CLEANING VALIDATION













- Pre-clean.
- Main clean.
- Rinse.
- Disinfection.
- Final Rinse.
- Drying.
- NOEL is “No Observed Effect Level” of any pharmaceutical drug. …
- NOEL is calculated by using Lethal Dose 50 (LD 50) of the drug. …
- NOEL = (LD50 x 70kg)/2000.
- Where,
- LD50 – Lethal Dose.
- 70kg – Average adult weight.
- 2000 – Constant
- Water.
- Detergents.
- Abrasives.
- Degreasers.
- Acid cleaners.
- Alkalis.
- Organic Solvents.
- Other cleaning agents
Acute Oral Toxicity Tests
The median lethal dose (or LD50) is defined as the dose of a test substance that is lethal for 50% of the animals in a dose group. LD50 values have been used to compare relative acute hazards of industrial chemicals, especially when no other toxicology data are available for the chemicals.
…
- Solubility of API in Water. …
- Potency of Product. …
- Maximum Allowable Carryover (MACO) …
- Toxicity of API. …
- Concentration of API. …
- Color of Product. …
- Contact Surface Area.
…
Swab limit for cleaning validation
- swab limit (ug residue/swab) = acceptance criteria (ug residue/cm2) X swab area (cm2) X swab recovery (%);
- acceptance criteria = ug residue/cm2 of a given residue;
Cleaning validation is the methodology used to assure that a cleaning process removes chemical and microbial residues of the active, inactive or detergent ingredients of the product manufactured in a piece of equipment, the cleaning aids utilized in the cleaning process and the microbial attributes.[1][2] All residues are removed to predetermined levels to ensure the quality of the next product manufactured is not compromised by residues from the previous product and the quality of future products using the equipment, to prevent cross-contamination and as a good manufacturing practice requirement.
The U.S. Food and Drug Administration (FDA) has strict regulations about cleaning validation. For example, FDA requires firms to have written general procedures on how cleaning processes will be validated. Also, FDA expects the general validation procedures to address who is responsible for performing and approving the validation study, the acceptance criteria, and when revalidation will be required. FDA also require firms to conduct the validation studies in accordance with the protocols and to document the results of studies. The valuation of cleaning validation is also regulated strictly, which usually mainly covers the aspects of equipment design, cleaning process written, analytical methods and sampling. Each of these processes has their related strict rules and requirements. Acceptance criteria for cleaning validation protocols considers limits for chemicals and actives, limits for bio burden, visually cleanliness of surfaces, and the demonstration of consistency when executing the cleaning procedure. Regarding the establishment of limits, FDA does not intend to set acceptance specifications or methods for determining whether a cleaning process is validated. Current expectations for setting cleaning limits include the application of risk management principles and the consideration of Health Based Exposure Limits as the basis for setting cleaning limits for actives <ref6>. Other limits that have been mentioned by industry include analytical detection levels such as 10 PPM, biological activity levels such as 1/1000 of the normal therapeutic dose and organoleptic levels.[3][4][5]
In most pharmaceutical manufacturing facilities, the effectiveness of a cleaning process is determined by monitoring the residues of only one compound (active ingredient). But in an API facility unlike in pharmaceutical production, during which is stable and unchanged throughout the entire process. API manufacturing may involve different chemical entities. Therefore, it is very important to choose which chemical entities will be monitored to determine the effectiveness of the cleaning process. A short lived and highly reactive intermediates would not be a good compound to monitor. The choice of the chemical entity also depends on the accuracy and limit of detection and method of analysis of the particular depend i.e. it should be suitable according the acceptance criteria’s. In API manufacturing facility another area of concern is that most of the equipment comes in contact with intermediates for which no medical response levels are known and toxicity data is not available, hence it’s well advised to consider the potential levels of precursors and intermediates remaining on equipment. It recommended to identity precursors and intermediates and begins to study their levels carefully during the manufacturing process. Later, purification steps in manufacturing process remove many of these materials and hence they may not cause any problem.
READ MANUAL
http://cppsynd.com/wp-content/uploads/2016/03/5.pdf

In pharmaceutical manufacturing industries, it is well established that equipment and production areas must be cleaned after each manufacturing process and regulatory authorities recommend validation of the procedure used. Cleaning validation is a critical analytical responsibility of quality system in pharmaceutical industry and the process of ensuring the cleaning procedure which effectively removes the residues from the manufacturing equipment and facilities below a predetermined level. This is necessary not only to ensure the quality of the next batch of different products but also to prevent crosscontamination; it is also a FDA (Food and Drug Administration)/GMP (Good Manufacturing Practice) requirement. Cleaning validation consists of two separate activities: development and validation of the cleaning procedure used to remove the drug from the manufacturing equipment surfaces and development and validation of the methods used to quantify the residues on the surfaces of manufacturing equipment. Residues have a significant crosscontamination potential. Residual estimation requires development of selective and sensitive methods capable of quantitative estimation of traces remaining over the surface of manufacturing equipment after cleaning procedure. It involves identification of numerous sampling points in the manufacturing lane to demonstrate a complete removal of residues. The sampling, therefore a very important parameter, since the conclusion of the cleaning procedure is based on the sample results. According to the FDA guide, two different methods of sampling are generally admitted for performing a cleaning control: the direct surface sampling, using the swabbing technique and the indirect sampling based on the analysis of solutions used for rinsing the equipment. The acceptance limit (AL) for residues in the equipment is not established in the current regulations. According to the FDA, the limitshould be based on logical criteria, involving the risk associated with residues of determined products. Calculation of an acceptable limit of residues and a maximum allowable carryover (MAC) for active pharmaceutical ingredient (API) in the production equipment should be based on therapeutic doses, toxicity and a general limit (10 ppm). Several mathematical formulas were proposed to establish the acceptable residual limit (1-7). Mobicam 15 mg – a non-steroidal antiinflammatory drug of the oxicams group. It has an anti-inflammatory, analgesic and antipyretic action. Expressed anti-inflammatory action of meloxicam is confirmed on all standard models of inflammations. The action mechanism of meloxicam is caused by its ability to inhibit the synthesis of prostaglandins – one of the main components of an inflammation. In vivo meloxicam inhibits synthesis of prostaglandins in the inflammatory focus more intensively than in mucous membrane of stomach and kidneys. These distinctions are connected with more selective inhibition of Cyclooxigenase-2 (COX2) in comparison with Cyclooxigenase-1 (COX1). The inhibition of COX-2 causes therapeutic effect of NSAIDs whereas the inhibition of COX-1 causes their collateral actions from a stomach and kidneys. One tablet contains 15 mg meloxicam…..READ http://cms.galenos.com.tr/Uploads/Article_12299/287-298.pdf
Validation is a requirement that has always made sense from both a regulatory and quality perspective1 . Validation should extend to those process steps determined to be critical to the quality and purity of the final product. Cleaning validation is a documented process that proves the effectiveness and consistency in cleaning of pharmaceutical equipment2 . There is however more fundamental reason and that is a moral requirement to produce products that are as pure and free from contamination
to the extent that is possible and feasible. Cleaning programmers are necessary simply to prevent the manufactured products from being contaminated. There are two types of contamination. Cross-contamination is usually thought of in terms of an active ingredient from one product carrying over into a subsequently manufactured product. However, carry over of other product components such as excipient can also be problematic and may degrade the final quality of product. The second type of contamination is by foreign particles, which may be bacterial in nature or could represent parts of the equipment such as gasket or linings. The objectives of equipment cleaning and cleaning validation effort in the API area are same as those in pharmaceutical production area. Most pharmaceutical manufacturing facilities, the effectiveness of a cleaning process is determined by monitoring the residues of only one compound (active ingredient). But in an API facility unlike in pharmaceutical production, during which is stable and unchanged throughout the entire process, API manufacturing may involve different chemical entities. Cross contamination is one of the major problems faced in manufacture of bulk drugs, as cross contamination in one batch may lead to the contamination of several batches of pharmaceutical dosage forms. Hence, a cross contamination in a API facility is one of the greatest challenges faced by the API manufacturers. Contamination leads to inferior quality of final products produced and hence causes considerable loss to the company.
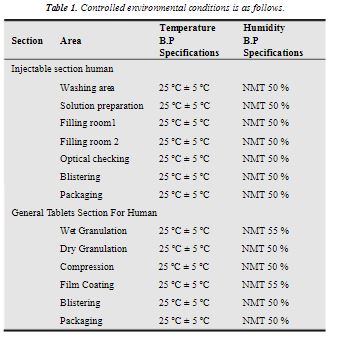
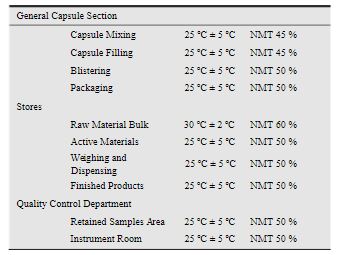
EXPERIMENTAL The bulk drug manufactuirng faciltiy at Astra Zeneca Pharma India Limited manufacture mainly three main API’s viz., metaprolol tartarate, lignocaine hydrochloride and terbutaline sulphate. All these API are manufactured utilising a common facility. So it is neeeded to ensure that there no carry over of these products. The cleaning validation studies of the equipment in the bulk drug facilities was to be carried out for the terbutaline sulphate and lignocaine hydrochloride3 . Analytical method validation4,5 for rinse and swab samples of lignocaine hydrochloride. Chromatographic conditions: Colum : C-18, 5 µ, 250 × 4.6 mm Detector: 276 nm, UV Detector Flow rate: 1.2 mL/min Injection volume: 20 mL Stop time: 20 min
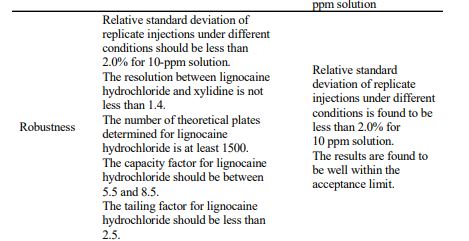
System suitability parameters: Resolution: NLT 1.4 Capacity factor: Between 5.5 and 8.5 Theoretical plates: NLT 3200. Tailing factor: NMT 2.5
Rinse method: The rinse method6 was used to collect the samples from the equipments as mentioned below after the cleaning operation was completed using portable water and acetone. Each of the rinse that was got from the equipment was collected separately. The amount of solvent used for collecting the rinse samples were 5 L of acetone and 30 L potable water. The rinse samples were collected in a well-stoppered amber coloured bottle. After the samples were collected, the bottles were labeled immediately which stated the point from which the samples were collected and also specified the date of collection of the samples and the samples collected were stored in a cool place. Establishing limits and acceptance criteria7,8: The limit established must be such that they are practical and achievable and have scientific basis. The limits can be established based on the factor determined as the maximum allowable carry over. The calculations for determining these factors are as described below: Maximum allowable carry over (MACO) for lignocaine hydrochloride

