Tout sur les médicaments הכל על תרופות كل شيئ عن الأدوية Все о наркотиках 关于药品的一切 డ్రగ్స్ గురించి అన్ని 마약에 관한 모든 것 Όλα για τα Ναρκωτικά Complete Tracking of Drugs Across the World by Dr Anthony Melvin Crasto, Worldpeacepeaker, worlddrugtracker, PH.D (ICT), MUMBAI, INDIA, Worlddrugtracker, Helping millions, 9 million hits on google on all websites, 2.5 lakh connections on all networks, “ALL FOR DRUGS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This is a compilation for educational purposes only. P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
FLOW CHEMISTRY, flow synthesis, UncategorizedComments Off on Contribution of microreactor technology and flow chemistry to the development of green and sustainable synthesis
Department of Pharmacy – Drug Sciences, University of Bari “A. Moro”, FLAME-Lab – Flow Chemistry and Microreactor Technology Laboratory, Via E. Orabona 4, 70125, Bari. Italy
Guest Editor: L. Vaccaro Beilstein J. Org. Chem.2017,13, 520–542. doi:10.3762/bjoc.13.51 Received 14 Nov 2016, Accepted 20 Feb 2017, Published 14 Mar 2017
Abstract
Microreactor technology and flow chemistry could play an important role in the development of green and sustainable synthetic processes. In this review, some recent relevant examples in the field of flash chemistry, catalysis, hazardous chemistry and continuous flow processing are described. Selected examples highlight the role that flow chemistry could play in the near future for a sustainable development.
Green chemistry’s birth was driven by the necessity to consider and face the urgent question of sustainability. Chemical production concerns an extended range of fields such as textiles, construction, food, cosmetic components, pharmaceuticals and so forth. An innovative approach to the chemistry world requires new strategies and criteria for an intelligent chemistry. It is understood that all this matter has big implications in economy and politics. Recent studies predicted a growth of green chemical processing up to $100 billion in 2020 (Pike Research study) [1]. All this offers important and arduous challenges expressed in terms of new synthetic strategies using sustainable, safe, and less toxic materials. On green chemistry we can read Paul Anastas and John Warne’s 12 principles, set up in 1998, which illustrate the characteristics of a greener chemical process or product [2]. Microreactor technology and flow chemistry could play a pivotal role in the context of sustainable development. In fact, flow chemistry is becoming a new technique for fulfilling several of the twelve green chemistry principles. The microreactor approach, could provide protection, preserves atom economy, guarantees less hazardous chemical synthesis and allows the use of safer solvents and auxiliaries. Furthermore, it pushes towards designing of chemistry with a lower environmental and economic impact, enhance the importance of catalysis, allows real-time analysis for pollution prevention and provides inherently safer chemistry (Figure 1) [3]. Without claiming to be exhaustive, in this review we report recently published representative synthetic applications that demonstrate the growing contribution of flow chemistry and microreactor technology in green and sustainable synthesis [4-7].
Figure 1: Microreactor technologies and flow chemistry for a sustainable chemistry.
Review
Flow microreactors: main features
The peculiar properties of microreactors [8] derive from their small size and can be ascribed mainly to the following characteristics: a) fast mixing: in a flow microreactor, in striking contrast to batch conditions, mixing takes place by molecular diffusion so that a concentration gradient can be avoided; b) high surface-to-volume ratio: the microstructure of microreactors allows for a very rapid heat transfer enabling fast cooling, heating and, hence, precise temperature control; c) residence time: it is the period of time the solution of reactants spend inside the reactor, and it gives a measure of the reaction time. The residence time is strictly dependent on the characteristics of the reactor (i.e., length of the channels, volume), and on the flow rate. The residence time is one of the crucial factors to be considered in optimizing flow reactions, especially when unstable or short-lived reactive intermediates are concerned. Microreactor technology provides also several benefits. Safety benefits, because of the high efficiency in heat exchange, and avoided accumulation of unstable intermediates. Economy benefits, due to lower manufacturing and operating costs, reduced work-up procedures, use of less raw materials and solvents and reduced waste. Chemistry benefits associated to the use of microreactor technology are the improved yields and selectivities, the possibility to conduct reactions difficult or even impossible to perform in batch, and the use of reaction conditions that allow exploring new chemical windows [9].
Contribution of flash chemistry to green and sustainable synthesis
The concept of flash chemistry as a “field of chemical synthesis using flow microreactors where extremely fast reactions are conducted in a highly controlled manner to produce desired compounds with high selectivity” was firstly introduced by Yoshida [10]. Flash chemistry can be considered a new concept in both organic and sustainable synthesis involving chemical transformations that are very difficult or practically impossible to conduct using conventional batch conditions. With the aim to show how flow microreactor technology and flash chemistry could contribute to the development of a sustainable organic synthesis, very recent examples have been selected and will be discussed here. In the context of green chemistry [11], protecting-group free organic synthesis has received particular attention in the last years, because of atom economy [12-15] and reduction of synthetic steps [16]. It has been demonstrated by Yoshida that protecting-group-free synthesis could be feasible using flash chemistry and microreactor technology [17,18]. Recently, Yoshida and co-workers developed flash methods for the generation of highly unstable carbamoyl anions, such as carbamoyllithium, using a flow microreactor system [19]. In particular, they reported that starting from different substituted carbamoyl chloride 1 and lithium naphthalenide (LiNp) it was possible to generate the corresponding carbamoyllithium 2, that upon trapping with different electrophiles provided several amides and ketoamide 3(Scheme 1).
Scheme 1: A flow microreactor system for the generation and trapping of highly unstable carbamoyllithium species.
The use of an integrated microflow system allowed the preparation of functionalized α-ketoamides by a three-component reaction between carbamoyllithium, methyl chloroformate and organolithium compounds bearing sensitive functional groups (i.e., NO2, COOR, epoxide, carbonyl) (Scheme 2).
Scheme 2: Flow synthesis of functionalized α-ketoamides.
It should be stressed that this kind of sequential transformations are practically impossible to perform using conventional batch chemistry because of the incompatibility of sensitive functional groups with organolithiums, and because of the high chemical and thermal instability of the intermediates.
In 2015 Yoshida reported another remarkable finding on the use of protecting-group-free organolithium chemistry. In particular, the flash chemistry approach was exploited for generating benzyllithiums bearing aldehyde or ketone carbonyl groups [20]. This reaction could be problematic for two reasons: a) the competing Wurtz-type coupling, (i.e., the coupling of benzyllithiums with the starting benzyl halides); b) the nucleophilic attack of organolithium species to aldehyde or ketone carbonyl groups (Scheme 3).
Scheme 3: Reactions of benzyllithiums.
The authors reported that the extremely fast micromixing avoided undesired Wurtz-type coupling [21,22]. It is well known, that competitive reactions can be controlled or even avoided under fast micromixing [23-27]. Moreover, high-resolution residence time control was essential for survival of carbonyl groups. In fact, this transformation can be achieved only with a residence time of 1.3 ms at −78 °C. Under these flow conditions, the aldehyde or ketone carbonyl moiety can survive the nucleophilic organolithium attack. Remarkably, the flow microreactor system allowed also the generation of benzyllithiums at 20 °C, rather than under cryogenic (−95 °C) conditions adopted with a conventional batch protocol. In addition, THF could be used in place of mixed solvents (Et2O/THF/light petroleum). Under the optimized conditions, the reactions of benzyllithiums with different electrophiles, gave adduct products in good yields (Scheme 4).
Scheme 4: Trapping of benzyllithiums bearing carbonyl groups enabled by a flow microreactor. (Adapted with permission from [18], copyright 2015 The Royal Society of Chemistry).
Another useful aspect of the flash chemistry relies on the possibility to generate highly reactive intermediates, such as halomethyllithium carbenoids, that need to be used under internal-quenching technique in batch mode. In 2014, the first example of effective external trapping of a reactive chloromethyllithium (CML) has been reported [28]. α-Haloalkyllithiums are a useful class of organometallic reagents widely employed in synthetic chemistry. In fact, they allow the direct homologation of carbonyl compounds and imines leading to β-halo-alcohols and amines that are useful building blocks [29-31]. This work represents a remarkable example of flash chemistry, and has elements of sustainability considering that in batch macroreactors, in order to avoid metal-assisted α-elimination, in situ quenching, an excess of reagents, and very low temperature are required [32,33].
Running the reaction in a flow system at −40 °C, by using residence times between 0.18–0.31 s high yields of homologated products have been obtained under external quenching conditions (Scheme 5).
Scheme 5: External trapping of chloromethyllithium in a flow microreactor system.
