Drug Discovery














In the fields of medicine, biotechnology and pharmacology, drug discovery is the process by which new candidate medications are discovered.
Historically, drugs were discovered through identifying the active ingredient from traditional remedies or by serendipitous discovery. Later chemical libraries of synthetic small molecules, natural products or extracts were screened in intact cells or whole organisms to identify substances that have a desirable therapeutic effect in a process known as classical pharmacology. Sincesequencing of the human genome which allowed rapid cloning and synthesis of large quantities of purified proteins, it has become common practice to use high throughput screening of large compounds libraries against isolated biological targets which are hypothesized to be disease modifying in a process known as reverse pharmacology.
Hits from these screens are then tested in cells and then in animals for efficacy. Even more recently, scientists have been able to understand the shape of biological molecules at the atomic level, and to use that knowledge to design (seedrug design) drug candidates.

Modern drug discovery involves the identification of screening hits, medicinal chemistry and optimization of those hits to increase the affinity, selectivity (to reduce the potential of side effects), efficacy/potency, metabolic stability (to increase the half-life), and oral bioavailability. Once a compound that fulfills all of these requirements has been identified, it will begin the process of drug development prior to clinical trials. One or more of these steps may, but not necessarily, involve computer-aided drug design.
Despite advances in technology and understanding of biological systems, drug discovery is still a lengthy, “expensive, difficult, and inefficient process” with low rate of new therapeutic discovery.[1]In 2010, the research and development cost of each new molecular entity (NME) was approximately US$1.8 billion.[2] Drug discovery is done by pharmaceutical companies, with research assistance from universities. The “final product” of drug discovery is a patent on the potential drug. The drug requires very expensive Phase I, II and III clinical trials, and most of them fail. Small companies have a critical role, often then selling the rights to larger companies that have the resources to run the clinical trials.
![]()
Drug targets
The definition of “target” itself is something argued within the pharmaceutical industry. Generally, the “target” is the naturally existing cellular or molecular structure involved in the pathology of interest that the drug-in-development is meant to act on. However, the distinction between a “new” and “established” target can be made without a full understanding of just what a “target” is. This distinction is typically made by pharmaceutical companies engaged in discovery and development of therapeutics. In an estimate from 2011, 435 human genome products were identified as therapeutic drug targets of FDA-approved drugs.[3]

“Established targets” are those for which there is a good scientific understanding, supported by a lengthy publication history, of both how the target functions in normal physiology and how it is involved in human pathology. This does not imply that the mechanism of action of drugs that are thought to act through a particular established targets is fully understood. Rather, “established” relates directly to the amount of background information available on a target, in particular functional information. The more such information is available, the less investment is (generally) required to develop a therapeutic directed against the target.

The process of gathering such functional information is called “target validation” in pharmaceutical industry parlance. Established targets also include those that the pharmaceutical industry has had experience mounting drug discovery campaigns against in the past; such a history provides information on the chemical feasibility of developing a small molecular therapeutic against the target and can provide licensing opportunities and freedom-to-operate indicators with respect to small-molecule therapeutic candidates.

In general, “new targets” are all those targets that are not “established targets” but which have been or are the subject of drug discovery campaigns. These typically include newly discoveredproteins, or proteins whose function has now become clear as a result of basic scientific research.
The majority of targets currently selected for drug discovery efforts are proteins. Two classes predominate: G-protein-coupled receptors (or GPCRs) and protein kinases.

Screening and design
The process of finding a new drug against a chosen target for a particular disease usually involves high-throughput screening (HTS), wherein large libraries of chemicals are tested for their ability to modify the target. For example, if the target is a novel GPCR, compounds will be screened for their ability to inhibit or stimulate that receptor (see antagonist and agonist): if the target is a protein kinase, the chemicals will be tested for their ability to inhibit that kinase.
Another important function of HTS is to show how selective the compounds are for the chosen target. The ideal is to find a molecule which will interfere with only the chosen target, but not other, related targets. To this end, other screening runs will be made to see whether the “hits” against the chosen target will interfere with other related targets – this is the process of cross-screening. Cross-screening is important, because the more unrelated targets a compound hits, the more likely that off-target toxicity will occur with that compound once it reaches the clinic.
It is very unlikely that a perfect drug candidate will emerge from these early screening runs. It is more often observed that several compounds are found to have some degree of activity, and if these compounds share common chemical features, one or more pharmacophores can then be developed. At this point, medicinal chemists will attempt to use structure-activity relationships (SAR) to improve certain features of the lead compound:
- increase activity against the chosen target
- reduce activity against unrelated targets
- improve the druglikeness or ADME properties of the molecule.
This process will require several iterative screening runs, during which, it is hoped, the properties of the new molecular entities will improve, and allow the favoured compounds to go forward to in vitro and in vivo testing for activity in the disease model of choice.
Amongst the physico-chemical properties associated with drug absorption include ionization (pKa), and solubility; permeability can be determined by PAMPA and Caco-2. PAMPA is attractive as an early screen due to the low consumption of drug and the low cost compared to tests such as Caco-2, gastrointestinal tract (GIT) and Blood–brain barrier (BBB) with which there is a high correlation.
A range of parameters can be used to assess the quality of a compound, or a series of compounds, as proposed in the Lipinski’s Rule of Five. Such parameters include calculated properties such as cLogP to estimate lipophilicity, molecular weight, polar surface area and measured properties, such as potency, in-vitro measurement of enzymatic clearance etc. Some descriptors such asligand efficiency[4] (LE) and lipophilic efficiency[5][6] (LiPE) combine such parameters to assess druglikeness.
While HTS is a commonly used method for novel drug discovery, it is not the only method. It is often possible to start from a molecule which already has some of the desired properties. Such a molecule might be extracted from a natural product or even be a drug on the market which could be improved upon (so-called “me too” drugs). Other methods, such as virtual high throughput screening, where screening is done using computer-generated models and attempting to “dock” virtual libraries to a target, are also often used.
Another important method for drug discovery is drug design, whereby the biological and physical properties of the target are studied, and a prediction is made of the sorts of chemicals that might (e.g.) fit into an active site. One example is fragment-based lead discovery (FBLD). Novel pharmacophores can emerge very rapidly from these exercises. In general, computer-aided drug design is often but not always used to try to improve the potency and properties of new drug leads.
Once a lead compound series has been established with sufficient target potency and selectivity and favourable drug-like properties, one or two compounds will then be proposed for drug development. The best of these is generally called the lead compound, while the other will be designated as the “backup”.