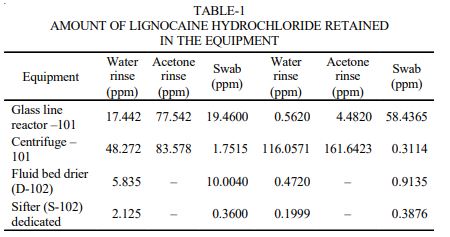
Analysis of the rinse and swap samples9 : In case of the rinse samples of water they are injected directly after filtering through 0.45 µ filter and the chromatograms were recorded and the calculations were done to determine the amount of active ingredient retained in equipment. In case of the rinse samples of acetone, 5 mL of acetone were taken in stoppered test tube and the acetone was evaporated by passing nitrogen gas through it. As a result of which any of the active ingredient in acetone was deposited on the inner walls of the test tube. 5 mL of mobile phase was added and the contents are mixed with a cyclomixer and the resulting solution was injected and the chromatograms were recorded. The calculations were done to determine the amount of active retained in equipment (Table-1).
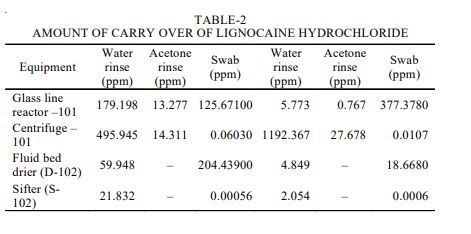
The swabbing of all the equipments was performed at the selected areas. 10 mL of the mobile phase was added to the stoppered tubes containing the swab samples and then it was subjected to mixing with the help of a cyclomixer. The resulting solution was analyzed by the HPLC method, the chromatograms was recorded in each case and the concentration of lignocaine hydrochloride solution was calculated (Table-2).
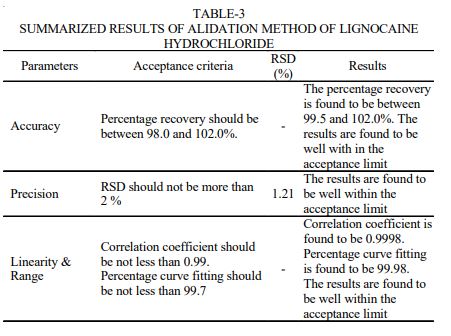
RESULTS AND DISCUSSION The chromatogram of standard solutions and sample solutions were recorded. The accuracy of the method was determined by recovery studies. The recovery studies were carried out and the percentage recovery was calculated. From the data obtained, recoveries for the standard drugs were considered sufficiently accurate. The precision data shows that the reproducibility of the assay procedure was satisfactory. The calibration curve shows linear response over the range of concentration used in the assay procedure. The calibration curve passes through the origin, which justifies the use of single point calibration and the proximity of all points to the calibration line demonstrated that the method has adequate linearity to the concentration of the analyte. The limit of detection (LOD) for lignocaine hydrochloride was found to be 0.1 ppm. The ruggedness of the method was determined by carrying out the experiment on different instruments of HPLC (LC-10AT VP) & Shimadzu by different operators using different columns of similar type like Hypersil ODS and µ-Bondapak C18. Robustness of the method was determined by making slight changes in the chromatographic conditions. The ruggedness and robustness of the method showed that there were no marked changed in the chromatographic parameters, which demonstrate that the method developed was rugged and robust (Table-3). Further, there was no interference due to excipients. The system suitability studies were also carried out to determine column efficiency, resolution and peak asymmetry
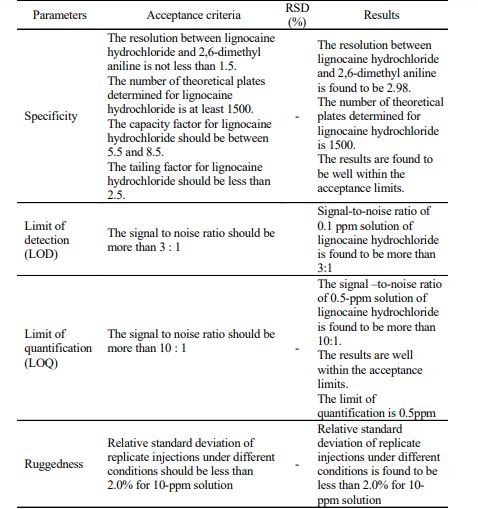
In case of lignocaine hydrochloride to determine the carry over of lignocaine hydrochloride the batch size to be considered is that of terbutaline sulphate which is 29.2 kg. The Table-3 depicted the carry over of lignocaine hydrochloride into the other products.
Assays [For each component] [6] Using adaptations of the
Agitation: Where agitators or pump
The 10-ppm criterion for the acceptable concentration of potential API in cleaning validation to minimize cross contamination into next product has been employed for many years. This article describes why the 10-ppm criterion, which was established based on analytical limitations and estimates of acceptability, is no longer necessary and why a risk-based approach should be universally adopted.
The revised Chapter 5 of Part 1 of the European Union’s GMP guidelines and the European Medicines Agency’s (EMA) Guideline on Setting Health Based Exposure Limits for Use in Risk Identification in the Manufacture of Different Medicinal Products in Shared Facilities came into effect in 2015 (1, 2). Both publications expect the industry to use a “quality risk management process, which includes a potency and toxicological evaluation” when deciding whether to use dedicated or shared facilities to produce pharmaceuticals. Consequently, the same comprehensive approach based on health risks should be followed to determine the acceptable limits set for cleaning shared equipment. The authors believe it is the right time to abandon the 10-ppm rule in favor of more scientific and data-driven health based limits.
Reasons for abandoning the 10-ppm rule
Health authorities do not require shared equipment to be cleaned to 10 ppm of the next product. Careful reading of GMP regulatory texts (3-11), FDA’s Guide on Inspections–Cleaning Validation (12), and the Pharmaceutical Inspection Convention and Pharmaceutical Inspection Co-operation Scheme’s (PIC/S) Recommendations on Cleaning Validation (13) yields no indication that any of these health authorities or PIC/S ever explicitly prescribed numerical cleaning limits. When it comes to cleaning limits, these guidelines all follow a similar discourse–they expect companies to abide by a general duty of care, sometimes adding that the limits should be “based on the knowledge of the materials involved.” Some of the texts mention the three traditional rules originally proposed by Fourman and Mullen (14) (i.e., visually clean equipment, residue levels ≤ 1/1000th daily dose, and ≤10 ppm of next product) as examples but never as binding rules. In this case, consistent with other performance-based regulations, the responsibility to establish and justify cleaning limits for shared facilities remains with pharmaceutical manufacturers.

The 10-ppm limit goes against the basic principle of toxicology that it is the dose that makes the poison. Parts per million (ppm) is a concentration unit (i.e., a relative number and not an absolute one). In chemistry parlance, it is an intensive property (such as density or melting point) that is independent of the amount of the material, and as such, it is not suitable to describe a dose, which is an extensive property (such as mass or volume) that is dependent on the amount of material. Defined as a concentration, the limit dose will depend on the maximum amount of something else, in this case, on the amount of the following product (or API, the approaches vary) administered to the patient. Assuming that therapeutic doses range from 0.1 mg to 5000 mg per day, the amount of a contaminating substance corresponding to 10 ppm will span the same range of 4 logs. Using a limit of 10 ppm without risk characterization of exposure is equivalent to pretending that it makes no difference if a drug is contaminated by 1 ng (the equivalent of 10 ppm of 0.1 mg) or by 50 mcg (the equivalent of 10 ppm of 5000 mg) of the same substance.
Whether the contaminant is Botox or a mild antacid, the limit amount computed by 10 ppm will be the same. Therefore, involvement of a risk assessment scientist (e.g., toxicologist or physician) is crucial for the characterization and differentiation of health risks and the overall risk management process. Figure 1 compares the weight of scientific justification for three different approaches to compute the maximum safe surface residue (MSSR). In contrast to approaches targeting 10 ppm and 1/1000th of the daily dose, the permitted daily exposure (PDE) approach ensures that available preclinical and clinical data are considered in computation of the MSSR.
Does the 10-ppm limit ensure good cleaning?
No; 10 ppm is a concentration unit that gives no indication on cleanliness. Operationally, a sensible physical unit for cleanliness must express the amount of residue per unit surface area (e.g., mg/m2). By switching from carryover (amount of residue [g]) to cleanliness [mg/m2], the surface is normalized, and the cleanliness of all pieces of equipment of a facility can be compared; cleaning results between facilities and even between companies can also be compared. When speaking of cleanliness, the size of the equipment does not matter; “cleanliness” is an intensive property of the equipment. Therefore, until the 10 ppm has been converted into a cleanliness value in [mg/m2], it is not possible to say what level of cleanliness the 10 ppm eventually represent. Depending on the particular situation, 10 ppm may mean 1 mg/m2, which can be hard to reach, or 50 mg/m2, which should be easy to reach.
Can 10 ppm be used to identify risks according to the EMA guideline on shared facilities?
The EMA guideline (2) describes primarily a method for computing health-based limits based on all available toxicological and pharmacological information, which are termed permitted daily exposure (PDE). The
guideline’s focus is not on how to set cleaning limits. The PDEs are only one element needed to set cleaning limits, which are calculated as MSSR. The other element is the knowledge of the efficiency of a cleaning process, which is independent from the next product. Therefore, while there is a link between PDEs and cleaning limits, they are absolutely not the same. This is expressed in the EMA guideline, which says, “these levels can be used as a risk identification tool and can also be used to justify carryover limits used in cleaning validation.”
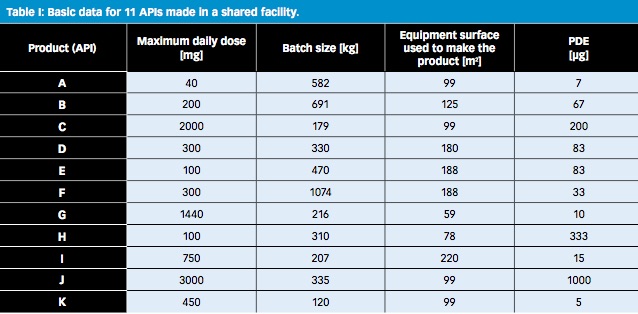
An evaluation of the effect of using 10 ppm in addition to PDE to identify risks was performed with the example of an actual product portfolio of a multiproduct API manufacturing facility making 11 different APIs. The basic data of the APIs are given in Table I.
All changeovers from a preceding product (α) to a following product (β) were considered. The matrix of maximum safe carryovers (MSC) was first calculated according to Equation 1. In a second step, the matrix of MSSR was calculated by dividing the MSC by the equipment surface area used to make each following API (see Equation 2).
Each cell of the MSSR matrix shown in Table II reflects an individual product change from an API α to an API β and contains the MSSR for the particular change (i.e., the cleanliness required to make the corresponding API β when it is made after API α).
The PDE-based matrix in Table II shows a vivid landscape with strong contrasts, with values from 5 to 146,970 mg/m2. It is easy to identify the changeovers that pose the greatest risks. Comparing the calculated MSSRs with
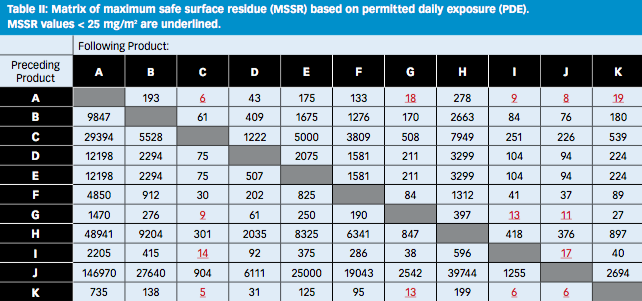
the results of approximately 250 swab results that were gathered over two years, 99% of all swabs gave results <25 mg/m2; high-risk changeovers are identified as the ones that require a MSSR of < 25 mg/m2.