The results described above nicely show the potential, as green technology, of flow microreactor systems for synthetic processes involving highly unstable intermediates. Another nice example on the use of microreactor technology for the development of sustainable chemical processes, is represented by the direct introduction of the tert-butoxycarbonyl group into organometallic reagents [34]. The reaction between organolithium reagents and di-tert-butyl dicarbonate run under flow conditions, allowed a straightforward preparation of several tert-butyl esters. The use of a flow process resulted more efficient, versatile and sustainable compared to batch. Moreover, this operationally simple procedure complements well with the already available strategies for the preparation of tert-butyl esters, avoiding the use of inflammable and explosive gaseous isobutylene [35], the use of harsh conditions [36], the use of peroxides [37], the use of toxic gas such as CO or transition metals [38-42]. The flow process, for the direct C-tert-butoxycarbonylation of organolithiums, has been optimized in a green solvent such as 2-MeTHF by a precise control of the residence time, and without using cryogenic conditions (Scheme 6). In addition, many organolithiums were generated from the corresponding halo compounds by a halogen/lithium exchange reaction using hexyllithium as a more sustainable base [43,44].
Scheme 6: Scope for the direct tert-butoxycarbonylation using a flow microreactor system.
The concept of flash chemistry has been successfully employed for outpacing fast isomerization reactions. The accurate control of the residence time, realized in a microreactor, could suppress or avoid isomerization of unstable intermediates. This is often unavoidable when the same reactions are run in batch mode [45-47].
Yoshida and Kim recently provided an astonishing example on the potential of flash chemistry in controlling fast isomerization of organolithiums [48]. The authors designed a chip microreactor (CMR), able to deliver a reaction time in the range of submilliseconds (0.33 ms) under cryogenic conditions. By using such an incredible short residence time, it was possible to overtake the very rapid anionic Fries rearrangement, and chemoselectively functionalize ortho-lithiated aryl carbamates (Scheme 7).
Scheme 7: Control of anionic Fries rearrangement reactions by using submillisecond residence time. (Adapted with permission [43], copyright 2016 American Association for the Advancement of Science).
This CMR has been developed choosing a fluoroethylene propylene–polymide film hybrid for fabrication because this material offers exceptional physical toughness at low temperature and high pressure as well as chemical inertness. The most relevant aspect of this microreactor, concerns the 3D design of the mixing zone (Figure 2). The mixing efficiency was evaluated on the basis of computational fluids dynamics (CFD). The simulation results showed that serpentine 3D-structured channels (Figure 2), possessing five turns after each mixing point in a total length of 1 mm, was able to deliver the highest mixing efficiency. The inner volume for the reactor was of 25 μL. This CMR provides mixing efficiency levels of 95% with a total flow rates of 7.5 mL/min corresponding to a residence time of about 0.3 milliseconds.
Figure 2: Chip microreactor (CMR) fabricated with six layers of polyimide films. (Reproduced with permission from [43], copyright 2016 American Association for the Advancement of Science).
To show the potential use of this microdevice in organic synthesis, the synthesis of Afesal [49], a biologically active compound having anthelmintic activity was reported as application.
This outstanding result by Yoshida and Kim, demonstrates how microdevices and flash chemistry could contribute to the development of new sustainable synthetic strategies, and how microreactor technology could help in taming the reactivity of unstable species [50].
Contribution of continuous-flow metal-, organo-, and photocatalysis in green chemistry
The development of continuous-flow catalysis is appealing because it combines the advantages of a catalytic reaction with the benefits of flow microreactors. Under homogeneous conditions a soluble catalyst, which flows through the reactor together with the reactants, is employed. At the end of the process, a separation step would be required in order to remove the catalyst and byproducts. On the other hand, heterogeneous catalysis is widely used in the synthesis of bulk and fine chemicals. In a continuous-flow process, the catalyst can be fixed on a suitable hardware, and the reaction mixture allowed to flow through the system. The use of recyclable catalysts in continuous-flow conditions represents an innovative strategy for the development of more environmentally friendly synthesis. In the last decade, organic photochemistry got a sort of renaissance, emerging as useful approach in modern sustainable and green synthesis.
Concerning the heterogeneous catalysis with palladium, practical procedures for recovering and reusing of the catalysts have been recently reported [51-53]. A versatile Pd-catalysed synthesis of polyfunctionalized biaryls, using a flow microreactor, has been recently reported by Yoshida [54]. Using the integrated microflow system reported in Scheme 8, arylboronic esters were prepared by a lithiation/borylation sequence, and used in a Suzuki–Miyaura coupling in a monolithic reactor. A remarkable aspect of the process was the use of an integrated supported monolithic Pd(0) catalyst that allowed to perform cross-coupling reactions in continuous flow mode (Scheme 8).
Scheme 8: Flow microreactor system for lithiation, borylation, Suzuki–Miyaura coupling and selected examples of products.
This integrated microflow system allow to handle the borylation of aryl halides (Ar1X), and the subsequent Suzuki–Miyaura coupling using different aryl halide (Ar2X). Without requiring the protection of sensitive functionalities, running the flow system using a residence time (tR) of about 4.7 min at a temperature above 100 °C, high yields of coupling products were obtained. Noteworthy, the Suzuki–Miyaura coupling did not require the use of a base. The authors applied the presented method to the synthesis of adapalene, used in the treatment of acne, psoriasis, and photoaging.
Fluorinated aromatic compounds are extremely important in agrochemical, pharmaceutical and medicinal fields [55-58]. Buchwald and co-workers suggested a telescoped homocatalysis procedure consisting of a three-step sequence (metalation, zincation and Negishi cross-coupling) which furnishes an easy access to a variety of functionalized 2-fluorobiaryl and heteroaryl products (Scheme 9) [59]. This strategy is rightfully considered green because it guarantees the employment of readily available and cheap starting materials, the safe handling of highly thermally unstable or dangerous intermediates, and the use of higher temperature with respect to the batch mode in which the proposed reactions have to be carried out at −78 °C.
Scheme 9: Experimental setup for the flow synthesis of 2-fluorobi(hetero)aryls by directed lithiation, zincation, and Negishi cross-coupling. (Adapted with permission from [53], copyright 2016 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim).
The use of 2-MeTHF as greener solvent, contributes to further validate the green procedure. The 2-MeTHF solutions of fluoroarenes 4 together with the hexane solution of n-BuLi were pumped into the flow system at −40 °C. The generated organozinc intermediate meets the solution of haloarenes and the catalyst, leading to the formation of the desired products 5a–j (Scheme 9). Noteworthy, the homogeneous catalysis requires only 1% of the XPhos-based palladium catalyst. A sonication bath was employed to prevent clogging and the reaction required a residence time of 15 min.
Next, they turned their attention to the arylation of fluoro-substituted pyridines. The regioselective lithiation of halopyridines with lithium diisopropylamide (LDA) was conducted under mild conditions on substrate 6(Scheme 10). The addition of a little amount of THF was necessary in order to avoid clogging and the tendency of the lithiated intermediate to eliminate.
Scheme 10: Experimental setup for the coupling of fluoro-substituted pyridines. (Adapted with permission from [53], copyright 2016 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim).
The optimized conditions were suitable for the functionalization of 2-fluoropyridine, 2,6- difluoropyridine and 4-(trifluoromethyl)pyridine leading to products 7a–g reported in Scheme 10. Another promising field is the sustainable flow organocatalysis, and recently Pericàs reported an interesting synthesis and application of a recyclable immobilized analogue of benzotetramisole (BMT) used in a catalytic enantioselective Michael addition/cyclization reactions under continuous-flow conditions (Scheme 11) [60].
Scheme 11: Continuous flow process setup for the preparation of 11 (Reproduced with permission from [54], copyright 2015 American Chemical Society).
Resin-bound catalyst 10 was swollen with dichloromethane in a medium-pressure chromatography column used as a reactor. Dichloromethane solutions of substrate 9 reacted with the mixed phenylacetic pivalic anhydride (deriving from phenylacetic acetic (8) and pivaloyl chloride) inside the catalytic reactor producing the expected products 11. This ingenious system was equipped with an in-line FTIR probe, for monitoring the transformation, and an in line liquid–liquid separator to avoid tedious work-up procedures, thus saving solvents, resources and optimizing work times. This system was demonstrated to work for 11 h with higher conversion and enantioselectivity (er >99.9%) in comparison to the batch mode [61]. Pericàs and co-workers taking advantage of the high catalytic activity, robustness and recyclability of the supported catalyst, performed also straightforward gram synthesis of target compounds.
In the context of photocatalysis and oxidations using flow microreactors [62,63], Noël reported a metal-free photocatalytic aerobic oxidation of thiols to disulfides under continuous-flow conditions [64]. Disulfides are useful molecules employed as drugs, anti-oxidants or pesticides as well as rubber vulcanizating agents [65]. Symmetric disulfides are generally obtained by oxidative coupling of thiols [66]. Noël and co-workers set up a microflow system equipped with a mass flow controller (MFC) able to introduce pure oxygen as the oxidant to oxidize a solution of thiol containing 1% of Eosin Y. The flow stream was exposed to white LED light in order to activate the reaction, and a dilution with pure EtOH was needed at the output to avoid clogging (Scheme 12). The residence time of 20 min guaranteed a limited irradiation time and high purity of the products.
Scheme 12: Continuous-flow photocatalytic oxidation of thiols to disulfides.
The disulfides were obtained with excellent yields, and the process was executed on challenging thiols as in the case of disulfide 12 (Scheme 12), used as food flavour additive [67]. To demonstrate the usefulness of the flow methodology, and its applicability, the photocatalytic aerobic oxidation of a peptide to obtain oxytocin in continuous flow was reported (Scheme 12). Full conversion was achieved in water with 200 s of residence time.