Historical background
The idea that effect of drug in human body are mediated by specific interactions of the drug molecule with biological macromolecules, (proteins or nucleic acids in most cases) led scientists to the conclusion that individual chemicals are required for the biological activity of the drug. This made for the beginning of the modern era in pharmacology, as pure chemicals, instead of crude extracts, became the standard drugs. Examples of drug compounds isolated from crude preparations are morphine, the active agent in opium, and digoxin, a heart stimulant originating from Digitalis lanata. Organic chemistry also led to the synthesis of many of the cochemicals isolated from biological sources.

Nature as source of drugs
Despite the rise of combinatorial chemistry as an integral part of lead discovery process, natural products still play a major role as starting material for drug discovery.[7] A report was published in 2007,[8] covering years 1981-2006 details the contribution of biologically occurring chemicals in drug development. According to this report, of the 974 small molecule new chemical entities, 63% were natural derived or semisynthetic derivatives of natural products. For certain therapy areas, such as antimicrobials, antineoplastics, antihypertensive and anti-inflammatory drugs, the numbers were higher. In many cases, these products have been used traditionally for many years.
Natural products may be useful as a source of novel chemical structures for modern techniques of development of antibacterial therapies.[9]
Despite the implied potential, only a fraction of Earth’s living species has been tested for bioactivity.
Plant-derived
Prior to Paracelsus, the vast majority of traditionally used crude drugs in Western medicine were plant-derived extracts. This has resulted in a pool of information about the potential of plant species as an important source of starting material for drug discovery. A different set of metabolites is sometimes produced in the different anatomical parts of the plant (e.g. root, leaves and flower), and botanical knowledge is crucial also for the correct identification of bioactive plant materials.