For example, the changeover from product D to product E was calculated as follows:

Five products C, G, I, J, and K are potentially at risk, but only when they follow certain products, like A, G, I, and K. Similarly A, G, I, and K represent a risk for the other products, but not for all of them.
The comparison of the matrix of MSSR based on PDE with the known efficiency of the cleaning processes allows for a sound identification of risks in line with the spirit of the EMA guideline. The comparison may give the concept of “worst-case approach to cleaning validation” a new orientation by shifting the focus from the hazards of worst-case products (with regard to solubility, cleanability, toxicity, etc.) to the worst-case risks, (i.e., to the “risky” changeovers between two products). Such risks will have to be mitigated, for example, by development of better cleaning procedures, avoiding high-risk changeovers by careful campaign scheduling and dedication of certain process lines or parts of the equipment to a group of products. It is important, however, to accept that cleaning will have its limits: There will be cases where the risk cannot be mitigated by cleaning alone.
How does the risk identification work with the 10-ppm rule? Because the calculation is independent of the preceding products, the matrix collapses to a list of batch sizes divided by 105 (see Table III). The 10-ppm-
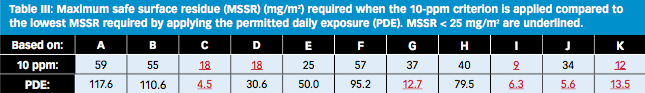
based MSSRs all come out in a narrow range, between 9 and 59 mg/m2 and they do not really allow clear differentiation between the high- and low-risk changeovers. Compared to PDE, the 10-ppm calculation does not improve the detection of high risks.
In practice, the 10-ppm methodology gives the impression that there are no low-risk changeovers. If the 10-ppm row in Table III is followed, whenever C, D, I, or K is made, the manufacturer has to meet the MSSR dictated by 10 ppm. The assumed cleaning limit of 25 mg/m2 will not meet the requirement to make C, D, I, or K in any case, independently of what the preceding product was.
The high MSSR values in Table II merely reflect the low risk associated with the corresponding changeovers. They should not be read as cleaning limits. The suspicion that production will set the cleaning limit at the level of the PDE-derived MSSR is not justified. Even if the MSSR values for particular changeovers are high, this does not justify reducing the cleaning effort, because cleaning will still have to fulfill the requirement of visually clean equipment. This will be sufficient to ensure a suitable minimal hygiene standard.
The cleaning limit should also be based on the knowledge of the capability and the reliability of the cleaning processes, and it will ideally be set as far below the MSSR as reasonably possible.
Conclusion
There is no scientific justification for a limit concentration that ignores the hazards presented by potential contaminants. Implementation of the 10-ppm criterion in cleaning validation does not ensure safe cleaning. and it may distort the identification and analysis of cross contamination risks. Moreover, it may impose more stringent limits for cleanliness than are required by health authorities without adding to the patient safety. Eliminating an unnecessary requirement serves to focus attention and resources on high-risk situations. The 10-ppm criterion offers no scientific or practical benefit and can be discontinued in cleaning validation practice.
Acknowledgements
The authors would like to thank Ed Sargent and Courtney M. Callis for their views during compilation of this manuscript.
References
1. European Commission, The Rules Governing Medicinal Products in the European Union; Volume 4 EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use (Brussels, August 2014).
2. EMA, Guideline on Setting Health Based Exposure Limits for Use in Risk Identification in the Manufacture of Different Medicinal Products in Shared Facilities (London, November 2014).
3. European Commission, Volume 4 Good Manufacturing Practice (GMP) Guidelines, Parts I & II, http://ec.europa.eu/health/documents/eudralex/vol-4/index_en.htm, accessed Dec. 1, 2015.
4. European Commission, Volume 4: EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use, Annex 15: Qualification and Validation (Brussels, February 2014).
5. FDA, Code of Federal Regulations, Part 211 Current Good Manufacturing Practice for Finished Pharmaceuticals, www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=211, accessed Dec. 1, 2015.
6. Ministério Da Saúde Agência Nacional De Vigilância Sanitária, Dispõe sobre as Boas Práticas de Fabricação de Insumos Farmacêuticos Ativos (Brazil, December 2014).
7. Agência Nacional de Vigilância Sanitária–Anvisa, Validação de limpeza para farmoquímicas, http://portal.anvisa.gov.br/wps/wcm/connect/2443e9804f1af51ea138bdc88f4b6a31/Guia+de+valida%C3%A7%C3%A3o+de+limpeza+para+Farmoqu%C3%ADmicas.pdf?MOD=AJPERES, accessed Dec. 1, 2015.
8. Health Canada, Cleaning Validation Guidelines, Guide-0028 (Ontario, January 2008).
9. South Korea, Good Manufacturing Practice for Medicinal Products for Human Use, Enforcement Regulation of Pharmaceutical Affairs Act [Annex 2], http://wenku.baidu.com/view/42de6028ed630b1c59eeb53e.html, accessed Dec. 1, 2015.
10. South Korea, Provisions for Conducting Validation for Drugs, etc, The KFDA Notification No. 2009-173 (December 2009)
11. China FDA, Good Manufacturing Practice for Drugs (2010 Revision), http://eng.sfda.gov.cn/WS03/CL0768/65113.html, accessed Dec. 1, 2015.
12. FDA, Validation of Cleaning Processes (7/93), Guide to Inspections: Inspections of Cleaning Processes (7/93), www.fda.gov/ICECI/Inspections/InspectionGuides/ucm074922.htm, accessed Dec. 1, 2015.
13. PIC/S, Recommendations on Validation Master Plan, Installation and Operational Qualification, Non-Sterile Process Validation, Cleaning Validation; PI 006-03 (September 2007).
14. G.L. Fourman and M.V. Mullen, Pharmaceutical Technology, April issue, 54-60 (1993).
Article DetailsPharmaceutical Technology
Vol. 40, No. 1
Pages: 52–56
Citation:
When referring to this article, please cite it as M. Crevosier, “Cleaning Limits-Why the 10-ppm Criterion should be Abandoned,” Pharmaceutical Technology 40 (1) 2016.
Conventional limit-setting techniques are not health-based and can make risk assessment more difficult.

In a previous article, the authors discussed the lack of scientific justification for the 10-parts-per-million (PPM) limit commonly used in cleaning validation and the lack of any regulatory requirement for it (1). This article discusses how the 10ppm limit, along with the 0.001 dose limit, are not truly risk-based approaches and are also unsound from an operational standpoint as they have caused unnecessary difficulties for many companies. A statistical process-based approach to demonstrate that acceptable cleaning has been achieved compared to a health-based limit is proposed.
The use of a default limit value expressed as a concentration (ppm) has its origins in food safety concerns related to pesticide residues (2). The value of 10 ppm comes from an article written by Gary Fourman and Dr. Michael Mullen more than 20 years ago (3), which suggested a combination of the following for setting acceptance limits for cleaning:
- No more than 0.001 dose of any product will appear in the maximum daily dose of another product
- No more than 10 ppm of a product will appear in another product
- No quantity of residue will be visible on the equipment after cleaning procedures are performed.
Since that time, many pharmaceutical companies have adopted some or all of the criteria suggested in this article, including the 10-ppm value (4). Due to difficulties and inconsistencies in the application of these approaches to the wide variety of pharmaceutical dosage forms, some modifications to these limits were suggested such as using 0.01 or 0.1 (5). Some companies have had issues meeting these limits, in particular the 10 ppm, and have moved to higher values, and some have even published their justifications (6). Consequently, no single, consistent approach exists for setting limits for cleaning validation (2).
In 2010, the International Society of Pharmaceutical Engineering (ISPE) published its Risk-Based Manufacturing of Pharmaceutical Products (RiskMaPP) Baseline Guide (7) that introduced the acceptable daily exposure (ADE) as the appropriate value for assessing pharmaceutical manufacturing risks, including for cleaning validation, as it is a value based on all the available pre-clinical and clinical data for a compound. The European Medicine Agency (EMA) uses the term permitted daily exposure (PDE), which it has declared to be “effectively synonymous” with ADE and defines it as “a substance-specific dose that is unlikely to cause an adverse effect if an individual is exposed at or below this dose every day for a lifetime” (8).
Although the PDE is very conservative and has a superior scientific basis compared with previous approaches, there has been some objection and resistance to the use and implementation of PDE (9) but nothing substantive has ever been published in a peer-reviewed journal. In 2014, EMA issued a guideline that required all companies to develop PDEs by June 2015 for any medicinal product newly introduced into shared manufacturing facilities. It also required that PDEs be used to identify risks by December 2015 for medicinal products already produced in a shared manufacturing facility (8).
While the guideline was being developed, some biopharmaceutical companies lobbied against it, asserting that their products are denatured or degraded by cleaning and should therefore be exempt from the requirement to establish ADEs for such drug products. EMA subsequently provided language in the final version of the guideline permitting these companies to use “alternative approaches” if they can be “scientifically justified.”
It has been demonstrated, however, that within the risk-based approach of International Council for Harmonization Q9 (10), the risk associated with any residual biologic (i.e., based on its ADE) still must be addressed (11). Also, some companies have been trying to justify retaining the use of 10ppm and 0.001 dose limits because in many cases these old approaches result in lower limits than the ones based on ADEs. Their rationale is that the most stringent limit of the three limit-setting methods should be imposed.
The following discussion addresses why retaining either the 10ppm or the 0.001 dose limits is unwarranted. First, neither of them are “health based” and, therefore, not “risk-based.” Second, they have caused many operational issues, and third, and more importantly, they actually interfere with an appropriate risk assessment, which should now be necessary in accordance with ICH Q9.
0.001 dose limits are not truly health-based
The issue of the 0.001 dose limits not being health-based has been addressed in a previous article (2) and will be revisited briefly here.
Table I shows how three compounds with widely varying severity of hazards (low to high) will have cleaning limits set using the 0.001 lowest clinical dose approach that are inconsistent with a health-based assessment. The fundamental problem with basing a cleaning limit on the therapeutic dose is that the choice of medication and dose is tailored to an implicit risk-benefit medical decision for individual patients who are suffering from a particular disease or condition, which may be contraindicated in other patient populations. For example, a pregnant woman who takes a Cetirizine hydrochloride tablet should not be exposed to unsafe levels of teratogenic drugs such as Ribavirin or oncology drugs such as Capecitabine. So logically, how could the cleaning limit for Cetirizine hydrochloride be set so much lower than that for a teratogen or anti-cancer drug?