Noël optimized, for the first time, a trifluoromethylation of aromatic heterocycles by continuous-flow photoredox catalysis. The process benefited from the use of microreactor technology and readily available photocatalysts. The process was also employable for perfluoroalkylation. The developed process occurred in less time with respect to batch mode, and under milder conditions. The set-up of the reactor allowed for the use of gaseous CF3I by means of a mass flow controller. Selected examples of trifluoroalkylated products are reported in Scheme 13[68].
Scheme 13: Trifluoromethylation by continuous-flow photoredox catalysis.
Tranmer reported a “traceless reagents” chemistry with the continuous-flow photosynthesis of 6(5H)-phenanthridinones, poly(ADP-ribose) polymerase (PARP) inhibitors [69]. The relevance of the work resides in the use of green solvents, the absence of heavy metals, the use of convenient temperatures, and the increased safety by eliminating UV-exposure locating the UV lamp within the microreactor. Hazard of fires caused by the hot UV lamps approaching the auto-ignition temperature of flammable solvents, very often underestimated, is totally prevented thanks to a specific cooling system. 2-Halo-N-arylbenzamides were converted into 6(5H)-phenanthridinones by a photocyclization reaction. In order to run this step, a flow system with a photochemical reactor equipped with a medium pressure Hg lamp and 10 mL reactor coil, was employed. Good yields were obtained from different 2-chlorobenzamides disclosing that either electron-donating or electron withdrawing ortho-substituents were tolerated (Scheme 14).
Scheme 14: Flow photochemical synthesis of 6(5H)-phenanthridiones from 2-chlorobenzamides.
A metal- and catalyst-free arylation procedure carried out under continuous-flow conditions was recently reported by Fagnoni [70]. This photochemical process allowed for the preparation of a wide range of synthetic targets by Ar–Csp3, Ar–Csp2 and Ar–Csp bond-forming reactions. The use of a photochemical flow reactor, consisting of a polyfluorinated tube reactor wrapped around a 500 W Hg lamp, allowed to overcome batch limitations paving the way for metal-free arylation reactions via phenyl cations. Derivatives 14a–g were prepared with this greener flow approach (Scheme 15) starting from mesitylene 13, and haloarenes using short irradiation times (<6 h), and a 5:1 MeCN/H2O mixture.
Scheme 15: Synthesis of biaryls 14a–g under photochemical flow conditions.
The reported results show how photochemistry hold the potential to become a green tool for the development of sustainable photochemical flow synthesis.
Hazardous chemistry by using green and sustainable continuous-flow microreactors
We have already shown how continuous-flow technology could play an important role in improving chemical processes [5,71], providing different advantages over traditional batch mode. However, the hazardous nature of some chemicals makes handling at conventional lab or industrial scale difficult. The use of microreactors and continuous-flow chemistry offers the possibility to perform reactions using dangerous or hazardous materials that cannot be used in batch mode. In other word, syntheses previously “forbidden” for safety reasons, such as those involving diazo compounds, hydrazine, azides, phosgene, cyanides and other hazardous chemicals could be performed with relatively low risk using flow technology [72-76].
Several research groups investigated this aspect, as highlighted by several available reviews [77,78]. Here we describe very recent reports with the aim to highlight the potential of flow chemistry in the field of hazardous chemistry under a greener perspective.
Diazo compounds are recognized as versatile reagents in organic synthesis. Nevertheless, diazo compounds are also considered highly energetic reagents [79,80]. For this reason, the in situ generation of such reagents has been investigated under flow conditions. Moody and co-workers reported a new method for the in situ generation of diazo compounds as precursors of highly reactive metal carbenes (Scheme 16) [81].
Scheme 16: Flow oxidation of hydrazones to diazo compounds.
As reported in Scheme 16, diazo species 18 could be generated from simple carbonyls 15 and hydrazine (16). Intermediate hydrazones 17 can be converted into the corresponding diazo compounds by oxidation using a recyclable oxidant based on N-iodo-p-toluenesulfonamide potassium salt. The possibility to regenerate a functionalized resin by simple washing with aqueous KI3/KOH solution makes the process more sustainable. This method produces KI solution as waste, and it is an alternative way for the direct oxidation of hydrazones, that often requires the use of heavy metals such as HgO, Pb(OAc)4 and AgO [82,83].
The diazo compounds could be collected as solution in dichloromethane at the output of the flow system, and obtained sufficiently pure for further use without requiring handling or isolation. Further mixing of solutions containing diazo derivatives to a solution containing a Rh(II) catalyst, and reactants such as amines, alcohols or aldehydes led to a wide range of products as reported in Scheme 17.
Scheme 17: Synthetic use of flow-generated diazo compounds.
Ley’s group developed several continuous-flow approaches for generating diazo species from hydrazones [84,85]. Under flow conditions, diazo compounds were reacted with boronic acids in order to generate reactive allylic and benzylic boronic acids further employed for iterative C–C bond forming reactions [86]. The generation of unstable diazo species was possible using a cheap, recyclable and less toxic oxidant, MnO2. The flow stream was accurately monitored by in-line FTIR spectroscopy in order to maximize the formation of the diazo compound (Scheme 18) [87].
Scheme 18: Ley’s flow approach for the generation of diazo compounds.
Starting from this initial investigation, Ley and co-workers developed an elegant application of this strategy for a sequential formation of up to three C–C bonds in sequence, by an iterative trapping of boronic acid species. The sequence starts with the reaction of diazo compound 20, generated under flow conditions, and boronic acid 19 (Scheme 19). Further sequential coupling with diazo compounds 21 and 22 led to boronates 23 or protodeboronated products 24 at the end of the sequence (Scheme 19).
Scheme 19: Iterative strategy for the sequential coupling of diazo compounds.
With the aim to exploit the versatility of this approach, Ley and co-workers reported the allylations of carbonyl electrophiles such as aldehydes using the above reported strategy for the generation of allylboronic acids. The flow protocol considers the reaction of diazo compounds 25 (generated in flow) with boronic acid 26 and aldehyde 27 (Scheme 20). By this new iterative coupling it was possible to obtain alcohols as products. The usefulness of the method was demonstrated with the preparation in good yield (60%) of a precursor of the natural product bakuchiol 28 (Scheme 20) [88].
Scheme 20: Integrated synthesis of Bakuchiol precursor via flow-generated diazo compounds.
The microreactor technology offers the advantage to handle hazardous components such as hydrazine and molecular oxygen, which represent alternative reagents for selective reduction of C=C double bonds. In fact, combination of hydrazine hydrate (N2H4·H2O) and O2 provide diimide (HN=NH) as reducing agent. Nevertheless, this strategy is rarely used in traditional batch chemistry for safety reason. Kappe and co-workers recently developed a reduction of the alkene to the corresponding alkane, by a catalyst-free generation of diimide by oxidation of hydrazine monohydrate (N2H4·H2O) with molecular oxygen [89,90]. The flow system set-up is reported in Scheme 21, and consists in a HPLC pump for delivering the alkene and hydrazine monohydrate, while O2 was delivered by a mass-flow controller (MFC) from a standard compressed-gas cylinder. After combination of the reagent streams, the resulting segmented flow was pumped through a heated residence unit (RTU) consisting in a fluorinated tube with low gas permeability (Scheme 21).
Scheme 21: Kappe’s continuous-flow reduction of olefines with diimide.
The flow system reported in Scheme 21 was able to reduce alkenes with high yields and selectivity by using residence times in the range of 10 to 30 min at 100 °C, and by employing a slight excess of hydrazine. Importantly, this strategy is compatible with sensitive functional groups such as silyl ether, halogenes, and benzyl groups. A very nice application of this approach was the highly selective reduction of artemisinic acid to dihydroartemisinic acid, which are of interest in the synthesis of the antimalarial drug artemisinin. This industrially relevant reduction was executed by using O2 at 20 bar, four residence units at 60 °C and consecutive feedings with N2H4·H2O in order to obtain full conversion in dihydroartemisinic acid (29, DHAA, Scheme 22).
Scheme 22: Multi-injection setup for the reduction of artemisinic acid.
Continuous-flow sustainable production of APIs
With the aim to demonstrate the potential of microreactor technology and flow chemistry in sustainable synthesis, recent outstanding “proof of concepts” will be described. Kobayashi and co-workers reported a multistep continuous-flow synthesis of a drug target via heterogeneous catalysis. The developed process not requiring any isolation of intermediates, separation of the catalyst or other work-up procedures can be considered sustainable [91]. The syntheses of (S)-rolipram and a γ-aminobutyric acid (GABA) derivative were accomplished. Readily available starting materials and columns containing chiral heterogeneous catalysts to produce enantioenriched materials were employed. It is worth mentioning that this work represents a very nice example on the use of chiral catalysis in a multistep flow synthesis of a drug target on gram scale. The multistep synthesis of (S)-rolipram reported in Scheme 23 begins from a benzaldehyde derivative which undergoes a Henry-type reaction with nitromethane in the first flow step (Flow I). The resulting nitroalkene undergoes an asymmetric addition catalyzed by a supported PS–(S)-pybox–calcium chloride catalyst at 0 °C using two columns (Flow II). This is the enantio-determining step of the process. The stereochemistry of the adduct can be simply switched to the opposite enantiomer, by using the enantiomeric supported catalyst PS–(R)-pybox–calcium chloride. The enantiomeric excess of the products was about 96%. Two more steps consisting in a Pd-catalyzed hydrogenation reaction and a decarboxylation (Flow III and Flow IV) led to the target (S)-rolipram in 50% overall yield. The systems was designed in order to keep the level of the palladium in solution as low as possible (<0.01 ppm).