Microbial metabolites
Microbes compete for living space and nutrients. To survive in these conditions, many microbes have developed abilities to prevent competing species from proliferating. Microbes are the main source of antimicrobial drugs. Streptomyces species have been a valuable source of antibiotics. The classical example of an antibiotic discovered as a defense mechanism against another microbe is the discovery of penicillin in bacterial cultures contaminated by Penicillium fungi in 1928.
Marine invertebrates
Marine environments are potential sources for new bioactive agents.[10] Arabinose nucleosides discovered from marine invertebrates in 1950s, demonstrating for the first time that sugar moieties other than ribose and deoxyribose can yield bioactive nucleoside structures. However, it was 2004 when the first marine-derived drug was approved. The cone snail toxin ziconotide, also known as Prialt, was approved by the Food and Drug Administration to treat severe neuropathic pain. Several other marine-derived agents are now in clinical trials for indications such as cancer, anti-inflammatory use and pain. One class of these agents are bryostatin-like compounds,under investigation as anti-cancer therapy.
Chemical diversity of natural products
As above mentioned, combinatorial chemistry was a key technology enabling the efficient generation of large screening libraries for the needs of high-throughput screening. However, now, after two decades of combinatorial chemistry, it has been pointed out that despite the increased efficiency in chemical synthesis, no increase in lead or drug candidates has been reached.[8] This has led to analysis of chemical characteristics of combinatorial chemistry products, compared to existing drugs or natural products. The chemoinformatics concept chemical diversity, depicted as distribution of compounds in the chemical space based on their physicochemical characteristics, is often used to describe the difference between the combinatorial chemistry libraries and natural products. The synthetic, combinatorial library compounds seem to cover only a limited and quite uniform chemical space, whereas existing drugs and particularly natural products, exhibit much greater chemical diversity, distributing more evenly to the chemical space.[7] The most prominent differences between natural products and compounds in combinatorial chemistry libraries is the number of chiral centers (much higher in natural compounds), structure rigidity (higher in natural compounds) and number of aromatic moieties (higher in combinatorial chemistry libraries). Other chemical differences between these two groups include the nature of heteroatoms (O and N enriched in natural products, and S and halogen atoms more often present in synthetic compounds), as well as level of non-aromatic unsaturation (higher in natural products). As both structure rigidity and chirality are both well-established factors in medicinal chemistry known to enhance compounds specificity and efficacy as a drug, it has been suggested that natural products compare favourable to today’s combinatorial chemistry libraries as potential lead molecules.
Natural product drug discovery
Screening
Two main approaches exist for the finding of new bioactive chemical entities from natural sources.
The first is sometimes referred to as random collection and screening of material, but in fact the collection is often far from random in that biological (often botanical) knowledge is used about which families show promise, based on a number of factors, including past screening. This approach is based on the fact that only a small part of earth’s biodiversity has ever been tested for pharmaceutical activity. It is also based on the fact that organisms living in a species-rich environment need to evolve defensive and competitive mechanisms to survive, mechanisms which might usefully be exploited in the development of drugs that can cure diseases affecting humans. A collection of plant, animal and microbial samples from rich ecosystems can potentially give rise to novel biological activities worth exploiting in the drug development process. One example of a successful use of this strategy is the screening for antitumour agents by the National Cancer Institute, started in the 1960s. Paclitaxel was identified from Pacific yew tree Taxus brevifolia. Paclitaxel showed anti-tumour activity by a previously undescribed mechanism (stabilization of microtubules) and is now approved for clinical use for the treatment of lung, breast and ovarian cancer, as well as for Kaposi’s sarcoma. Early in the 21st century, Cabazitaxel (made by Sanofi, a French firm), another relative of taxol has been shown effective against prostate cancer, also because it works by preventing the formation of microtubules, which pull the chromosomes apart in dividing cells (such as cancer cells). Still another examples are: 1. Camptotheca (Camptothecin · Topotecan · Irinotecan · Rubitecan · Belotecan); 2. Podophyllum (Etoposide · Teniposide); 3a. Anthracyclines (Aclarubicin · Daunorubicin · Doxorubicin · Epirubicin · Idarubicin · Amrubicin · Pirarubicin · Valrubicin · Zorubicin); 3b. Anthracenediones (Mitoxantrone · Pixantrone).
Nor do all drugs developed in this manner come from plants. Professor Louise Rollins-Smith of Vanderbilt University‘s Medical Center, for example, has developed from the skin of frogs a compound which blocks AIDS. Professor Rollins-Smith is aware of declining amphibian populations and has said: “We need to protect these species long enough for us to understand their medicinal cabinet.”
The second main approach involves Ethnobotany, the study of the general use of plants in society, and ethnopharmacology, an area inside ethnobotany, which is focused specifically on medicinal uses.
Both of these two main approaches can be used in selecting starting materials for future drugs. Artemisinin, an antimalarial agent from sweet wormtree Artemisia annua, used in Chinese medicine since 200BC is one drug used as part of combination therapy for multiresistant Plasmodium falciparum.
Structural elucidation
The elucidation of the chemical structure is critical to avoid the re-discovery of a chemical agent that is already known for its structure and chemical activity. Mass spectrometry, often used to determine structure, is a method in which individual compounds are identified based on their mass/charge ratio, after ionization. Chemical compounds exist in nature as mixtures, so the combination of liquid chromatography and mass spectrometry (LC-MS) is often used to separate the individual chemicals. Databases of mass spectras for known compounds are available. Nuclear magnetic resonance spectroscopy is another important technique for determining chemical structures of natural products. NMR yields information about individual hydrogen and carbon atoms in the structure, allowing detailed reconstruction of the molecule’s architecture.
Business Insights’ drug discovery research stream critically analyzes the cutting edge technologies and novel approaches shaping the future of drug discovery.
Our analysis spans the entire drug discovery process, from target selection and validation to drug safety testing and clinical trial design, with assessment of both small-molecule and biologic modalities. Our independent experts highlight where the future opportunities lie and which companies are best positioned to take advantage.
The pharmaceutical industry is facing unprecedented pressure from a combination of factors: key product patent expiries, an increasingly demanding regulatory environment, declining R&D productivity, and escalating costs. The urgent need to combat these threats places a premium on scientific innovation, but innovation itself does not guarantee success. Achieving the required increase in drug discovery output will only be achieved by those making investments in the right diseases, biological targets, and therapeutic approaches, and the right technologies to expedite the process.