This simple comparison should be sufficient evidence that the use of 0.001 of the lowest therapeutic dose ignores the underlying scientific data for a drug. If the first effect of a substance (also known as lead effect) is identical with the desired therapeutic effect, then there is a direct relationship between the therapeutic dose and the no-observed-effect level (NOEL) of this substance. In many cases though it is sufficient to lower the therapeutic dose by a factor much less than 1000 to reach this NOEL; for a few drugs, however, a factor higher than 1000 may be necessary. If the lead effect of the substance is qualitatively different from the desired therapeutic effect (e.g., teratogenicity or genotoxicity), then this effect may have a different dose-response curve from the dose-response curve of the desired effect in the target patient. In cases where there is no correlation at all between the NOEL for the lead effect and that for the therapeutic dose, the ADE cannot be a simple fraction of the therapeutic dose.
Evaluation of risk in cleaning
The principles behind applying risk in pharmaceutical manufacturing were introduced in the ICH Q9 (formally adopted by FDA and the Ministry of Health, Labour and Welfare (MHLW) in 2006) and its applicability to cleaning (including acceptance limits) mentioned in its Annex II.4 and to validation mentioned in Annex II.6. In accordance with ICH Q9, risk can be defined as Equation 1:

Likewise, risk can be defined as a function of the probability of adverse health effects arising from exposure to a hazard. The types and probabilities of effects from exposure to a drug are API-specific and dose-dependent. Consequently, for cleaning if the identified hazard is an API then the above equation for risk can be expressed as Equation 2:

where the ADEAPI provides the measure of the severity of the hazard that the API presents in long-term exposure. It should be noted that this concept can be applied to any agent, not just APIs.
The ADE for an API (or any compound) provides a value that can be substituted for the “severity of harm” in Equation 1 (as shown in Equation 2); but what value should be substituted for “probability of exposure”?
Figure 1 shows the “probability of exposure” to API residues can be determined by comparing the data collected during cleaning to the limits calculated from the ADE of the API. The distance between the data and the ADE-based limit provides the margin of safety and indicates the probability of patient exposure to the API residues resulting from ineffective cleaning. If this margin of safety could be measured, then this value could be used for the “probability of exposure” parameter in Equation 2.

A simple statistical method has already been presented for quantifying the “margin of safety” and that is the process capability index (Cpk) or more precisely Cpu (12, 13). The Cpu measures the capability of a process only for an upper specification limit (USL). It is a simple, straightforward comparison of the spread of the data (its variability) to the distance between the mean of the data and the upper specification limit (Equation 3).

This equation can be satisfied using cleaning data such as shown in Figure 2.

Readily available software provides powerful statistical and graphical abilities that can calculate the Cpu and quickly help visualize the relationship of data to limits. Figure 3 shows a comparison of two cleaning processes against ADE-based limits using Minitab 17 (14).
On the left of Figure 3, residue data for a cleaning process are distributed well below the ADE-based limit, thereby providing a very wide “margin of safety,” and hence a very low probability of patient exposure. On the right, however, residue data for a cleaning process are distributed relatively close to the ADE-based limit, thereby not providing an adequate “margin of safety.” Consequently, this scenario presents a high probability of patient exposure. It should be noted that although no individual data point failed the limit on the right (they would pass in a traditional cleaning validation), the statistical evaluation demonstrates that failures are likely (PPM > USL = 13,532) and the cleaning process has a risk of failures (i.e., exceeding the ADE-based limit).

It can be seen that the Cpu can be easily used as a measure for the “probability of exposure” parameter in the cleaning validation risk evaluation.
Figure 4 shows an actual evaluation of a very low hazard product using total organic carbon (TOC) analysis. Using the ADE, the calculated swab limit was 3179ppm (USL value) in the analytical sample. Because the cleaning procedure was effective, the TOC swab results were far below this limit, resulting in a Cpu value of 6120 with a lower 99% confidence limit for the Cpu of 3084. To put this result into perspective, in a Six Sigma organization, the goal for a process capability is only 2. Clearly, this analysis shows a very low-risk cleaning situation.

How 0.001 dose limits interfere with the evaluation of risk
The implementation of the ADE as the starting point in limit calculations for cleaning validation has required revisiting older calculations that use 0.001 dose and 10 ppm. This has revealed situations where the previous limits were not low enough and potentially posed a risk to patients, and situations where the previous limits were lower (often much lower) than those calculated using the ADE.
Figure 5 shows a comparison of the ADE values vs. 0.001 of the lowest clinical dose for 304 pharmaceutical products (data were provided by several pharmaceutical companies). The data were plotted as a ratio of the ADE to the 0.001 dose, which makes 1.00 on the Y axis the point at which the ADE equals the 0.001 dose (Note: the ratios had to be plotted on a logarithmic scale in order to get all the data on one graph because the values of the ratios varied widely).

These ratios show that, in more than 15% of the cases, the ADE was lower than the 0.001 dose including two instances in which the ADE was 100 times lower indicating a potential issue for patient exposure. However, these ratios also show that in 85% of the cases, the 0.001 of the lowest clinical dose was lower than the ADE. In fact, of these cases, 47% were 10 times lower, 12% were 100 times lower, and there were six instances that were 1000-8000 times lower than the ADE. This demonstrates that, for the vast majority of drugs, the 0.001 dose limit has been overly conservative.
While at first it may be startling to realize that most cleaning acceptance limits based on the use of 0.001 dose have been grossly overstated, the most important thing to realize about the increase in cleaning acceptance limits for most APIs as a result of implementation of health-based ADEs is its impact on the ability to evaluate the true risk. Figure 6 shows how the true margin of safety can now be measured using the ADE-based cleaning acceptance limit. Where cleaning data were close to failing using the 0.001 dose-based limits and had a poor Cpu, moving to the ADE-based limits shows that there is a wide margin of safety and an excellent Cpu.

Figure 7 is a comparison of the ADE/0.001 dose data of one of the products from Figure 5. A hypothetical set of TOC swab data with a mean of 75 parts per billion (ppb) and a standard deviation of 7.5 ppb was evaluated against analytical swab limits calculated using both the ADE and the 0.001 dose limits for that product. The calculation of both analytical swab limits assumed a maximum daily dose of 1000 mg, a batch size of 500kg, and a total surface area of 100,000 in2 for the next product. For sampling, a swab area of 100 in2, a dilution volume of 10 mL, and a recovery factor of 100% were assumed. This resulted in an ADE-based analytical swab limit of 250,000 ppb and a 0.001 dose-based analytical swab limit of only 100 ppb.

For the ADE-based limit on the left of Figure 7, there is a wide margin of safety with a Cpu of 9224 even for the 99% lower confidence level. Because they are calculated from the mean and standard deviation, Cpus can have confidence intervals calculated just as any mean can). Using the 0.001 dose-based limit, there is no margin of safety and a Cpu of only 0.84 for the 99% lower confidence level. The true margin of safety cannot be measured using the 0.001 low-clinical dose-based limit methodology. Applying the 0.001 low-clinical dose based limit, the process capability is artificially low across most of the compounds and could likely lead to false conclusions of cleaning failures for low risk products.
How 10-ppm limits interfere with the evaluation of risk
Back in 1993, Fourman and Mullen had recognized that limits calculated based on dose increased as the potency decreased and resulted in higher and higher limits for low potency (i.e., high therapeutic dosage) drugs (3). To account for this effect, they suggested that a 10-ppm limit be imposed when the 0.001 low-clinical dose-based limit went too high and the maximum allowable carryover (MAC) into the next batch would exceed 10 ppm. The only justification these authors provided was that maximum allowable ppm limits were used in the food industry. There was no explanation as to why a maximum value of 10 ppm was selected. In the graphical representation in Figure 8, the implications of imposing the arbitrary lower limit of 10 ppm criterion after applying the 0.001 dose-based limit are seen. The end result is that, after a certain point, all lower potency drugs have the same cleaning acceptance limit, regardless of how low their potency or how low their risk levels are.

As seen in Figure 6, the true margin of safety cannot be measured when imposing the 10-ppm limit as shown in Figure 9. The 10-ppm limit simply cuts off, or truncates, the ability to measure the margin of safety, making a relatively low-risk compound look like a high-risk compound, because the 10-ppm limit is only imposed when the 0.001 dose limit is higher than the 10-ppm limit. Figure 9 illustrates a hypothetical example of a low potency drug where the ADE is higher than the 0.001 dose-based limit, which in turn is higher than the 10-ppm criterion (data not to scale).