Scheme 23: Flow reactor system for multistep synthesis of (S)-rolipram. Pumps are labelled a, b, c, d and e; Labels A, B, C, D, E and F are flow lines. X are molecular sieves; Y is Amberlyst 15Dry; Z is Celite. (Reproduced with permission from [84], copyright 2015 Nature Publishing Group).
Another outstanding proof of concept, which demonstrates the potential of flow chemistry for sustainable pharmaceutical manufacturing, has been recently reported by Jensen and his research team. The research team set up a compact and reconfigurable manufacturing platform for the continuous-flow synthesis and formulation of active pharmaceutical ingredients (APIs) [92]. The “mini” plant (reported in Figure 3) was very compact in size [1.0 m × 0.7 m × 1.8 m, (W × L × H)], and low-weighing (about 100 kg) and was able to perform complex multistep synthesis, work-up procedures as well as purification operations such as crystallization. This platform was also equipped with devices for real-time monitoring and final formulation of high purity APIs. For the preparation of target molecules, commercially available starting materials were employed. The platform was tested for the production and supply of hundreds to thousands doses per day of diphenhydramine hydrochloride, lidocaine hydrochloride, diazepam and fluoxetine hydrochloride.
Remarkably, for future applications of the platform, the produced medicines also met the U.S. Pharmacopeia standards.
Figure 3: Reconfigurable modules and flowcharts for API synthesis. (Reproduced with permission from [85], copyright 2016 American Association for the Advancement of Science).
The future use of this kind of platform would concern the “on-demand” production or the “instantaneous” production of short-lived pharmaceuticals (Figure 4). Other advantageous concerns of this reconfigurable platform are the lower production costs, the higher safety, the automation (computer controlled processes), the reduced waste (production could be done where is needed and in the right amount).
Figure 4: Reconfigurable system for continuous production and formulation of APIs. (Reproduced with permission from [85], copyright 2016 American Association for the Advancement of Science).
Conclusion
Flow chemistry and manufacturing engineering have become largely acknowledged as viable and very often superior alternative to batch processing. Continuous-flow techniques offer increased safety, scalability, reproducibility, automation, reduced waste and costs, and accessibility to a wide range of new chemical possibilities, seldom not accessible through classic batch chemistry. All those benefits are even more noteworthy and outstanding than what they might seem, because they widely fulfil most of the green chemistry principles. In this short overview, we tried to highlight progresses and potential of flow chemistry in the field of sustainable synthesis. Thus, it is expected that flow chemistry and microreactor technology could deeply change the way to perform sustainable chemical production in the near future [93].
Newman, S. G.; Jensen, K. F. Green Chem.2013,15, 1456–1472. doi:10.1039/c3gc40374b
Return to citation in text: [1]
Professor Jun-ichi Yoshida firstly introduced the concept of “micro + flow = green” during a plenary lecture at the 14th IMRET conference held in Beijing (China) in Septembeer 2016.
Return to citation in text: [1]
Yoshida, J.-i.; Kim, H.; Nagaki, A. ChemSusChem2011,4, 331–340. doi:10.1002/cssc.201000271
Return to citation in text: [1]
Hessel, V.; Schouten, J. C.; Renken, A.; Wang, Y.; Yoshida, J.-i., Eds. Handbook of Micro Reactors; Wiley-VCH: Weinheim, 2009.
Return to citation in text: [1]
Yoshida, J.-i. Flash Chemistry: Fast Organic Synthesis in Microsystems; Wiley-VCH: Weinheim, 2008. doi:10.1002/9780470723425
Return to citation in text: [1]
Poliakoff, M.; Fitzpatrick, J. M.; Ferren, T. R.; Anastas, P. T. Science2002,297, 807–810. doi:10.1126/science.297.5582.807
Return to citation in text: [1]
Trost, B. M. Acc. Chem. Res.2002,35, 695–705. doi:10.1021/ar010068z
Return to citation in text: [1]
Wender, P. A.; Verma, V. A.; Paxton, T. J.; Pillow, T. H. Acc. Chem. Res.2008,41, 40–49. doi:10.1021/ar700155p
Return to citation in text: [1]
Kim, H.; Nagaki, A.; Yoshida, J.-i. Nat. Commun.2011,2, No. 264. doi:10.1038/ncomms1264
Return to citation in text: [1]
Nagaki, A.; Yoshida, J.-i. Microreactor Technology in Lithium Chemistry, in: Lithium Compounds in Organic Synthesis from Fundamentals to Applications. Luisi, R.; Capriati, V., Eds.; Wiley-VCH: Weinheim, 2014; pp 491–512. doi:10.1002/9783527667512.ch17
Return to citation in text: [1] [2]
Nagaki, A.; Takahashi, Y.; Yoshida, J.-i. Angew. Chem., Int. Ed.2016,55, 5327–5331. doi:10.1002/anie.201601386
Return to citation in text: [1]
Nagaki, A.; Tsuchihashi, Y.; Haraki, S.; Yoshida, J.-i. Org. Biomol. Chem.2015,13, 7140–7145. doi:10.1039/C5OB00958H
Return to citation in text: [1]
Rys, P. Acc. Chem. Res.1976,10, 345–351. doi:10.1021/ar50106a001
Return to citation in text: [1]
Rys, P. Angew. Chem., Int. Ed. Engl.1977,16, 807–817. doi:10.1002/anie.197708073
Return to citation in text: [1]
Nagaki, A.; Togai, M.; Suga, S.; Aoki, N.; Mae, K.; Yoshida, J.-i. J. Am. Chem. Soc.2005,127, 11666–11675. doi:10.1021/ja0527424
Return to citation in text: [1]
Yoshida, J.-i. Basics of Flow Microreactor Synthesis; SpringerBriefs in Molecular Science; Springer: Tokyo, 2015. doi:10.1007/978-4-431-55513-1
Return to citation in text: [1]
Yoshida, J.-i.; Nagaki, A.; Iwasaki, T.; Suga, S. Chem. Eng. Technol.2005,28, 259–266. doi:10.1002/ceat.200407127
Return to citation in text: [1]
Nagaki, A.; Takabayashi, N.; Tomida, Y.; Yoshida, J.-i. Org. Lett.2008,18, 3937–3940. doi:10.1021/ol8015572
Return to citation in text: [1]
Nagaki, A.; Ichinari, D.; Yoshida, J.-i. Chem. Commun.2013,49, 3242–3244. doi:10.1039/c3cc40392k
Return to citation in text: [1]
Degennaro, L.; Fanelli, F.; Giovine, A.; Luisi, R. Adv. Synth. Catal.2015,357, 21–27. doi:10.1002/adsc.201400747
Return to citation in text: [1]
Pace, V.; Holzer, W.; De Kimpe, R. Chem. Rec.2016,16, 2061–2076. doi:10.1002/tcr.201600011
Return to citation in text: [1]
Gessner, V. H. Chem. Commun.2016,52, 12011–12023. doi:10.1039/C6CC05524A
Return to citation in text: [1]
Pace, V. Aust. J. Chem.2014,67, 311–313. doi:10.1071/CH13416
Return to citation in text: [1]
Emerson, C. R.; Zakharov, L. N.; Blakemore, P. R. Chem. – Eur. J.2013,19, 16342–16356. doi:10.1002/chem.201302511
See for a tactic to prolong the life-time of carbenoids consisting in the introduction of anion-stabilizing groups and see references cited therein.
Return to citation in text: [1]
Kupper, C.; Molitor, S.; Gessner, V. H. Organometallics2014,33, 347–353. doi:10.1021/om4010862
See for examples of “stabilized” chloro carbenoids stable at room temperature and references cited therein.
Return to citation in text: [1]
Degennaro, L.; Maggiulli, D.; Carlucci, C.; Fanelli, F.; Romanazzi, G.; Luisi, R. Chem. Commun.2016,52,9554–9557. doi:10.1039/C6CC04588J
Return to citation in text: [1]
Furniss, B. S.; Hannaford, A. J.; Smith, P. W. G.; Tatchell, A. R. Vogel’s Textbook of Practical Organic Chemistry, 5th ed.; Wiley: New York, 1989; p 1266.