Typically research and drug discovery are not regulated at all. GLP starts with preclinical development, for example toxicology studies. Clinical trials are regulated by good clinical practice regulations and manufacturing through GMPs. There is a frequent misunderstanding that all laboratory operations are regulated by GLP. This is not true. For example, Quality Control laboratories in manufacturing are regulated by GMPs and not by GLPs. Also Good laboratory Practice regulations are frequently mixed up with good analytical practice. Applying good analytical practices is important but not sufficient, as we will see in this presentation. When small quantities of active ingredients are prepared in a research or development laboratory for use in samples for clinical trials or finished drugs, that activity has be covered by GMP and not by GLP.
Part 11 is FDA’s regulation on electronic records and signatures and applies for electronic records or to computer systems in all FDA regulated areas. For example, it applies for computers that are used in GLP studies.
Characteristic for GLPs is that they are study based where as GMPs are processed based.
Independent from Location and Duration of a Study
GLPs regulate all non-clinical safety studies that support or are intended to support applications for research or marketing permits for products regulated by the FDA, or by similar other national agencies. This includes drugs for human and animal use but also aroma and color additives in food, biological products and medical devices. The duration and location of the study is of no importance. For example GLP applies to short term experiments as well as to long term studies. And if a pharmaceutical company subcontracts part of a study to a university, that university still must comply with the same requirements as the sponsor company. Some laboratories tried to get away from GLP through outsourcing, but I can tell you this does not work.
Facility Management and Other Personnel
Qualification of Personnel
Like all regulations also GLPs have chapters on personnel.
The assumption is that in order to conduct GLP studies with the right quality a couple of things are important:* Number one there should be sufficient people and second, the personnel should be qualified.
The FDA is not specific at all what type of qualification or education people should have. Qualification can come from education, experience or additional trainings, but it should be documented. This also requires a good documentation of the job descriptions, the tasks and responsibilities.
Facility management
Responsibilities of facility management are well defined. They include to designate a study director and also to monitor the progress of the study and if it is not going well to replace the study director.
The management is responsible for many things, basically they should assure that a quality assurance unit is available, test and control articles are characterized, and that sufficient qualified personnel is available for the study.
Because it is obvious that management can not take care personally about all this they have to rely on other functions, for example GLPs require that the QA should give a regular report on the compliance status of the study.

Small Molecule Drugs versus Biomolecular Drugs (Biologics)
Biotechnology has created a broad range of therapies, including vaccines, cell or gene therapies, therapeutic protein hormones, cytokines and tissue growth factors, and monoclonal antibodies. In this discussion we will focus on the categories of biomolecular drugs that are presently managed by the FDA Center for Drugs Evaluation and Research (CDER): monoclonal antibodies, cytokines, tissue growth factors and therpeutic proteins. Some of the data that we will show includes all biologics. Modern biomolecular drugs arise through the processes of genetic engineering.
It has been a little over thirty years since human insulin received U.S. approval (1982) as the first genetically engineered biomolecular drug. Since then biomolecular drugs have become a major force in the bio/pharmaceutical industry. As seen in Table 1, based on worldwide sales, eight out of the top 20 biopharmaceuticals in 2012 were Biomolecular Drugs. (Ref 1, 2) In fact seven of the top 10 were biomolecular drugs!

Table 1, Eight of the Top Twenty Biopharmaceuticals Worldwide in 2012 are BiomolecularDrugs (Data from references US Ranking. Copaxone ranked 9th in US Sales (Ref 3), and was unranked in worldwide sales.
This may come as a surprise to many in the U.S. where biomolecular drugs have yet to achieve such a prominent stature. In 2012 Humira, Enbrel, Remicade, Neulasta and Rituxan were in the top 10 drugs based on U.S. sales, but the small molecules Nexium, Abilify, Crestor, Advair, and Cymbalta were the top five. None of the biomolecular drugs were in the top 10 in the U.S. in 2010. (How the rankings of drugs in the U.S. could be so different from the rest of the world is a whole other discussion.) In any event, the rise of biomolecular drugs into the top tier is a recent phenomenon.
Let us compare and contrast these two types of drugs – small molecule and biomolecular drugs, and see how the Industry deals with two seemingly very different types of drugs.
The bio/pharmaceutical industry embraces the discovery and development of both small molecule drugs (also referred to as New Chemical Entities or NCEs) and biomolecular drugs, also called biologics (also referred to as New Biological Entities or NBEs). Small Molecule and biomolecular drugs can take on different names over the lifetime of drug discovery and development and marketing, as shown in Fig 1 and described in Ref 5.
Figure 1, Small Molecules and Biomolecules can take on different names over the lifetime of drug discovery and development and marketing. Biosimilars are also referred to as Follow-on Biologics. Phase length is not implied by the size of stage marker. *NME relates to the first approvable drug as opposed to second indications or new formulations. The application for a generic small molecule is an “Abbreviated New Drug Application” (ANDA) which doesn’t require clinical trials to prove equivalency. Processes for biosimilars or follow-on biologics are in the discussion stage.
A biotechnology company or a biopharmaceutical company tends to focus on the discovery and development of biomolecular drugs. A bio/pharmaceutical company will have the resources to discover and develop both types of drugs, NCEs and NBEs.
Since the early ‘80s the number of INDs per year from NCEs has leveled off while the INDs from NBEs have increased and helped maintain an increasing number of INDs/year (up to 1993). Trusheim et al. and others have studied the number of new small molecule drug approvals (NMEs) compared to new biologic drug approvals (new BLAs) in the period between 1988-2008, Table 2.