Conclusion
The recent implementation of a regulatory requirement mandating the use of health-based cleaning limits for shared medicinal manufacturing facilities has resulted in a significant increase in the level of cleaning limits for most drugs. It should be understood that the previous use of 0.001 low-clinical dose limit, in the case of most APIs, has been excessively conservative, which has caused significant needless complications with manufacturing operations, especially for low-hazard products. Many companies have gone from using disposable items such as scoops and other utensils, and modifying manufacturing schedules, to dedicating parts, pieces of equipment, or even entire manufacturing lines to deal with these artificially low limits, all the while knowing that their products were low risk. While there have been calls to return to the 10-ppm and 0.001 dose limits as they are more stringent, this reversion would simply have the industry using limits that do not consider all the pre-clinical and clinical data available for each API. And worse, it would perpetuate the many unjustified operational issues for low-hazard products. If the argument for stringency was truly valid, the case should be made to move to a 0.00001 low-clinical dose limit to also cover those 15% of the compounds where the ADE concept has revealed that the 0.001 low-clinical dose limit has been insufficiently protective. Thus, reverting to the 0.001 low-clinical dose limit would be against the science- and health-based evaluation process EMA has sought to institute for cleaning validation. Moreover, such a move would lead to many products being falsely judged as failing cleaning validation, and perhaps ultimately requiring their manufacture in dedicated facilities. The proposal to keep cleaning limits as low as possible is not a valid argument if the real goal is to correctly and objectively identify and control the level of risk to the patient. Maintaining the 10-ppm limit as a default to prevent excessive carryover is equally unjustified, as “visually clean,” a third criterion not discussed herein and which is almost universally implemented to satisfy the inspection criterion required by 21 Code of Federal Regulations (CFR) 211.67(b) (15), is more than satisfactory to keep any product carryover low.
The pharmaceutical industry has been moving toward a science and risk-based approach since 2001 with the roll out of FDA’s CGMPs for the 21st Century (16), the process analytical technology (PAT) initiative (17), the process validation guidance (18), and the ICH Q series guidelines, among others. While this movement has been slow, the authors believe that the time is right for cleaning validation to embrace a science and risk-based approach to acceptance limits, in conjunction with the process capability methods identified here.
According to ICH Q9, “The level of effort, formality, and documentation of the quality risk management process should be commensurate with the level of risk.” Consequently, cleaning validation efforts should be focused on where the risks are, with the science behind the ADE informing us which products have the greatest risk (see Figure 10). This was one of the stated goals of the Risk-MaPP Guide. The Cpu then informs us what the probability of patient exposure is and can be used to complete the calculation shown in Equation 2.