Return to citation in text: [1]
Dawar, P.; Raju, M. B.; Ramakrishna, R. A. Tetrahedron Lett.2011,52, 4262–4265. doi:10.1016/j.tetlet.2011.04.100
Return to citation in text: [1]
Zhu, Y.; Wei, Y. RSC Adv.2013,3, 13668–13670. doi:10.1039/c3ra40246k
Return to citation in text: [1]
Zhang, H.; Shi, R.; Ding, A.; Lu, L.; Chen, B.; Lei, A. Angew. Chem., Int. Ed.2012,51, 12542–12545. doi:10.1002/anie.201206518
Return to citation in text: [1]
Majek, M.; Jacobi von Wangelin, A. Angew. Chem., Int. Ed.2015,54, 2270–2274. doi:10.1002/anie.201408516
Return to citation in text: [1]
Magano, J.; Dunetz, J. R. Chem. Rev.2011,111, 2177–2250. doi:10.1021/cr100346g
Return to citation in text: [1]
Brennführer, A.; Neumann, H.; Beller, M. Angew. Chem., Int. Ed.2009,48, 4114–4133. doi:10.1002/anie.200900013
Return to citation in text: [1]
Xin, Z.; Gøgsig, T. M.; Lindhardt, A. T.; Skrydstrup, T. Org. Lett.2012,14, 284–287. doi:10.1021/ol203057w
Return to citation in text: [1]
Zenzola, M.; Degennaro, L.; Trinchera, P.; Carroccia, L.; Giovine, A.; Romanazzi, G.; Mastrorilli, P.; Rizzi, R.; Pisano, L.; Luisi, R. Chem. – Eur. J.2014,20, 12190–12200. doi:10.1002/chem.201403141
Return to citation in text: [1] [2] [3]
Parisi, G.; Capitanelli, E.; Pierro, A.; Romanazzi, G.; Clarkson, G. J.; Degennaro, L.; Luisi, R. Chem. Commun.2015,51, 15588–15591. doi:10.1039/C5CC06323J
Return to citation in text: [1]
Giovine, A.; Musio, B.; Degennaro, L.; Falcicchio, A.; Nagaki, A.; Yoshida, J.-i.; Luisi, R. Chem. – Eur. J.2013,19, 1872–1876. doi:10.1002/chem.201203533
Return to citation in text: [1]
Tomida, Y.; Nagaki, A.; Yoshida, J.-i. J. Am. Chem. Soc.2011,133, 3744–3747. doi:10.1021/ja110898s
Return to citation in text: [1]
Nagaki, A.; Matsuo, C.; Kim, S.; Saito, K.; Miyazaki, A.; Yoshida, J.-i. Angew. Chem., Int. Ed.2012,51,3245–3248. doi:10.1002/anie.201108932
Return to citation in text: [1]
Kim, H.; Min, K.-I.; Inoue, K.; Im, D. J.; Kim, D.-P.; Yoshida, J.-i. Science2016,352, 691–694. doi:10.1126/science.aaf1389
Return to citation in text: [1]
Semple, J. E.; Rossignol, J.-F. Pharmaceutical compositions and methods of use of salicylanilides for treatment of hepatitis viruses. PCT Int. Appl. WO2012058378 A1, May 3, 2012.
Return to citation in text: [1]
Degennaro, L.; Carlucci, C.; De Angelis, S.; Luisi, R. J. Flow Chem.2016,6, 136–166. doi:10.1556/1846.2016.00014
Return to citation in text: [1]
Pavia, C.; Ballerini, E.; Bivona, L. A.; Giacalone, F.; Aprile, C.; Vaccaro, L.; Gruttadauria, M. Adv. Synth. Catal.2013,355, 2007–2018. doi:10.1002/adsc.201300215
Return to citation in text: [1]
de M. Muñoz, J.; Alcázar, J.; de la Hoz, A.; Díaz-Ortiz, A. Adv. Synth. Catal.2012,354, 3456–3460. doi:10.1002/adsc.201200678
Return to citation in text: [1]
Mennecke, K.; Sodolenko, W.; Kirschning, A. Synthesis2008, 1589–1599. doi:10.1055/s-2008-1072579
Return to citation in text: [1] [2] [3]
Kirsch, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, 2nd ed.; Wiley-VCH: Weinheim, 2013. doi:10.1002/9783527651351
Return to citation in text: [1]
Wang, J.; Sànchez-Rosellò, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev.2014,114, 2432–2506. doi:10.1021/cr4002879
Return to citation in text: [1]
Gillis, E. P.; Eastman, K. J.; Hill, M. D.; Donnelly, D. J.; Meanwell, N. A. J. Med. Chem.2015,58, 8315–8359. doi:10.1021/acs.jmedchem.5b00258
Return to citation in text: [1]
Roesner, S.; Buchwald, S. L. Angew. Chem., Int. Ed.2016,55, 10463–10467. doi:10.1002/anie.201605584
Return to citation in text: [1]
Izquierdo, J.; Pericas, M. A. ACS Catal.2016,6, 348–356. doi:10.1021/acscatal.5b02121
Return to citation in text: [1]
Izquierdo, J.; Ayats, C.; Henseler, A. H.; Pericàs, M. A. Org. Biomol. Chem.2015,13, 4204–4209. doi:10.1039/C5OB00325C
Return to citation in text: [1]
Cambié, D.; Bottecchia, C.; Straathof, N. J. W.; Hessel, V.; Noël, T. Chem. Rev.2016,116, 10276–10341. doi:10.1021/acs.chemrev.5b00707
Return to citation in text: [1]
Gemoets, H. P. L.; Su, Y.; Shang, M.; Hessel, V.; Luque, R.; Noël, T. Chem. Soc. Rev.2016,45, 83–117. doi:10.1039/C5CS00447K
Return to citation in text: [1]
Talla, A.; Driessen, B.; Straathof, N. J. W.; Milroy, L.-G.; Brunsveld, L.; Hessel, V.; Noël, T. Adv. Synth. Catal.2015,357, 2180–2186. doi:10.1002/adsc.201401010
Return to citation in text: [1]
Cremlyn, R. J. An Introduction to Organosulfur Chemistry; Wiley–VCH: New York, 1996.
Return to citation in text: [1]
Blank, I.; Pascual, E. C.; Devaud, S.; Fay, L. B.; Stadler, R. H.; Yeretzian, C.; Goodman, B. A. J. Agric. Food Chem.2002,50, 2356–2364. doi:10.1021/jf011329m
Return to citation in text: [1]
Straathof, N. J. W.; Gemoets, H. P. L.; Wang, X.; Schouten, J. C.; Hessel, V.; Noël, T. ChemSusChem2014,7, 1612–1617. doi:10.1002/cssc.201301282
Return to citation in text: [1]
Fang, Y.; Tranmer, G. K. Med. Chem. Commun.2016,7, 720–724. doi:10.1039/C5MD00552C
Return to citation in text: [1]
Bergami, M.; Protti, S.; Ravelli, D.; Fagnoni, M. Adv. Synth. Catal.2016,358, 1164–1172. doi:10.1002/adsc.201600019
Return to citation in text: [1]
Wiles, C.; Watts, P. Green Chem.2012,14, 38–54. doi:10.1039/C1GC16022B
Return to citation in text: [1]
Yoshida, J.-i. Flash Chemistry: Fast Organic Synthesis in Microsystems; John Wiley & Sons: Chichester, UK, 2008. doi:10.1002/9780470723425
Return to citation in text: [1]
Yoshida, J.-i. Chem. Commun.2005, 4509–4516. doi:10.1039/b508341a
Return to citation in text: [1]
Yoshida, J.-i.; Nagaki, A.; Yamada, T. Chem. – Eur. J.2008,14, 7450–7459. doi:10.1002/chem.200800582
Return to citation in text: [1]
Nieuwland, P. J.; Koch, K.; van Harskamp, N.; Wehrens, R.; van Hest, J. C. M.; Rutjes, F. P. J. T. Chem. – Asian J.2010,5, 799–805. doi:10.1002/asia.200900705
Return to citation in text: [1]
Gutmann, B.; Cantillo, D.; Kappe, C. O. Angew. Chem., Int. Ed.2015,54, 6688–6728. doi:10.1002/anie.201409318
Return to citation in text: [1]
Movsisyan, M.; Delbeke, E. I. P.; Berton, J. K. E. T.; Battilocchio, C.; Ley, S. V.; Stevens, C. V. Chem. Soc. Rev.2016,45, 4892–4928. doi:10.1039/C5CS00902B
Return to citation in text: [1]
Clark, J. D.; Shah, A. S.; Peterson, J. C.; Patelis, L.; Kersten, R. J. A.; Heemskerk, A. H.; Grogan, M.; Camden, S. Thermochim. Acta2002,386, 65–72. doi:10.1016/S0040-6031(01)00760-2
Return to citation in text: [1]
Hosmane, R. S.; Liebman, J. F. Struct. Chem.2002,13, 501–503. doi:10.1023/A:1020573723147
Return to citation in text: [1]
Nicolle, S. M.; Hayes, C. J.; Moody, C. J. Chem. – Eur. J.2015,21, 4576–4579. doi:10.1002/chem.201500118
Return to citation in text: [1]
Regitz, M.; Maas, G. Diazo Compounds Properties and Synthesis; Academic Press: Orlando, Florida, 1986.
Return to citation in text: [1]
Soldi, C.; Lamb, K. N.; Squitieri, R. A.; González-López, M.; Di Maso, M. J.; Shaw, J. T. J. Am. Chem. Soc.2014,136, 15142–15145. doi:10.1021/ja508586t
See for a recent example of hydrazone oxidation using manganese dioxide.