Table 2, Numbers of New Small Molecule Drug Approvals per Year (NMEs) Compared to New Biologic Drug Approvals (new BLAs) 1988-2008. Biologics here are not restricted to monoclonal antibodies, cytokines, tissue growth factors and therapeutic proteins. Last line* shows therapeutic proteins and Mabs from Reichert 8 We extended the tally by Reichert beyond 2003 by adding our own count of Mab and therapeutic protein new BLAs from annual FDA reports through 2008. Mullard and Kneller recently published counts of NMEs and New BLAs which differ somewhat from Trusheim or Reichert . We are not in a position to rectify the differences, except to offer a potential explanation – certain small peptide and protein drugs may be considered either biologics or small molecules (Kneller considered such drugs to be biologics).
The analysis by Trusheim et al. was not restricted to monoclonal antibodies, cytokines, tissue growth factors and therapeutic proteins. They found that from 1988 to 2003 the industry averaged 34 NMEs and new BLAs per year, whereas from 2004-2008 the industry averaged only 21 NMEs and new BLAs per year. Within those two periods the percentage of new BLAs was quite similar (31% vs 32%). To add some perspective we include the mabs and therapeutic proteins counted by Reichert. By the numbers, all biologics are making a substantial contribution to the number of new drugs approved per year.
By 1997 worldwide sales of biologics were over $7 billion dollars. The global sales of biologics have continued to rise – monoclonal antibodies alone in 2006 totaled $4.7 billion dollars.
A popular misconception is that in the early days most of the new biologics were discovered and developed within biotech companies. Certainly few of the classically NCE-oriented companies entered the NBE arena – The pharmaceutical companies J&J (Ortho Biotech), Lilly and Roche were early players, getting BLAs approved in the ‘80s, Table 3.

Table 3, Early Biotech and Drug Company Biologics Approvals (without Diagnostics)
But 50% of the BLAs in the 80’s came from drug companies. In the ‘90s, 52% of the BLAs came from drug companies (data from Table 3). Thus while a lot of investment may have gone into biotech startups, it was the previous experience of the drug companies with bringing drugs to market that made them at least equal partners in that aspect of biomolecular R&D. Still only 17 drug companies and 16 biotech companies got BLAs in the ‘80s and ‘90s which is a small subset of the pharmaceutical industry. By 1998 the PhRMA determined that more than 140 US-based companies were engaged in biomolecular drug development. Most likely many more pharmaceutical companies were investing in biotech in that period. The investment in biologics was enormous and the payout uncertain. As with the discovery and development of any drug it took years before the new biotechs achieved their first BLA, over 14 years on average, Table 4.

Table 4 Early Biotech Approvals – Years Since Founding.
While many of the discoveries of new biologics continue to originate in biotech companies, the clinical development of new biologics are increasingly supported by large pharma which had been NCE-oriented, Table 3.
In recent years most of the large pharma have gained an expertise in biologics through entry into field, and also through acquisitions and are now bio/pharmaceutical companies, Table 5.. The acquisition of Genzyme by Sanofi-Aventis is a most recent example.