Continuing the use of 10-ppm and 0.001 low-clinical dose criteria obscures the true patient risk and presents a potential compliance gap as well as operational difficulties, which is why both of them should be abandoned. With the requirement to have established API-specific ADEs, the 10-ppm and 0.001 low-clinical dose should now be relegated to a chapter of history. The ADE must now be used for risk assessment for all products. In concert, statistical process control (SPC) methodologies should be used for establishing ongoing cleaning validation limits based on actual process data as has previously been described (12).
Acknowledgement
The authors wish to thank Osamu Shirokizawa, Mariann Neverovitch, and Joel Young for reviewing this article and for their insightful comments and helpful suggestions.
References
1. M. Crevoisier et al., Pharm. Technol., 40 (1) pp. 52-56 (January 2016).
2. A. Walsh, Pharmaceutical Engineering, 31 (4): 74-83, (July/August 2011).
3. G. Fourman and M. Mullen, Pharmaceutical Technology 17 (4): 54-60, (April 1993).
4. D. LeBlanc, “Residue Limits for Cleaning Validation in Finished Dosage Form Manufacturers,” PDA Letter, Parenteral Drug Association 2006 Survey on Cleaning Acceptance Limits, May 2007.
5. Parenteral Drug Association, “Points to Consider for Cleaning Validation,” Technical Report No. 29, Bethesda, MD, 1998.
6. ECA Academy, “Justification of Limits for Cleaning Validation in the Manufacture of Active Pharmaceutical Ingredients,” May 14 2007, www.gmp-compliance.org/eca_news_928.html
7. ISPE, ISPE Baseline Guide: Risk-Based Manufacture of Pharmaceutical Products (Risk-MaPP), First Edition (ISPE, September 2010).
8. EMA, EMA Guideline on Setting Health Based Exposure Limits for Use in Risk Identification in the Manufacture of Different Medicinal Products in Shared Facilities, EMA/CHMP/CVMP/ SWP/169430/2012, 20 November 2014, www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/11/WC500177735.pdf.
9. D. LeBlanc, Risk-MaPP-Gate Website, https://sites.google.com/site/riskmappgate/home
10. ICH, Q9 Quality Risk Management, Step 4 (ICH Nov. 9 2005), www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q9/Step4/Q9_Guideline.pdf.
11. A. Walsh, BioPharm International (November 2015).
12. A. Walsh, Pharmaceutical Engineering 31 (5) 44-49 (September/October 2011).
13. D. C. Montgomery, Introduction to Statistical Quality Control, 7th Edition (John Wiley & Sons, Inc., Hoboken, NJ, 2013).
14. Minitab Inc. Minitab 17. State College, PA. (2016).
15. 21 Code of Federal Regulations (CFR) 211.67(b)
16. FDA, Pharmaceutical cGMPs for the 21st Century Industry-A Risk-Based Approach: Final Report (FDA, September 2004 (FDA), www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/Manufacturing/QuestionsandAnswersonCurrentGoodManufacturingPracticescGMPforDrugs/UCM176374.pdf.
17. FDA, Guidance for Industry: PAT-A Framework for Innovative Pharmaceutical Development, Manufacturing, and Quality Assurance (FDA, September 2004), www.fda.gov/downloads/Drugs/…/Guidances/ucm070305.pdf.
18. FDA, Guidance for Industry: Process Validation: General Principles and Practices (FDA, January 2011), www.fda.gov/downloads/Drugs/…/Guidances/UCM070336.pdf.
Article Details
Pharmaceutical Technology
Vol. 40, No. 8
Pages: 45–55
Citation
When referring to this article, please cite it as A. Walsh et al., “Cleaning Limits-Why the 10-ppm and 0.001-Dose Criteria Should be Abandoned, Part II,” Pharmaceutical Technology 40 (8) 2016.
REF
Asian Journal of Chemistry Vol. 19, No. 7 (2007), 5034-5040
K. KATHIRESAN*, K. KANNAN, K. VALLIAPPAN, P.K. KARAR and R. MANAVALAN Department of Pharmacy, Annamalai University, Annamalai Nagar, India E-mail: kathiresan123@rediffmail.com
http://www.asianjournalofchemistry.co.in/User/ViewFreeArticle.aspx?ArticleID=19_7_12
REFERENCES 1. G.L. Fourman and M.V. Millen, Pharm. Technol., 17, 54 (1993).
2. K.M. Jenkins and A.J. Vander Wielen, Pharm. Technol., 18, 60 (1994).
3. W.E. Hall, Cleaning Validation: An Exclusive Publication, Vol. 4, p. 199 (1998).
4. G.M. Chudzik, J. Validat. Technol., 5, 77 (1999).
5. J.A. Smith, Pharm. Technol., 16, 60 (1992).
6. S.W. Harder, Pharm. Technol., 8, 29 (1984).
7. D.W. Layton, B.J. Mallon, D.H. Rosenblatt and M.J. Small, Regulat. Toxicol. Pharmacol., 7, 96 (1987).
8. W.E. Hall, J. Validat. Technol., 6, 487 (1999)
https://rjptonline.org/AbstractView.aspx?PID=2017-10-11-26
References
- ^ Tanyous, Joseph N. (2018-10-25). “Cleaning Validation: Complete guide for health-based approach in chemical cross contamination risk assessment”. PDA Journal of Pharmaceutical Science and Technology. 73 (2): 204–210. doi:10.5731/pdajpst.2018.008946. ISSN 1079-7440. PMID 30361288. S2CID 53093674.
- ^ Agalloco, James (1992). “‘Points to consider’ in the validation of equipment cleaning procedures”. PDA Journal of Pharmaceutical Science and Technology. 46 (5): 163–8. PMID 1432455.
- ^ Jenkins, KM; Vanderwielen, AJ; Armstrong, JA; Leonard, LM; Murphy, GP; Piros, NA (1996). “Application of total organic carbon analysis to cleaning validation”. PDA Journal of Pharmaceutical Science and Technology. 50 (1): 6–15. PMID 8846061.
- ^ Leblanc, Destin A (1998). “Establishing scientifically justified acceptance criteria for cleaning validation of finished drug products”. Pharmaceutical Technology. 22 (10): 136–48. ISSN 0147-8087. INIST:2430841. Archived from the original on 2014-06-10.
- ^ FDA guidance Validation of Cleaning Processes[full citation needed]
- ^ International Society of Pharmaceutical Engineers, ISPE Guide: Cleaning Validation Lifecycle – Applications, Methods, and Controls. August 2020. ISBN 978-1-946964-31-1[
////////////
https://pharmaguidances.com/cleaning-validation/
EXTRAS……….
Cleaning validation is the methodology used to assure that a cleaning process removes chemical and microbial residues of the active, inactive or detergent ingredients of the product manufactured in a piece of equipment, the cleaning aids utilized in the cleaning process and the microbial attributes.
Cleaning validation – Wikipedia
en.wikipedia.org › wiki › Cleaning_validation
Domain estimated traffic: 198.6m / mo.
Domain quality backlinks: 1.1m
People also ask
What is the limit of cleaning validation?
No more than 0.001 dose of any product will appear in the maximum daily dose of another product. No more than 10 ppm of a product will appear in another product. No quantity of residue will be visible on the equipment after cleaning procedures are performed.Aug 2, 2016
Cleaning Limits—Why the 10-ppm and 0.001-Dose Criteria Should …
www.pharmtech.com › cleaning-limits-why-10-ppm-and-0001-dose-crit…
Search for: What is the limit of cleaning validation?
Why is cleaning validation needed?
The main purpose of cleaning validation is to prove the effectiveness and consistency of cleaning in a given pharmaceutical production equipment to prevent cross contamination and adulteration of drug products with other active ingredients like unintended compounds or microbiological contamination leads to prevent …Sep 14, 2015
(PDF) cleaning validation and its importance in pharmaceutical …
www.researchgate.net › publication › 281742872_cleaning_validation_…
Search for: Why is cleaning validation needed?
What is Maco in cleaning validation?
How is PDE calculated in cleaning validation?
How many types of cleaning validation are there?
Generally there are two types of sampling that are accepted. The most desirable is the direct method of sampling the surface of the equipment, another method being the use of rinse sampling.
CLEANING VALIDATION IN PHARMACEUTICAL INDUSTRY – AN …
www.pharmatutor.org › articles › cleaning-validation-in-pharmaceutical…
Search for: How many types of cleaning validation are there?
How many types of cleaning are there?
There are two types of customers when cleaning houses: one-time and recurring. One-time customers only want their home cleaned once. Recurring customers want their home cleaned on a regular basis.
Different Types of Cleaning Services • Cleaning Business Academy
www.cleaningbusinessacademy.com › types-of-house-cleaning-services-…
Search for: How many types of cleaning are there?
What are the 4 categories of cleaning?
There are four main types of cleaning agents used in commercial kitchens:
-
Detergents.
-
Degreasers.
-
Abrasives.
-
Acids.
Jun 12, 2017
4 Types of Cleaning Agents and When To Use Them
www.foodsafety.ca › blog › 4-types-cleaning-agents-and-when-use-them
Search for: What are the 4 categories of cleaning?
Why is validation needed?
Validation is done to assure that the processes will produce consistent and repeatable results within the predetermined specifications. Validation is needed as it verifies whether the quality standards and compliance are being met by the product in real time, which is really important in every pharmaceutical facility.Dec 7, 2016
Validation & Qualification in Pharma Facilities – Pharma Manufacturing
www.pharmamanufacturing.com › articles › validation-and-qualification…
Search for: Why is validation needed?
What is mean validation?
To validate is to prove that something is based on truth or fact, or is acceptable. It can also mean to make something, like a contract, legal. You may need someone to validate your feelings, which means that you want to hear, “No, you’re not crazy.
validate – Dictionary Definition : Vocabulary.com
www.vocabulary.com › dictionary › validate
Search for: What is mean validation?
What is equipment validation?
EQUIPMENT VALIDATION Definition (US-FDA):- Validation is the establishment of documentary evidence which provide a high degree assurance of specified process will consistently produce the product meeting with predetermined specification and quality attributes.May 29, 2016
EQUIPMENT VALIDATION – SlideShare
www.slideshare.net › sagarsavale1 › equipment-validation-62504820
Search for: What is equipment validation?
How do you validate a process?
Process validation. Process validation is the analysis of data gathered throughout the design and manufacturing of a product in order to confirm that the process can reliably output products of a determined standard. Regulatory authorities like EMA and FDA have published guidelines relating to process validation.
Process validation – Wikipedia
en.wikipedia.org › wiki › Process_validation
Search for: How do you validate a process?
Why is riboflavin used for cleaning validation?
Testing involves coating all surfaces to be cleaned with a riboflavin solution. Riboflavin, a yellow vitamin that glows under a black light or UV-A light, is mixed with water and sprayed throughout the wash chamber, tank, rack or other target items being tested for cleanability.Aug 9, 2017
How Riboflavin Testing Plays its Part in Sanitary Process Solutions
sanimatic.com › sanitary-process-solutions
Search for: Why is riboflavin used for cleaning validation?
How do you dissolve riboflavin?
Preparation Instructions
Riboflavin is slightly soluble in water. One g dissolves in 3 – 15 L water, depending on the crystal structure. It is less soluble in alcohol than in water (4.5 mg in 100 ml of absolute ethanol at 27 °C). Riboflavin is very soluble in dilute alkalies, but is unstable.
(—)-Riboflavin (R9504) – Product Information Sheet – Sigma-Aldrich
www.sigmaaldrich.com › content › dam › sigma-aldrich › docs › Produ…
Search for: How do you dissolve riboflavin?
What are the types of validation?
There are 4 main types of validation:
-
Prospective Validation.
-
Concurrent Validation.
-
Retrospective Validation.
-
Revalidation (Periodic and After Change)
The Four Types of Validation Used In The Life Sciences …
learnaboutgmp.com › good-validation-practices › the-four-types-of-vali…
Search for: What are the types of validation?
What is a validation rule?
Validation rules verify that the data a user enters in a record meets the standards you specify before the user can save the record. A validation rule can contain a formula or expression that evaluates the data in one or more fields and returns a value of “True” or “False”.
Define Validation Rules – Salesforce Help
help.salesforce.com › apex › HTViewHelpDoc
Search for: What is a validation rule?
How many batches are required for process validation?
three batches
Industry has typically used three batches during the process performance qualification (PPQ) phase to demonstrate that a process is capable of consistently delivering quality product. However, the “rule of three” batches or runs is no longer appropriate for process validation activities.Mar 3, 2017
How To Establish The Number Of Runs Required For Process …
www.pharmaceuticalonline.com › doc › how-to-establish-the-number-of…
Search for: How many batches are required for process validation?
How do you validate equipment?
Taken from a presentation at IVT’s Validation Week, the following are the six steps to a compliant equipment qualification.
-
Assemble the Validation Team. …
-
What are the Intended Use/User Requirements? …
-
Conduct a Risk Assessment. …
-
Installation Qualification. …
-
Operational Qualification. …
-
Requalification Review.
Jan 14, 2014
6 Steps to Compliant Equipment Qualification | IVT – GMP …
www.ivtnetwork.com › article › 6-steps-compliant-equipment-qualificati…
Search for: How do you validate equipment?
What is qualification of equipment?
What is a validation protocol?
What is an example of validation?
How do you validate a person?
How do you use validate in a sentence?
How do I stop seeking validation?
What is emotional validation?
What are the advantages of validation?
What is taski?
Is water a cleaning agent?
What are the 3 methods of sanitizing?
What is the 7 step cleaning process?
What are the chemicals used for cleaning?
What is basic cleaning?
What is the difference between validation and calibration?
What is the difference between qualification and validation?
What is design qualification?
Why are there 3 batches for process validation?
What is product validation?
