ANDA applicant submitted a paragraph IV certification
205 that gives rise to the eligibility for 180-day exclusivity is referred to in this draft guidance as the qualifying patent(s).
3,053 total views, 2 views today