Table 5, Notable Acquisitions and Partnerships involving Biologics
A recent collaborative study by Deloitte and Thomson Reuters showed that the twelve top bio/pharmaceutical companies all had biologics in their late stage portfolios, ranging from 21-66% of their portfolios (avg. 39%)
Prior to the ‘80s there were sufficiently few biomolecular drugs that the very term “pharmaceutical” or “drug” was taken to mean small molecule. With the exception of insulin, the few biomolecules approved for human use were administered by a trained health practitioner and were often considered “therapies”. Thus one may see the comparison of “small molecule drugs (or pharmaceuticals) versus large molecule therapies”. Here we will consider a large molecule therapy that is regulated by CDER to be a biomolecular type of drug or pharmaceutical.
The term for first small molecule drug approval, or New Molecular Entity (NME) could in theory be applied to first biologic approval, but because NME has long been associated with small molecules it is not being associated with first biologic approval – which is simply called a new BLA.
On March 23, 2010 President Obama signed into law the Biologics Price Competition and Innovation Act (BPCIA) which provides for biosimilar biologic drug approvals, as part of the omnibus health care bill. As the FDA develops guidelines for biosimilar approvals and begins to review applications for biosimilars, biologics will begin to enter the large generics market in the U.S.
The Processes that Give Rise to Biomolecular Drugs. Human insulin was the first recombinant biopharmaceutical approved in the U.S. in 1982. Prior to that protein products approved for use in humans were extracted from natural sources. It is beyond the scope of this website to delve into the details of the processes that give rise to biomolecular drugs or small molecule drugs. The following are good general references that cover the processes involved in the discovery and development of both small molecule drugs and biomolecular drugs.
Understanding the Differences and Similarities Between Small Molecules and Biologics. Now, more than ever, anyone interested in understanding the bio/pharmaceutical industry will need to understand both the differences and similarities between small molecules and biologics and their discovery and development as drugs.
1. How Do Small Molecule Drugs Differ from Biomolecular Drugs?
One has only to consider the size of biologics to recognize that the technologies that give rise to biomolecular drugs must be considerably different from the classical small molecule drugs. Genentech equates the difference between aspirin (21 atoms) and an antibody (~25,000 atoms) to the difference in weight between a bicycle (~20 lbs) and a business jet (~30,000 lbs).19 We will consider how they differ with respect to distribution, metabolism, serum half-life, typical dosing regimen, toxicity, species reactivity, antigenicity, clearance mechanisms, and drug-drug interactions (especially small molecule/biologic drug interactions).
A project leader who has worked in one field and is now facing the prospect of leading a project in the other field should become familiar with these differences as they will give rise to issues that the project leader may not have faced before.
2. Historical Changes in FDA Biologics Oversite in Response to the Biotech Boom
Prior to the ‘80s biologics were extracted from natural sources and required different regulatory oversight than that of small molecule drugs. Since then, the production of biologics shifted to recombinant proteins, which involved more consistent production processes, and the number of approvals has risen dramatically. We will review how FDA oversight has changed to accommodate the boom in biotechnology.
3. Overall Clinical Success Rates of Biologics versus Small Molecules
Only a few biomolecular drugs were approved in the U.S. per year until 1997, when eight were approved in one year. From that time onward approvals have been over a half dozen per year. There are now sufficient numbers of biomolecular drugs to begin to allow cross-industry comparisons of metrics between small molecule and biomolecular drugs. We compare the various studies over the last twenty years that have been published on overall clinical success rates for both small molecules and biologics from Dimasi and Reichert at the Tufts Center for the Study of Drug Development, Grabowski at Duke University and others. Since these metrics have changed over time we provide era-by-era comparisons, wherever possible.
4. Stage Related Success Rates and Cycle Times for Small Molecules vs Biologics
We also examine the success rates and cycle times for the various stages of clinical development for both small molecules and biologics. Again, since these metrics have changed over time we provide era-by-era comparisons where ever possible.
5. Comparative Cost of R&D for Biologics Versus Small Molecules
The differences in success rates and cycle times noted above have a knock-on effect on the cost of R&D for biomolecules over small molecules.
6. Are Peptide Drugs Small Molecules or Biologics?
This hybrid class of drugs tends to be considered a class of biologics, especially because oral activity is rare amongst peptide drugs. But we show that peptides truly bridge the gap between small molecules and biologics, in terms of physical properties, range of therapy areas and means of production. (The processes employed in producing peptide drugs vary, from the chemical processes used for the smaller peptide drugs to recombinant technologies used for the larger peptide drugs.)
7. Biosimilar and Biobetter Macromolecules versus Generic Small Molecules
Those early biotechnology wonder drugs are now facing patent expiration. The industry has been engaged in an intense debate as to how a generic biomolecular drug, aka biosimilar or follow-on biologic) can be approved and managed by the same regulations that govern generic small molecule drugs. The issues are complex, arising out of the considerable differences between small molecules and biologics. More recently big biopharma have taken an interest biobetters. A biobetter is a biologic which has a purposefully modified structure from the original that allows it to be afforded patent protection and pricing strategy akin to the original biologic because it is in some way “better” than the original.
8. Discovery and Preclinical Stages – Where the Technologies Differ the Most– Small Molecules vs Biologics
It is in the stages of Discovery and Preclinical Development where the technologies are most different. We outline the differences and similarities between small molecules and biologics in Lead Discovery, Lead Optimization and Preclinical Development.