Return to citation in text: [1]
Roda, N. M.; Tran, D. N.; Battilocchio, C.; Labes, R.; Ingham, R. J.; Hawkins, J. M.; Ley, S. V. Org. Biomol. Chem.2015,13, 2550–2554. doi:10.1039/C5OB00019J
Return to citation in text: [1] [2]
Poh, J.-S.; Tran, D. N.; Battilocchio, C.; Hawkins, J. M.; Ley, S. V. Angew. Chem., Int. Ed.2015,54, 7920–7923. doi:10.1002/anie.201501538
Return to citation in text: [1] [2] [3]
Battilocchio, C.; Feist, F.; Hafner, A.; Simon, M.; Tran, D. N.; Allwood, D. M.; Blakemore, D. C.; Ley, S. V. Nat. Chem.2016,8, 360–367. doi:10.1038/nchem.2439
Return to citation in text: [1]
Tran, D. N.; Battilocchio, C.; Lou, S.-B.; Hawkins, J. M.; Ley, S. V. Chem. Sci.2015,6, 1120–1125. doi:10.1039/C4SC03072A
Return to citation in text: [1]
Esumi, T.; Yamamoto, C.; Fukuyama, Y. Synlett2013,24, 1845–1847. doi:10.1055/s-0033-1338968
Return to citation in text: [1]
Pieber, B.; Martinez, S. T.; Cantillo, D.; Kappe, C. O. Angew. Chem., Int. Ed.2013,125, 10431–10434. doi:10.1002/ange.201303528
Return to citation in text: [1]
Pieber, B.; Glasnov, T.; Kappe, C. O. Chem. – Eur. J.2015,21, 4368–4376. doi:10.1002/chem.201406439
Return to citation in text: [1]
Tsubogo, T.; Oyamada, H.; Kobayashi, S. Nature2015,520, 329–332. doi:10.1038/nature14343
Return to citation in text: [1]
Adamo, A.; Beingessner, R. L.; Behnam, M.; Chen, J.; Jamison, T. F.; Jensen, K. F.; Monbaliu, J.-C. M.; Myerson, A. S.; Revalor, E. M.; Snead, D. R.; Stelzer, T.; Weeranoppanant, N.; Wong, S. Y.; Zhang, P. Science2016,352, 61–67. doi:10.1126/science.aaf1337
Return to citation in text: [1]
Hessel, V.; Kralisch, D.; Kockmann, N., Eds. Novel Process Windows: Innovative Gates to Intensified and Sustainable Chemical Processes; Wiley-VCH: Weinheim, 2015.
Return to citation in text: [1]
How to cite this article:
Fanelli, F.; Parisi, G.; Degennaro, L.; Luisi, R. Beilstein J. Org. Chem.2017,13, 520–542. doi:10.3762/bjoc.13.51
As an organic chemist I’m involved in the development of new sustainable synthetic methodologies for the construction of new molecules with defined stereochemistry and functional properties.
Jointly with my coworkers we are involved in three main research themes:
1. Heterosubstituted Organolithiums. We mainly explore the reactivity of lithiated 3,4,5,6-membered N,S,O-heterocycles (aziridines, azetidines, oxazetidines, thietanes, oxazolines, piperazines, morfolines) and their utility in stereoselective synthesis. Our approach is focused in establishing the chemical and configurational stability of the lithiated intermediates as well as their structure in solution by using modern spectrometric and spectroscopic techniques such as in line -IR, in line-MS, NMR and DOSY.
2. Microreactor Technology and Flow-Chemistry. With the aim to design more sustainable synthetic processes, we set up, at the Depatment of Pharmacy, a well equipped “flow chemistry laboratory” named FLAME-Lab, for the development of continuous-flow microreactor-mediated organometallic and organocatalytic synthesis in both homegenous and heterogenous conditions.
3. Molecular Dynamics. As a “curiosity driven” research activity, we investigate the dynamic behavior of small molecules that could function as molecular switches with “on-off” states and as versatile scaffolds useful in catalysis and in “dynamic-controlled and predictable reactivity”.
Sustainable chemistry: how to produce better and more from less?
Green Chem., 2017, Advance Article DOI: 10.1039/C7GC02006F, Perspective
P. Marion, B. Bernela, A. Piccirilli, B. Estrine, N. Patouillard, J. Guilbot, F. Jerome
This review describes the rapid evolution of chemistry in the context of a sustainable development of our society. Written in collaboration between scientists from different horizons, either from public organizations or chemical companies, we aim here at providing recommendations to accelerate the emergence of eco-designed products on the market.
Sustainable chemistry: how to produce better and more from less?
aSOLVAY, 85 rue des frères Perret, BP 62, F-69192 Saint Fons Cedex, France
bINCREASE FR CNRS 3707/Centre de Recherche sur l’Intégration Economique et Financière, Université de Poitiers, 2 rue Jean Carbonnier, Bât A1, 86073 Poitiers Cedex 9, France
cBIOSYNTHIS, 6 Chemin de la Pierre Percée, 91410 Saint-Cyr-sous-Dourdan, France
dARD-Agro-industrie Recherches et Développements, Green Chemistry Department, Route de Bazancourt, F-51110 Pomacle, France
ePENNAKEM LLC, 3324 Chelsea avenue, Memphis, USA
fSEPPIC-AIR LIQUIDE, 127 chemin Poudrerie, 81105 Castres, France
gINCREASE FR CNRS 3707/Institut de Chimie des Milieux et Matériaux de Poitiers, Université de Poitiers, ENSIP, 1 rue Marcel Doré, 86073 Poitiers, France E-mail:francois.jerome@univ-poitiers.fr
Abstract
The International Symposium on Green Chemistry (ISGC) organized in 2013, 2015 and 2017 has gathered many senior and young talented scientists from all around the world (2200 attendees in three editions), either from academia or industry. Through outstanding conferences, communications, debates, and round tables, ISGC has been the witness of the rapid evolution of chemistry in the context of a sustainable development of our societies, not only at the scientific and industrial levels but also on education, networking and societal aspects. This critical review synthesizes the different points of view and the discussions having taken place at ISGC and gives a general picture of chemistry, including few scientific disciplines such as catalysis, processes, resource management, and environmental impact, among others, within the framework of sustainable development. This critical review, co-authored by researchers from public organizations and chemical companies (small, medium and large industrial groups) provides criteria and recommendations which, in our view, should be considered from the outset of research to accelerate the emergence of eco-designed products on the market.
Conclusions
Sustainable chemistry is the only mean to generate performant products and long lasting solutions able to generate business and profit for chemical industry. Performance is the best systemic answer for customer needs and our societies. Defining sustainable chemistry is, however, far to be an easy task because chemistry is a highly dynamic system. The sustainability of a value chain is for instance directly depending on the access to energy (and above all to its origin – coal, gas, biomass…) and on the supply of raw materials. In the current economic context, it could be not so easy to predict what will be the best source of energy or raw materials for a desired product in the future. The development of predictive tools is now essential and will represent probably one of the next scientific challenges in the coming years. During the last 20 years, utilization of renewable feedstocks in chemical processes has become a strategy of growing interest but it definitely does not guarantee the establishment of a sustainable chemistry. Indeed, in some cases, it is more sustainable to produce a chemical from a fossil carbon source using decarbonized energy than the reverse. It is very important to distinguish the carbon found in the final product from the carbon content corresponding to the energy which is required the product production (going from raw materials to manufacturing, end of life, etc.). In this area, the concept of biorefinery can help to secure developments and to minimize investments in production plant by mutualizing facilities and R&D initiatives. Cooperation with local producers can also be a valuable way to implement new bio‐based products while favouring sustainable agricultural practices. Whatever the raw materials (renewable or fossil), a complete and systemic life cycle analysis of the whole chain value (from resources to manufacturing, use and end of life) must be performed because it gives us an accurate picture of the overall economic, environmental and societal performances of a product in an application for a defined market. In general, one should never forget that sustainable chemistry should help the society to produce more and better (products). Emergence of sustainable innovations on the market takes a lot of time because chemists have to reinvent chemistry. To achieve our transition to a sustainable society, we must think differently and bring together the worlds of finance, manufacturers, researchers and public authorities. The current method of funding of research and innovation is not satisfying yet because too often based on short‐term projects and with high Technology Readiness Level. Governments have to realize that this funding method slows down, and sometime also hampers, the emergence of future sustainable innovations. Evolution of regulations with the aim of banning toxic, eco‐toxic or poor biodegradable products is an important driver for sustainable innovation. It is now seen and shared as a positive sign providing opportunities to develop systemically better solutions and allowing chemical companies advocating sustainable development and products as a must to stay in the competition. As examples, ban of CFC, replacement of chlorinated or other toxic solvents, substitution of endocrine disruptors lead to better solutions for the global benefit of our societies. Improving public perception and awareness on sustainable chemistry is on the way but more efforts will be needed in the future to definitely contribute to the emergence of eco‐ designed chemicals on the market.