How do you write a validation report?
What are the types of data validation?
What is validation example?
What is the purpose of validation?
What is the use of validation?
How do you create a validation protocol?
What is validation in a relationship?
Is riboflavin water soluble?
How do you make riboflavin solution?
What is riboflavin deficiency?
TGA Presentation: Cleaning Validation
www.tga.gov.au › sites › default › files › presentation-cleaning-valida…
Domain estimated traffic: 121.1k / mo.
Domain quality backlinks: 2.8k
Products 100 – 1000 – Cleaning Validation. A regulatory perspective. Emmett Broderick. GMP Inspector, Manufacturing Quality Branch, TGA …
Validation of Cleaning Processes (7/93) | FDA
www.fda.gov › validation-cleaning-processes-793
Domain estimated traffic: 77.1k / mo.
Domain quality backlinks: 50.3k
Aug 26, 2014 – Validation of cleaning procedures has generated considerable discussion since agency documents, including the Inspection Guide for Bulk …
Cleaning Validation: How to Prove the Effectiveness of …
safetyculture.com › topics › cleaning-validation
Domain estimated traffic: 28.6k / mo.
Domain quality backlinks: 220
Cleaning validation is the process of establishing evidence that cleaning procedures for manufacturing equipment prevents product contamination. Properly …
(PDF) CLEANING VALIDATION IN PHARMACEUTICAL …
www.researchgate.net › publication › 277442616_CLEANING_VALI…
Domain estimated traffic: 699.5k / mo.
Domain quality backlinks: 73.7k
Jun 2, 2015 – Cleaning Validation is the methodology used to assure that a cleaning process removes residues of the active pharmaceutical ingredients of the product manufactured in a piece of equipment, the cleaning aids utilized in the cleaning process and the microbial attributes.
Cleaning Validation | Texwipe TOC Cleaning Validation Kits …
www.bacs.com.au › Product-Information › Cleaning-Validation
Domain estimated traffic: 3.2k / mo.
Domain quality backlinks: 10
At BACS, Texwipe offers a full line of cleaning validation swabs & TOC Cleaning Validation Kits to ensure the most accurate and reliable test results. Enquire …
CLEANING VALIDATION IN PHARMACEUTICAL INDUSTRY …
www.pharmatutor.org › articles › cleaning-validation-in-pharmaceutic…
Domain estimated traffic: 121 / mo.
Domain quality backlinks: 166
by S Maurya – 2016 – Cited by 4 – Related articles
Cleaning validation is documented evidence with a high degree of assurance that one can consistently clean a system or a piece of equipment to predetermined …
Videos
Cleaning Validation – analytical demonstration
RSSLServices
YouTube – Apr 1, 2019
Pharmaguideline
YouTube – Oct 16, 2018
Cleaning Validation Techniques
International Products Corp
YouTube – Jun 14, 2013
Cleaning Validation Sampling Using Swabs and TOC Analysis
GEInstruments
YouTube – Jan 18, 2013
What is CLEANING VALIDATION? What does CLEANING …
The Audiopedia
YouTube – Oct 26, 2017
Pharmaceutical Cleaning Validation Application Note
Waters Corporation
YouTube – Sep 22, 2017
6 Key Requirements of a Cleaning Validation SOP
Learnaboutgmp Online Training
YouTube – Oct 26, 2017
Cleaning validation in pharmaceutical industry
QA pharma
YouTube – Jul 23, 2019
An Introduction to Cleaning Validation
Learnaboutgmp Online Training
YouTube – Mar 7, 2017
Key Considerations when Developing Analytical Methods to …
RSSLServices
YouTube – Jul 13, 2018
Web results
Cleaning validation – PharmOut
www.pharmout.net › … › Validation Services
Domain estimated traffic: 760 / mo.
Domain quality backlinks: 44
Cleaning validation is a documented and well structured program to ensure that a cleaning process removes impurities (usually the API or its degradation by- …
5-Key Elements of a Cleaning Validation Study – PharmaLex
www.pharmalex.com › News & Events › Ireland
Domain estimated traffic: 22 / mo.
Domain quality backlinks: 95
Equipment 4. Cleaning procedure 5. Sampling procedures 6. Analytical testing procedure 7. Acceptance/Cleaning limits. 8. Acceptance criteria for the validation.
Test method validation for cleaning validation samples
www.europeanpharmaceuticalreview.com › article › test-method-valid…
Domain estimated traffic: 358 / mo.
Domain quality backlinks: 320
Mar 19, 2008 – Cleaning validation is a critical function in pharmaceutical manufacturing. Regulatory agencies have placed great emphasis on demonstrating …
Common Pitfalls During Implementation of a Cleaning … – ISPE
ispe.org › pharmaceutical-engineering › common-pitfalls-during-impl…
May 30, 2019 – Since the initial discussions on Cleaning Validation in the early 1990’s and even after the FDA Guidance was published in 1993, there has …
Cleaning Validation Steps for GMP Plant | Pharmaceutical …
www.gmpsop.com › cleaning-validation-requirements
Oct 27, 2017 – Procedures used to clean the facilities and equipment between products are required to be validated. Prior to the initiation of cleaning validation …
What Is Pharmaceutical Cleaning Validation?
www.gallay.com.au › blog › what-is-pharmaceutical-cleaning-validati…
Nov 19, 2013 – Did you know that pharmaceutical cleaning validation is a necessary and time-consuming part of the manufacturing process? It is designed to …
guidance on aspects of cleaning validation in … – APIC – Cefic
apic.cefic.org › pub › APICCleaningValidationGuide-updateSeptembe…
The main changes were introduced in Chapter 4, Acceptance. Criteria. The subject of cleaning validation in active pharmaceutical ingredient manufacturing plants.
WHO Supplementary Training Modules: Validation, Water, Air …
Dec 1, 2019 – Validation. Cleaning validation protocols (2). The cleaning validation protocol should include: – objectives, responsible people;. – description …
Contamination Control “Cleaning Validation”
www.pda.org › default-source › chapters › presentations › australia
Aug 12, 2014 – 1. Contamination Control. “Cleaning Validation”. Ravi Hattarki. Manufacturing Projects Manager. CSL Behring, Australia. 17th June 2014 …
Cleaning Validation in Pharmaceuticals – Ankur Choudhary …
medium.com › cleaning-validation-in-pharmaceuticals-5213ffb1953c
The manufacturing process of an active pharmaceutical industry typically consists of various chemical reactions and purification steps followed by physical …
Microbiological Cleaning Method Validation
www.raci.org.au › document › item
Don’t claim this in Cleaning Validation Policies or Protocols. If microbial bioburden were at an unacceptable level prior to cleaning – the last batch produced must …
What You Should Know About Pharmaceutical Cleaning …
www.drugdiscoveryonline.com › doc › what-you-should-know-about…
What Is Cleaning Validation? Cleaning validation is a requirement in industries such as pharmaceutical manufacturing which adhere to Good Manufacturing …
www.cleaningvalidation.com › glossary
Cleaning Validation – Documented evidence with a high degree of certainty that a cleaning process will consistently produce product meeting its predetermined …
Pharmaceutical Cleaning Validation
www.rsc.org › events › detail › pharmaceutical-cleaning-validation
After an introduction to the overall objectives, the course then covers process design, pitfalls and associated validation issues including sampling and testing. The …
Validating cleaning methods used for production equipment
www.octapharma.com › news › our-employees › validating-cleaning-…
Mar 1, 2018 – As part of the international regulatory framework in which Octapharma operates, the Cleaning Validation department contributes to patient …
Rethinking Limits in Cleaning Validation | Pharmaceutical …
www.pharmtech.com › rethinking-limits-cleaning-validation
by RJ Forsyth – 2015 – Cited by 1 – Related articles
Oct 2, 2015 – Abstract: Cleaning validation programs must have cleaning limits, worst-case residues to validate, and recovery factors to accurately determine …
Lifecycle Approach Application to Cleaning Validation | IVT …
www.ivtnetwork.com › article › lifecycle-approach-application-cleani…
Dec 1, 2015 – A definition of cleaning validation based on the 2011 FDA definition for process validation is proposed. Cleaning validation activities are …
Cleaning Validation Guidelines – A Complete List [Updated …
www.leucinetech.com › post › cleaning-validation-guidelines-a-compl…
Feb 29, 2020 – Cleaning Validation is a critical component of an effective GMP Compliance program at any regulated drug manufacturing facility. In fact …
www.dcvmn.org › IMG › pdf › 402___cleaning_validation
Cleaning valida+on is necessary to demonstrate that the methods used to clean manufacturing equipment are adequate to ensure that the risk of cross-‐.
Cleaning Validation of Manufacturing Equipment …
www.pharmaguideline.com › Quality Assurance › Validation
1.4 The objective of cleaning validation is to prove that the equipment is consistently cleaned of product, detergent and microbial residues to an acceptable level, …
Cleaning Validation | Ofni Systems
www.ofnisystems.com › services › validation › cleaning-validation
The FDA expects that regulated companies will have: Written general procedures on how cleaning processes will be validated. General cleaning validation …
Basics of cleaning validation – LinkedIn
www.linkedin.com › pulse › basics-cleaning-validation-srinivas-m
Dec 7, 2016 – Cleaning validation is primarily applicable to the cleaning of process manufacturing equipment in the pharmaceutical industry. The focus of …
Cleaning Validation Products – Fisher Scientific
www.fishersci.com › Products › Laboratory Wipes and Cleaners
Designed to simplify sampling for cleaning validation. Kits compatible for use with Sievers Model 800/900, Anatel ANATOC™, Teledyne Tekmar, OI Analytical …
Equipment Cleaning Validation Within a Multi-Product …
www.biopharminternational.com › equipment-cleaning-validation-wit…
Feb 1, 2006 – Before designing cleaning procedures, it’s vital to know all physical and chemical characteristics of the product ingredients.
Cleaning Validation Guidelines (GUIDE-0028) – Canada.ca
www.canada.ca › services › good-manufacturing-practices › cleaning-…
Jan 1, 2008 – 3.1 The objective of the cleaning validation is to verify the effectiveness of the cleaning procedure for removal of product residues, degradation …
Validation & Engineering Group Cleaning Validation Specialist
jobs.smartrecruiters.com › ValidationEngineeringGroup › 743999681…
Coordinate cleaning validation activities. Prepare and execute protocols and reports for cleaning development and validation. Perform investigation of …
Cleaning Validation, Pharmaceuticals – Journal of Innovations …
www.jipbs.com › VolumeArticles › FullTextPDF
by B Lodhi – Cited by 19 – Related articles
Cleaning validation for the pharmaceuticals, biopharmaceuticals, cosmetic and neutraceuticals industries. Babita Lodhi*1, Poonam Padamwar1, Arif Patel1.
Alconox releases cleaning validation white paper
www.cleanroomtechnology.com › news › article_page › Alconox_rel…
May 9, 2018 – Validation is a documented guarantee that cleaning can be performed reliably and repeatedly to satisfy a predetermined level of cleanliness.
Components of a Cleaning Validation Protocol | STERIS
www.sterislifesciences.com › resources › documents › technical-tips
It is essential that a cleaning validation protocol contains the necessary components to appropriately challenge a cleaning process.
Cleaning Validation: The Critical Steps Involved …
learnaboutgmp.com › good-manufacturing-practices-cgmp › cleaning…
To avoid this contamination during manufacturing and Packaging process of pharmaceutical areas cleaning validation should be implemented. The benefit of …
Cleaning Validation and Its Regulatory Aspects in the …
www.sciencedirect.com › science › article › pii
by SL Prabu – 2015 – Cited by 2 – Related articles
The purpose of cleaning validation is to prevent contamination and cross-contamination in pharmaceutical dosage forms. The pharmaceutical products’ …
Ecolab Cleaning Validation: Home public area
www.ecolab-cleaning-validation.com
This website has been developed to help Ecolab’s clients with their cleaning validation processes or revalidation in the pharmaceutical industry.
Cleaning of Dedicated Equipment: Why Validation is Needed …
www.processdevelopmentforum.com › articles › cleaning-of-dedicate…
by C Baccarelli – Cited by 2 – Related articles
Aug 13, 2015 – This article discusses cleaning validation of equipment dedicated to the production of a single API. By Cristina Baccarelli, Paola Bernard, Teresa …
Cleaning Validation The Basics | Ecolab | Life Sciences | Ecolab
www.ecolab.com › articles › 2019/10 › cleaning-validation-the-basics
Oct 29, 2019 – Cleaning validation is a documented evidence providing assurance that an equipment train and/or piece of equipment can be cleaned reliably …
Developing A Science- Risk- Statistics-Based Approach To …
www.pharmaceuticalonline.com › doc › developing-a-science-risk-sta…
Jun 28, 2017 – Cleaning validation incorporates the traditional preapproved protocol, with predetermined acceptance criteria and a three-run process validation …
What does the FDA expect from Cleaning Validation today …
www.gmp-compliance.org › gmp-news › what-does-the-fda-expect-fr…
Apr 10, 2019 – FDA’s regulations regarding cleaning validation are now relatively old. A GUIDE TO INSPECTIONS VALIDATION OF CLEANING PROCESSES …
How Clean is Clean in Drug Manufacturing: Cleaning …
www.americanpharmaceuticalreview.com › Articles › Article Archives
Feb 5, 2017 – This article will continue the discussion about a risk based and lifecycle approach to Cleaning Validation. We will discuss assessment of …
Cleaning validation – SlideShare
www.slideshare.net › vinayjain1048 › cleaning-validation-26237202
Sep 16, 2013 – CLEANING VALIDATION Presented by G.Vinay jain M.Pharmacy 1st year 2nd sem Department of Pharmaceutical analysis and QA Malla reddy …
Medical Device Cleaning Validation References
www.meddeviceonline.com › doc › medical-device-cleaning-validatio…