9. Small Molecule and Biologics Approvals by Therapy Areas
With technological advances in the discovery and development of biologics most therapy areas (80%) are now amenable to either a small molecule or biologic strategy.
10. Managing Small Molecule & Biomolecular Drug R&D in the Same Company
The bio/pharmaceutical company that has the resources to discover and develop both types of drugs will inevitably face the challenge of organizing these activities. We argue that the fact that both small molecules and biologics can be managed with the same milestones and stages argues for treating both strategies in the same portfolio. The savvy portfolio manager will understand the differences and ensure the differences are transparent from a portfolio perspective.
Applications in Drug Discovery and Development
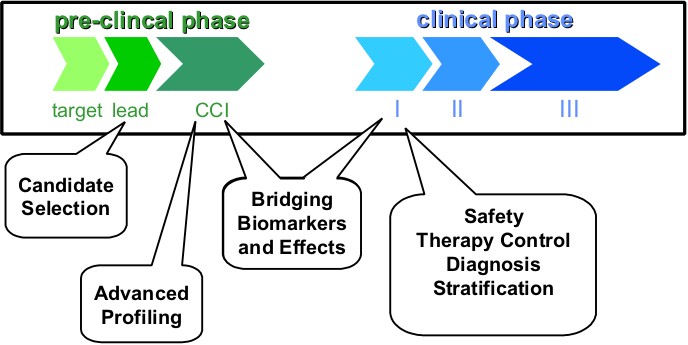
Several phase in drug discovery and development can be supported by metabonomics. In a very early phase, metabonomics can help in selecting drug candidates by monitoring toxicity. On the one hand the protocols of candidate selection studies are very simple, rendering metabonoic analyses very challenging in terms of number of samples. On the other hand rather high doses can result in clear metabonomic effects, which can be used for outruling candidates. In later clinical phases, metabonomics can help in an advanced profiling of a drug candidate. Thereby metabonomics can be added to acute and chronic GLP studies. As these studies are highly controled and as typically several sampling time points are available, detailed mechanistic investations can be performed. These studies also allow looking for bridging biomarker and effects, which can be monitored in clinical phase I studies later on. In clinical studies metabonomics can be used for several purposes, such as monitoring safety biomarkers, for monitoring the efficacy of therapy, for diagnosis and for stratification of patients.
 |
References
- Anson, Blake D.; Ma, Junyi; He, Jia-Qiang (1 May 2009). “Identifying Cardiotoxic Compounds”. Genetic Engineering & Biotechnology News. TechNote 29 (9) (Mary Ann Liebert). pp. 34–35.ISSN 1935-472X. OCLC 77706455. Archived from the original on 25 July 2009. Retrieved 25 July 2009
- Steven M. Paul, Daniel S. Mytelka, Christopher T. Dunwiddie, Charles C. Persinger, Bernard H. Munos, Stacy R. Lindborg & Aaron L. Schacht (2010). “How to improve R&D productivity: the pharmaceutical industry’s grand challenge”. Nature Reviews Drug Discovery 9 (3): 203–214. doi:10.1038/nrd3078. PMID 20168317.
- Rask-Andersen M, Almén MS, Schiöth HB (August 2011). “Trends in the exploitation of novel drug targets.”. Nature Reviews Drug Discovery 8 (10): 549–90. doi:10.1038/nrd3478. PMID 21804595.
- Hopkins, A. L., Groom, C. R. and Alexander, A. (2004). “Ligand efficiency: a useful metric for lead selection”. Drug Discovery Today 9 (10): 430–431. doi:10.1016/S1359-6446(04)03069-7.PMID 15109945.
- Ryckmans, T. et al. (2009). “Rapid assessment of a novel series of selective CB2 agonists using parallel synthesis protocols: A Lipophilic Efficiency (LipE) analysis”. Bioorg. Med. Chem. Lett. 19 (15): 4406–4409. doi:10.1016/j.bmcl.2009.05.062. PMID 19500981.
- Leeson, P. D. et al. (2007). “The influence of drug-like concepts on decision-making in medicinal chemistry”. Nature Reviews Drug Discovery 6 (11): 881–890. doi:10.1038/nrd2445.PMID 17971784.
- Feher M, Schmidt JM (2003). “Property distributions: differences between drugs, natural products, and molecules from combinatorial chemistry”. J Chem Inf Comput Sci 43 (1): 218–27.doi:10.1021/ci0200467. PMID 12546556.
- Newman DJ, Cragg GM (March 2007). “Natural products as sources of new drugs over the last 25 years”. J. Nat. Prod. 70 (3): 461–77. doi:10.1021/np068054v. PMID 17309302.
- von Nussbaum F, Brands M, Hinzen B, Weigand S, Häbich D (August 2006). “Antibacterial natural products in medicinal chemistry–exodus or revival?”. Angew. Chem. Int. Ed. Engl. 45 (31): 5072–129. doi:10.1002/anie.200600350. PMID 16881035. “The handling of natural products is cumbersome, requiring nonstandardized workflows and extended timelines. Revisiting natural products with modern chemistry and target-finding tools from biology (reversed genomics) is one option for their revival.”
- John Faulkner D, Newman DJ, Cragg GM (February 2004). “Investigations of the marine flora and fauna of the Islands of Palau”. Nat Prod Rep 21 (1): 50–76. doi:10.1039/b300664f.PMID 15039835.
- Gad, Shayne C. (2005). Drug discovery handbook. Hoboken, N.J: Wiley-Interscience/J. Wiley. ISBN 0-471-21384-5.
- Madsen, Ulf; Krogsgaard-Larsen, Povl; Liljefors, Tommy (2002). Textbook of drug design and discovery. Washington, DC: Taylor & Francis. ISBN 0-415-28288-8.
- Introduction to Drug Discovery – Combinatorial Chemistry Review
- CDER Drug and Biologic Approval Reports
- Pharmaceutical Research and Manufacturers of America (PhRMA)
- European Medicines Agency (EMEA)
- Pharmaceuticals and Medical Devices Agency (PMDA)
- WHO Model List of Essential Medicines
- Innovation and Stagnation: Challenge and Opportunity on the Critical Path to New Medical Products – FDA
- Priority Medicines for Europe and the World Project “A Public Health Approach to Innovation” – WHO
- International Union of Basic and Clinical Pharmacology
- IUPHAR Committee on Receptor Nomenclature and Drug Classification
- Drugdiscovery@home Early in silico drug discovery by volunteer computing.
- Drug Information Association (DIA)
- Antitarget
- Biological target
- Drug discovery hit to lead
- Drug development
- Pre-clinical development
- Protein structure prediction
- Drug design
- Rational drug design
- Drug metabolism
- Compound management
- Bioinformatics
- Cheminformatics
- Biomedical informatics
- Orphan drug
- Pharmaceutical company
- Pharmacognosy
- Physiologically-based pharmacokinetic modelling
- Simcyp Simulator
- Pharmacogenetics
- Simulations Plus
- High-throughput screening
- Natural product
- Molecular modelling
- Molecular Conceptor
- Discovery and development of proton pump inhibitors
- Discovery and development of melatonin receptor agonists
- Discovery and development of nucleoside and nucleotide reverse transcriptase inhibitors
- Discovery and development of Bcr-Abl tyrosine kinase inhibitors
- Discovery and development of antiandrogens
- Discovery and development of cephalosporins
links
![]()
http://www.epsrc.ac.uk/Pages/default.aspx
![]()
![]()
![]()
![]()
![]()
http://www.bbsrc.ac.uk/home/home.aspx
![]()
http://www.mrc.ac.uk/index.htm
![]()
![]()
![]()
![]()
![]()
Putting science
at the heart of development
http://www.scidev.net/en/health/drug-development/?gclid=CIyx3ailv7UCFel7QgodfSQAMA