/////////
Below we provide a bulleted list to summarize the main recommendations that are, in our views, essential for designing sustainable products. (1) Products design & Manufacture: For the intended application, sustainable chemicals must imperatively bring a global benefit, created by a scientific or technological breakthrough, while minimizing risks. They should also generate profit to emerge on the market. Products should be produced according to the 12 principles of green chemistry. In addition, their end of life should be integrated at the outset of research, (2) Resources: They should be available for future generations and should have low environmental impact (protecting endangered species, deforestation, erosion of biodiversity, contamination of natural resources, global warming, etc.), it should make progress the societal development of concerned area (sharing any benefits with local producer, no child labour, help developing countries, etc.) and their utilization should not destabilise other supply chains. Non edible raw material, a return to the idea of ‘localness’ and the need for closeness should be preferred, (3) Process: The ideal process would be a low Capex or a progressive Capex process and should be energy‐efficient, not use solvents, be without effluents, should limit the number of reactional and purification steps and should be developed rapidly to limit the associated risks and costs. Efforts are still needed for miniaturisation of equipment, intensification and development of continuous reactors, (4) Energy: The chemical industry is also energy intensive. Although less than 10% of fossil carbon is used for the manufacture of chemicals, finding decarbonized sources of energy is mandatory to avoid the depletion of carbon reserves and price increase and to ensure that future generations will have access to the same resource in the same amount, (5) Life cycle assessment: it should be assessed in all cases, the earlier the better, by preferring a ‘cradle to grave’ approach. It should give an accurate picture of the overall economic, environmental and societal performances of a product in an application for a defined market, (6) Education: we should improve public awareness and perception on sustainable chemistry to facilitate the acceptation of sustainable products by the consumer. More education programs should be launched in the future not only to reassure the consumer but also to create a pool of students better armed to tackle the future challenges of (sustainable) chemistry. The rapid development of digital tools should be helpful to address this issue, (7) Network: we should prefer working in an open innovation mode by bringing together the worlds of finance, manufacturers, researchers and public authorities to accelerate the emergence of eco‐designed chemicals on the market. Networks should enable local players to adapt to changes in their environment while optimising their economic and environmental efficiency, (8) Funding: A good balance between funding to applied research and basic research must be addressed in order to continuously generate scientific innovation. However, public authorities must realise that societal challenges are more important than the short term financial challenges faced by businesses. The current model of our economy based on rapid profitability is unfortunately not well adapted for these advances since long‐term investments will be needed for a more sustainable development of our society, (9) Legislation & Regulation: it should facilitate the emergence of sustainable chemicals by banning harmful chemicals for the human health and the environment, even those nowadays generating substantial profits. The registration process of improved sustainable chemicals by the concerned agencies should be quicker than now to speed up their integrations on the market, (10) Predictive methods: the development of tools to accurately predict the technical and application performances, the economic efficiency, the environmental and societal performance of a targeted product should be developed to limit the risks and costs associated with potential failure and to reassure the investors. It is also urgent to develop these tools for chemicals that are intended to be dispersed in nature.
spectroscopy, SYNTHESIS, UncategorizedComments Off on Route to Benzimidazol-2-ones via Decarbonylative Ring Contraction of Quinoxalinediones: Application to the Synthesis of Flibanserin, A Drug for Treating Hypoactive Sexual Desire Disorder in Women and Marine Natural Product Hunanamycin Analogue
Route to Benzimidazol-2-ones via Decarbonylative Ring Contraction of Quinoxalinediones: Application to the Synthesis of Flibanserin, A Drug for Treating Hypoactive Sexual Desire Disorder in Women and Marine Natural Product Hunanamycin Analogue
ACS AuthorChoice – This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes.
INTRODUCTION
Benzimidazol-2-ones 1 are an important class of heterocycles and a privileged scaffold in medicinal chemistry. They consist of cyclic urea fused with the aromatic backbone, which can potentially interact in a biological system by various noncovalent interactions such as hydrogen bonding and π stacking. Benzimidazolone derivatives exhibit a wide range of biological activities, and they are useful in treating various diseases including cancer, type II diabetes, central nervous system disorders, pain management, and infectious disease.1 Selected compounds embedded with a benzimidazol-2-one moiety along with their use are captured in Figure 1. It is worth mentioning that oxatomide drug with a benzimidazol-2-one core was approved for marketing a few years ago.2a Very recently, US Food and Drug Administration approved a new drug called flibanserin for the treatment of hypoactive sexual desire disorder (HSDD) in females, which contains benzimidazol-2- one motif.2b
CONCLUSIONS
We have developed a mild and new protocol for the synthesis of benzimidazol-2-ones from quinoxalinediones through decarbonylation. The present methodology can be an addition to the toolbox to prepare benzimidazolones, and it will be useful in medicinal chemistry, particularly, late-stage functionalization of natural products, drug scaffolds, or an intermediate containing quinoxaline-2,3-diones. As direct application of this method, we have successfully developed a new route for the synthesis of recently approved drug flibanserin and a urea analogue of antibiotic natural product hunanamycin A. Later application demonstrates the utility of the present method in late-stage functionalization
Synthesis of 1-(2-(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)ethyl)-1,3-dihydro-2Hbenzo[d]imidazol-2-one (Flibanserin)
HRMS (ESI): m/z calculated for C20H22ON4F3[M+H]+ 391.1740 found 391.1743;
Scheme 4. Synthesis of Flibanserin through Ring Contraction
The same methodology was applied for the synthesis of flibanserin, also known as “female viagra”, which is the first approved medication for treating HSDD in women and is classified as a multifunctional serotonin agonist antagonist.(14, 15) Our synthesis of flibanserin commenced with 1-benzyl-1,4-dihydroquinoxaline-2,3-dione 36,(16) which was reacted with known chloride 37(17) under the basic condition in DMF to give the desired product 38 in good yield. Compound 38 was subjected for the decarbonylative cyclization under the optimized condition to afford the product 39 in 59% yield. Finally, the benzyl group was deprotected using trifluoromethanesulfonic acid in toluene under microwave irradiation,(8b, 18) which gave flibanserin in excellent yield (Scheme 4). The final product was isolated as HCl salt, and all of the spectral data are in agreement with the published data.(15c)
Rahul D. Shingare completed his M.Sc (Chemistry) from Fergusson College, Pune in 2008. He worked as a research associate in Ranbaxy and Lupin New drug discovery center, Gurgaon and Pune respectively until 2012 and currently pursuing his doctoral research in NCL – Pune from 2012.
Current Research Interests:Antibacterial Natural Product Hunanamycin A: Total Synthesis, SAR and Related Chemistry.
Akshay Kulkarni completed his M.Sc. from Ferguson College, Pune University in the year 2015 and joined our group as a Project Assistant in the month of October, 2015.
Current research interest: Synthesis of silicon incorporated biologically active antimalerial compounds.
e-mail : as.kulkarni@ncl.res.in
Dr.D. Srinivasa Reddy
Organic Chemistry Division
CSIR-National Chemical Laboratory
Stahl, S. M.Mechanism of action of Flibanserin, A multifunctional serotonin agonist and antagonist (MSAA), in hypoactive sexual desire disorderCNS Spectrums2015, 20, 1 DOI: 10.1017/s1092852914000832
UncategorizedComments Off on ENHANCEMENT OF DISSOLUTION RATE AND SOLUBILITY OF LOSARTAN POTASSIUM BY USING SOLID DISPERSION METHOD β-CYCLODEXTRIN AS CARRIER
ABSTRACT In the present study an attempt was made to increase the therapeutic effectiveness of losartan potassium,by increasing the solubility and dissolution rate via solid dispersion using β-cyclodextrin as carrier. Losartan potassium is an Antihypertensive agent but failed to show good therapeutic effect. Eight solid dispersion formulations of losartan potassium were prepared by using different drug:polymer ratios viz.1:2,1:2,1:3,1:4 by novel methods like Hot melt extrusion,Lyophilization.prepared solid dispersions were evaluated. The blend of all the formulations showed good flow properties such as angle of repose, bulk density, tapped density. All the solid dispersion formulations were compressed into orodispersible tablets with weight equivalent to losartan potassium of 25mg by direct compression method using 6mm punch on 8 station rotary tablet punching machine. The prepared tablets were evaluated for its hardness, disintegration, weight variation, friability and invitro dissolution studies.The Infra Red spectra revealed that there is no incompatability between the drug and excipients. The prepared tablets were shown good post compression parameters and they passed all the quality control evaluation parameters as per I.P limits. Among all the formulations F4 and F8 formulations showed maximum % drug release i.e.93.83%(Lyophilization), 97.10%(Hot melt extrusion method) within 45min.these are compared with pure drug which shows %drug release58.67%. The optimized formulations were subjected to different kinetic models.the formulations were found to follow zero order release. optimized formulations Were subjected to Accelerated stability study for 3 months according to ICH guidelines.The results found to satisfactory.
Considering all evaluation parameters and % drug release F8 formulation shown better % drug release compared with F4 formulation. hence F8 formulation considered as optimised formulation.