A cleaning validation involves testing for acceptable residues on pharmaceutical manufacturing or medical device surfaces. The validation involves: Identifying …
Cleaning Validation Equipment & Facility … – HPRA
www.hpra.ie › docs › default-source › default-document-library › 5-2…
Slide 1. Cleaning Validation. Equipment & Facility considerations & potent materials. IMB Information Day , 27th September 2012. Victor Garvin,. GMP Inspector.
Cleaning validation of ofloxacin on pharmaceutical … – NCBI
by MS Arayne – 2008 – Cited by 3 – Related articles
2008 Sep-Oct;62(5):353-61. Cleaning validation of ofloxacin on pharmaceutical manufacturing equipment and validation of desired HPLC method. Arayne MS(1), …
Cleaning Validation Swabs – Texwipe
www.texwipe.com › food-processing-cleaning-validation-swabs
Engineered to deliver consistent and accurate sample recovery for cleaning validation for both specific (HPLC, UV-Vis, IMS) and nonspecific (TOC) analytical …
life cycle approach to cleaning validation – IJPSR
ijpsr.com › bft-article › life-cycle-approach-to-cleaning-validation
Apr 1, 2017 – The guidelines on “cleaning validation in Active pharmaceutical Ingredients manufacturing plants” was released by APIC (Active Pharmaceutical …
Cleaning Validation Tests for Reuse Devices | Nelson Labs
www.nelsonlabs.com › testing › cleaning-validation-reusable-devices
Cleaning validations evaluate the recommended cleaning procedure for a reusable device according to AAMI TIR12, AAMI TIR30, ISO 17664, and the FDA …
Pharmaceutical Cleaning Validation – Photon Systems
photonsystems.com › applications › pharmaceutical-cleaning-validation
Current cleaning validation cost are dominated by high labor cost of swabbing and ~90 hours of manufacturing equipment down time due to exhaustive lab (HPLC) …
The Shirokizawa Matrix Determining The Level Of Effort …
www.bioprocessonline.com › doc › the-shirokizawa-matrix-determini…
Dec 11, 2019 – Part of the Cleaning Validation For The 21st Century Series. The International Congress on Harmonization Quality Risk Management Guidance …
Determination of Anthracycline Drug Residual in Cleaning …
www.hindawi.com › journals › jspec
by Z Slivová – 2015 – Cited by 1 – Related articles
Jul 12, 2015 – The cleaning validation involves identification of numerous sampling points in the manufacturing line to demonstrate complete removal of …
Cleaning Validation Analysis Support for Pharma Production
www.intertek.com › pharmaceutical › analysis › cleaning-validation
Intertek Pharmaceutical Services (Whitehouse, NJ) supports the pharmaceutical industry with Cleaning Validation expertise and services.
Cleaning validation of pharmaceutical process equipment …
Define your best cleaning validation strategy – New GMP requirements – Methods and techniques.
Cleaning Validation – Documentation Requirements | inglasia …
inglasia.com › cleaning-validation-documentation-requirements
Cleaning Validation – Documentation Requirements. Introduction. The cleaning of pharmaceutical equipment is an area of regulatory importance. The potential …
Cleaning Validation | Independent Solutions | Limerick
www.independentsolutions.ie › services › lean-validation › cleaning-v…
Independent Solutions provide validation of cleaning processes, in accordance with site-specific requirements, FDA and EU standards.
Cleaning Validation of Pharmaceutical Manufacturing …
www.lucideon.com › healthcare › pharmaceuticals › pharmaceutical-…
Outsourcing the cleaning validation of your pharmaceutical manufacturing equipment can speed up the process, minimize disruption and provide a greater …
Cleaning validation of clean – Pharmacie des HUG
pharmacie.hug-ge.ch › ens › conferences › fs_cleaning_validation_kr…
Cleaning validation of clean- rooms and preparation equipments. Dr Farshid SADEGHIPOUR. Head of production. Central Pharmacy, Geneva University …
Equipment Cleaning Validation| Integrated Analytical Labs …
www.ialonline.com › pharmaceutical-services › cleaning-validation
Cleaning validation is a multi-step process that begins with focusing on the objective of the validation process. Typically, the objective is to prevent the adulteration …
cleaning validation in the food industry – EHEDG
www.ehedg.org › fileadmin › guidelines › DOC_45_E_2016
THE ENGLISH VERSION OF THIS EHEDG DOCUMENT IS THE OFFICIAL VERSION. THE RESPONSIBILITY. FOR THE PREPARATION, DEVELOPMENT AND …
Pharmaceutical Cleaning & Validation | Alconox, Inc.
alconox.com › pharmaceutical-cleaning-validation
Pharma validations assist with controlling pharmaceutical microbiology, maintenance of pharmaceutical clean rooms, pharmaceutical process validation, and …
Standard Operating Procedure Title: Procedure for Cleaning …
manoxblog.com › 2018/02/08 › standard-operating-procedure-title-pr…
Feb 8, 2018 – Cleaning Validation, cleaning validation is a validation program to verify that the processes and procedures used to clean product residue from …
Cleaning Validation – Contract Pharma
www.contractpharma.com › contract-manufacturing › cleaning-valida…
Cleaning Validation – Our Contract Services Directory contains listings for all of your outsourcing needs, covering manufacturing, packaging, formulation, clinical …
Cleaning Validation Manual: A Comprehensive Guide for the …
www.crcpress.com › Pharmaceutical Science › Cleaning & Sterilization
Cleaning Validation Manual: A Comprehensive Guide for the Pharmaceutical and Biotechnology Industries – CRC Press Book.
Cleaning validation instruments – Matrix Process Solutions
www.matrixps.com › product › cleaning-validation-instruments
Cleaning validation instruments. The Alfa Laval Sanitary Rotacheck System consisting of a Universal Relay and a Sensor designed for confirmation of correct …
Cleaning validation – GMP services & consulting | testotis.com
www.testotis.com › services › validation › cleaning-validation
Clean does not always mean spotless – cleaning validation verifies the effectiveness and reproducibility of the entire cleaning process. Testo Industrial Services …
Effective and reliable optical system for cleaning validation in …
Nov 23, 2015 – Cleaning validation in the pharmaceutical industry is the verification that the cleaning procedure used to remove contaminants from equipment …
“Health-based approach” implementation for setting limits in …
a3p.org › health-based-approach-implementation-for-setting-limits-in-…
Cleaning – Process validation principle The key point before setting limits in cleaning is that cleaning is now considered as a process on its own subject to the …
What Is Cleaning Validation? – International Products …
Sep 6, 2018 – Any residue must be removed to a predetermined level of cleanliness. Cleaning validation processes protect against the cross-contamination of …
Pharmaceutical Cleaning Validation – RSSL
www.rssl.com › research-and-development-support › pharmaceutical-…
Our cleaning validation services will: Test and challenge your cleaning regime; Demonstrate compliance with GMP requirements; Allow you to manufacture with …
Cleaning Validation Services for the Biopharmaceutical …
www.manganbio.com › cleaning-validation
Dec 4, 2018 – A cleaning validation program will provide documented evidence that residuals from previous processing and cleaning do not adulterate and …
Cleaning Validation – Samedan Ltd
www.samedanltd.com › uploads › pdf › white_paper
Authors: Steve Alley, Technical Specialist. Brian Hammond, RSSL Consultant. Darlington Nwodo, Microbiology Laboratory Manager. Cleaning Validation.
Risk-based Automated Cleaning Validation Software for Life …
www.valgenesis.com › cleaning-validation
ValGenesis maintains the entire cleaning validation process electronically through a web interface. And our VLMS is risk-based fully automates the entire …
Cleaning Validation Guidelines – PHARMAPEDIA
pharmapedia.wikidot.com › cleaning-validation-guidelines
Jun 23, 2009 – The document is intended to cover validation of equipment cleaning for the removal of contaminants associated with previous products, residues …
Cleaning Validation, Priscilla Browne – Amazon.com
www.amazon.com › Cleaning-Validation-Priscilla-Browne-ebook
Cleaning Validation – Kindle edition by Priscilla Browne. Download it once and read it on your Kindle device, PC, phones or tablets. Use features like bookmarks …
Cleaning validation in pharmaceuticals – Hilaris Publishing SRL
www.hilarispublisher.com › proceedings › cleaning-validation-in-pha…
Cleaning validation in pharmaceuticals. 6th International Conference and Exhibtion on Pharmaceutical Regulatory Affairs and IPR September 29-30, 2016 …
FDA Requirements for Cleaning Validation – GMP news
gmpnews.net › 2019/06 › fda-requirements-for-cleaning-validation
Jun 5, 2019 – This is particularly the case with Warning Letters. Here, even details of deficiencies regarding cleaning validation are included in the Warning …
CLEANING VALIDATION PROTOCOL Cleaning Validation …
CV VMP 001 Cleaning Validation Master Plan. SOP #145. CIP Cleaning of the Bulk Tank and the Filling Line – Version # 5. SOP #003. COP (Manual) Cleaning …
Cleaning validation – Pharma Solutions Ltd
pharmsolution.org › cleaning-validation
Cleaning validation is documented evidence that an approved cleaning procedure will reproducibly remove the previous product or cleaning agents used in the …
Process Cleaning Validation Jobs, Employment | Indeed.com
www.indeed.com › q-Process-Cleaning-Validation-jobs
1570 Process Cleaning Validation jobs available on Indeed.com. Apply to Senior Process Engineer, Process Development Specialist, Technician and more!
Cleaning Validation | ERA-certified reference materials …
www.eraqc.com › Catalogs › catalogid › categoryid
Cleaning validation has received increasing attention not only in manufacturing but also in research and development. To ensure that your sampling techniques …
Cleaning Validation Tool Products | Medline Industries, Inc.
www.medline.com › category › Cleaning-Validation-Tool › cat3610003
Filters. Manuf / Supplier. Neogen Corporation (1). Sealed Air (1). Measurement Type. Digital (1). Product Use. ATP Monitoring (1). Cleaning Validation Tool …
Pharmaceutical cleaning validation simplified with new software
www.digitaljournal.com › tech-and-science › science › article
Jul 12, 2017 – Cleaning validation is a key process, and it avoids cross-contamination. Washington – The main purpose of Quascenta’s recently launched …
Clearing Up Cleaning Validation : Modern Machine Shop
www.mmsonline.com › articles › clearing-up-cleaning-validation
Examining common cleaning validation methods. water break test. The water break test is a visual inspection method. The clean tile on …
Sampling Techniques for Cleaning Validation from Cole-Parmer
www.coleparmer.com › tech-article › effective-swabbing-techniques
Read our article to learn how to succeed in the search for nothing with effective swabbing techniques for cleaning validation.
Medical Device Cleaning Validation and Disinfection …
pacificbiolabs.com › Medical Device Tests › Microbiology
Medical device cleaning validations are critical component to the overall reprocessing validation, it proves that the device can be cleaned following the …
Cleaning Validation – THE FORCE
the-force.org › cleaning-validation
From our own experience Cross-Contamination Control and Cleaning Validation a topics that many pharmaceutical companies struggle with. This is confirmed …
Cleaning validations for the pharmaceutical sector | UFAG …
www.ufag-laboratorien.ch › cleaning-validation
Cleaning Validation. Certain to be clean. The manufacturing of pharmaceutical active ingredients and medicines takes place in a strongly regulated environment.
Continued process verification methods in cleaning validation
fedegari.com › wp-content › uploads › 2019/03 › Continued-process-…
by S Ferretti – 2014
It follows that the cleaning validation protocol must include non-specific assay methods to measure residues of deactivated products [2]. Conductivity analysis is a …
www.bd.com › products › diagnostics-systems › medicinal-products
Cleaning Validation. PLATED MEDIA – Sterile Pack Plated Media. Catalog, Description, Quantity. 292229, CDC Anaerobe Blood Agar RODAC™, 10. 221232 …
Best Practices for Cleaning Validation Swab Sampling and …
This Web Seminar will present a detailed overview of best practices for cleaning validation swab recovery and sampling for Active Pharmaceutical Ingredients …
Validation of cleaning – Higiene Ambiental
higieneambiental.com › files › pdf › cleaning_validation_white_paper
The cleaning methods shall be validated to ensure they are effective and the effectiveness of the procedure routinely verified. Cleaning equipment used to clean …
How to Improve Cleaning Processes – Eurofins
cdnmedia.eurofins.com › eurofins-us › media › how-to-improve-clea…
Often, this can be the most daunting step for pharmaceutical manufacturing facilities throughout the clean– ing validation process simply because of how specific …
Cleaning Validation Considerations for Biopharmaceuticals …
www.avomeen.com › lifesciences-cleaning-validation-whitepaper
Cleaning validation is a company’s documented guarantee that the cleaning processes are performed reliably and consistently with the FDA’s cleaning validation …
Cleaning Validation for the 21st Century – Stevens Institute of …
web.stevens.edu › ses › documents › fileadmin › documents › pdf › Cle…
by A Walsh – 2011 – Cited by 12 – Related articles
This new ISPE Guide, “Science and Risk-. Based Cleaning Process Development and. Validation,” will describe how to implement cleaning programs, using …
Cleaning Validation for Medical Device Manufacturing
vertassets.blob.core.windows.net › download › cleaning_validation_f…
Cleaning validation is documentation establishing that a cleaning process will consistently result in devices that are clean to a predetermined acceptable level of …
9,450 total views, 8 views today