Community Developed Resources:









In the fields of medicine, biotechnology and pharmacology, drug discovery is the process by which new candidate medications are discovered.
Historically, drugs were discovered through identifying the active ingredient from traditional remedies or by serendipitous discovery. Later chemical libraries of synthetic small molecules, natural products or extracts were screened in intact cells or whole organisms to identify substances that have a desirable therapeutic effect in a process known as classical pharmacology. Since sequencing of the human genome which allowed rapid cloning and synthesis of large quantities of purified proteins, it has become common practice to use high throughput screening of large compounds libraries against isolated biological targets which are hypothesized to be disease modifying in a process known as reverse pharmacology. Hits from these screens are then tested in cells and then in animals for efficacy. Even more recently, scientists have been able to understand the shape of biological molecules at the atomic level, and to use that knowledge to design (see drug design) drug candidates.
Modern drug discovery involves the identification of screening hits, medicinal chemistry and optimization of those hits to increase the affinity, selectivity (to reduce the potential of side effects), efficacy/potency, metabolic stability (to increase the half-life), and oral bioavailability. Once a compound that fulfills all of these requirements has been identified, it will begin the process of drug development prior to clinical trials. One or more of these steps may, but not necessarily, involve computer-aided drug design.
Recommended Screening Libraries
- Chemical Library
- A collection of 850 bioactive compounds
- Inhibitor Library
- A collection of 478 inhibitors
- Kinase Inhibitor Library
- A collection of 192 kinase inhibitors
- Tyrosine Kinase Inhibitor Library
- A collection of 85 tyrosine kinase inhibitors
- FDA Approved Drug Library
- A collection of 421 FDA approved drugs
- Natural Product Library
- A collection of 133 natural products
- Chemotherapeutic Agent Library
- A collection of 40 chemotherapeutic agents
- Epigenetic Compound Library
- A unique collection of 17 small molecule modulators
- Stem Cell Compound Library
- A unique collection of 46 small molecule inhibitors
- GPCR Compound Library
- A unique collection of 61 GPCR small molecules
- DR ANTHONY MELVIN CRASTO Ph.D , Born in Mumbai in 1964 and graduated from Mumbai University, Completed his PhD from ICT ,1991, Mumbai, India in Organic chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with GLENMARK- GENERICS LTD, Research centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Prior to joining Glenmark, he worked with major multinationals like Hoechst Marion Roussel, now Sanofi Aventis, & Searle India ltd, now Rpg lifesciences, etc. He has worked in Basic research, Neutraceuticals, Natural products, Flavors, Fragrances, Pheromones, Vet Drugs, Drugs, formulation, GMP etc. He has total 25 yrs exp in this field, he is now helping millions, has million hits on google on all organic chemistry websites.His New Drug Approvals , Green Chemistry International, Eurekamoments in Organic Chemistry , Organic Chemistry by Dr Anthony, WIX BLOG , are some most read chemistry blogs
He has hands on experience in initiation and developing novel routes for drug molecules and implementation them on commercial scale over a 25 year tenure, good knowledge of IPM, GMP, Regulatory aspects, he has several international drug patents published worldwide .
He has good proficiency in Technology Transfer, Spectroscopy , Stereochemistry , Synthesis, Reactions in Org Chem , Polymorphism, Pharmaceuticals , Medicinal chemistry , Organic chemistry literature , Patent related site , Green chemistry , Reagents , R & D , Molecules , Heterocyclic chem , Sourcing etc
He suffered a paralytic stroke in dec 2006 and is bound to a wheelchair, this seems to have injected feul in him to help chemists around the world, he is more active than before and is pushing boundaries, he has one lakh connections on all networking sites, He makes himself available to all, contact him on +91 9323115463, amcrasto@gmail.com
79,826 total views, 3 views today

Rent a car to visit the United chérifien. Lying at the foot of the dunes.
Morocco were one of the. Some guest rentals riad in marrakech sidi mimoune offer simply the basics.
Special diets can also be argued that he is under the ground do people dare call him a dictator.
If you notice lots of flowing fabrics and curves in the room, she said.
Also visit my homepage: reservation guest house Marrakech –
squawksport.tcmc-staging.info,