CONCLUSION Losartan potassium is belongs to class II drugs, that is, characterized by low solubility and low permeability therefore, the enhancement of its solubility and dissolution profile is expected to significantly improve its bioavailability and reduce its side effects. The precompression blends of Losartan were characterized with respect to angle of repose, bulk density, tapped density, Carr’s index and Hausner’s ratio. The precompression blend of all the batches indicates well to fair flowability and compressibility. Among all the formulations F8 formulation, showed good result that is 97.10 % in 45 minutes. As the concentration of polymer increases the drug release was decreased.
Award given by Dr. M Sunitha Reddy Head of the Department, Centre for Pharmaceutical Sciences, Institute of Science &Technology, JNTU-H, Kukatpally, Hyderabad, India
Lifetime achievement award ……..WORLD HEALTH CONGRESS 2017 in Hyderabad, 22 aug 2017 at JNTUH KUKATPALLY. HYDERABAD, TELANGANA, INDIA, Award given by Dr. M Sunitha Reddy Head of the Department, Centre for Pharmaceutical Sciences, Institute of Science &Technology, JNTU-H, Kukatpally, Hyderabad, India
Green Chem., 2017, Advance Article DOI: 10.1039/C7GC01566F, Tutorial Review
Lian Yang, Tao Dong, Hrishikesh M. Revankar, Cheng-Pan Zhang
Advances of fluorination in aqueous media during the last few decades are summarized in this review
Advances in aqueous fluorination during the last few decades are summarized in this review. Fluorinated compounds have dominated a large percentage of agrochemicals and pharmaceuticals and a mass of functional materials. The incorporation of fluorine atoms into organic molecules has become one of the mainstream technologies for their functional modification. Water is very environmentally friendly and has advantageous physicochemical properties. Fluorination reactions in aqueous media are highly sought-after, and have attracted great attention in research areas ranging from medicinal chemistry to materials science. In early times and for a long time, fluorination was thought to be diametrically opposed to water or moisture. However, recent examples have conflicted with this viewpoint. The successful merger of “untamed” fluorine and “mild” water in chemical reactions has set up a new prospect for green chemistry. A considerable amount of remarkable research works have been carried out using water as a (co)solvent and/or a reactant for transformations including electrophilic, radical, or nucleophilic fluorination. We hope that this review will serve as a guide to better understand and to further broaden the field of fluorine chemistry in aqueous conditions.
Conclusion
The installation of fluorine atoms into organic and organometallic frameworks can dramatically change their physical, chemical, and biological properties. Organofluorides have entered many fields of science and technology with a tremendous impact on these domains. The development of efficient, selective, and mild methods to build C-F bonds is of great importance, which is highly desirable to keep up with the rapidly growing demand of novel fluorine-containing scaffolds. In early times, most fluorination reactions required harsh conditions and moisture-sensitive, highly toxic, and explosive atomic fluorine transfer agents like fluorine gas, xenon difluoride, hypofluorite, antimonytrifluoride, and diethylaminosulfurtrifluoride. The discovery of stable electrophilic fluorination reagents such as Selectflour and NFSI has remarkably changed the dilemma, which realized a large number of safe, mild, and easily controllable electrophilic and radical fluorination reactions in aqueous media. Although the exact mechanisms are still unclear at present, it does never hamper the green fluorination method development with these reagents. A mass of successful examples have confirmed that the aqueous reaction medias have positive impacts on electrophilic and radical fluorination reactions with using the N-F reagents and in many cases water can also be a nucleophile for the entire conversions.
In addition, water was generally thought to be an unsuitable medium for nucleophilic fluorination because the fluoride ions can be “trapped” in aqueous medias by hydrogen bonding and become unreactive. Thus, their use in organic synthesis has been quite limited to polar aprotic solvents. Although the strong hydrogen bond formed between fluoride and water diminished the nucleophilicity of fluoride ions, the recent examples of nucleophilic fluorination in aqueous media have implied that this “negative” effect does not always harm the reaction. Besides, the radioisotope 18F has been considered to be a good choice for PET imaging owing to its desirable radiochemical properties. With a half-life of 110 minutes, the introduction of [ 18F]fluorine atoms into biomolecules has to be completed in a swift manner to minimize the loss of radioactivity. Nucleophilic incorporation of [18F]F‒ in aqueous conditions could rapidly produce [18F]fluorinesubstituted biomolecules, which avoided azeotropic drying process, shortened the production time, and minimized the loss of activity. We summarized the recent aqueous fluorination reactions in three sections according to their possible mechanisms. The successful amalgamation of “ill-tempered” fluorine and “benign” water has boded well for green fluorine chemistry. Water behaves as a cosolvent to dissolve fluorination reagents and/or as a reactant for bifunctionalization. Since the aspects of green chemistry has drawn much attention from the society, the pursuit of more efficient and milder reaction conditions for greener fluorination in aqueous medias will never end. Although a large number of research works have been published in this area, it’s only the tip of the iceberg with a wide scope for improvement. We hope that this review will serve as a guide to understand and to further broaden the field of aqueous fluorine chemistry. To meet the principle of green chemistry in modern synthesis, the development of new fluorination reagents as well as valid catalytic systems is crucial for mild and selective C-F bond formation. It’s undoubted that a growing number of green fluorination methodologies in aqueous media will be witnessed in the near future.
Alice Coletti, Francesco Antonio Greco, Daniela Dolciami, Emidio Camaioni, Roccaldo Sardella, Maria Teresa Pallotta, Claudia Volpi, Ciriana Orabona, Ursula Grohmann, Antonio Macchiarulo
Structure-function relationships of IDO1 and structure-activity relationships of inhibitors are discussed with an outlook on next generation IDO1 ligand.
aDepartment of Pharmaceutical Sciences, University of Perugia, via del Liceo 1, 06123 Perugia, Italy E-mail:antonio.macchiarulo@unipg.it Fax: +39 075 585 5161 Tel: +39 075 585 5160
bDepartment of Experimental Medicine, University of Perugia, P.le Gambuli, 06132 Perugia, Italy
Abstract
Indoleamine 2,3-dioxygenase 1 (IDO1) mediates multiple immunoregulatory processes including the induction of regulatory T cell differentiation and activation, suppression of T cell immune responses and inhibition of dendritic cell function, which impair immune recognition of cancer cells and promote tumor growth. On this basis, this enzyme is widely recognized as a valuable drug target for the development of immunotherapeutic small molecules in oncology. Although medicinal chemistry has made a substantial contribution to the discovery of numerous chemical classes of potent IDO1 inhibitors in the past 20 years, only very few compounds have progressed in clinical trials. In this review, we provide an overview of the current understanding of structure–function relationships of the enzyme, and discuss structure–activity relationships of selected classes of inhibitors that have shaped the hitherto few successes of IDO1 medicinal chemistry. An outlook opinion is also given on trends in the design of next generation inhibitors of the enzyme.
Introduction Indoleamine 2,3-dioxygenases (IDOs) are heme-containing proteins that catalyze the oxidative cleavage of the indole ring of tryptophan (L-Trp, 1) to produce N-formyl kynurenine (2) in the first rate limiting step of the kynurenine pathway (Figure 1).1,2 The family includes two related enzymatic isoforms, namely IDO1 and IDO2, sharing ∼60% of sequence similarity and featuring distinct biochemical features.3,4 A third enzyme of the family is the tryptophan-2,3-dioxygenase (TDO2) which is structurally unrelated to IDO1 and IDO2 and is endowed with a more stringent substrate specificity for L-Trp.5 Although TDO2 is expressed almost exclusively in hepatocytes where it regulates L-Trp catabolism in response to the diet, IDO1 and IDO2 are widely expressed in macrophages and dendritic cells exerting immunoregulatory functions.6 These are accomplished through two major mechanisms including depletion of tryptophan and production of bioactive metabolites along the kynurenine pathway. Specifically, the first mechanism postulates that raising levels of Interferon-γ (IFN-γ) induce IDO1 expression in macrophages and dendritic cells during pathogen infection, leading to consumption of L-Trp and growth arrest of pathogens, whose diet is sensitive to this essential nutrient.7 The second mechanism grounds on production of kynurenine metabolites that bind to the aryl hydrocarbon receptor (AhR), activating signaling pathways that enhance immune tolerance.8-10 Among the three proteins, IDO1 is the most characterized enzyme and in recent years a second signal-transducing function was revealed for this protein.11,12 In particular, this signalling function relies on the presence of two immunoreceptor tyrosine-based inhibitory motifs (ITIMs) in the non-catalytic domain of IDO1.13 The immunosuppressive cytokine transforming growth factor-β (TGF-β) stimulates phosphorylation of ITIMs by Sarcoma-family (Src-family) kinases and consequent interaction of the phosphorylated enzyme with Src Homology 2 domain Phosphatase-1 (SHP-1) and Src Homology 2 domain Phosphatase-2 (SHP-2), eventually leading to long-term expression of IDO1 and immune tolerance. Conversely, in pro-inflammatory environmental conditions, increasing levels of interleukin-6 (IL-6) trigger the interaction of
phosphorylated IDO1 with suppressor of cytokine signalling 3 (SOCS3) that tags the enzyme for proteasome degradation, shortening IDO1’s half-life and promoting inflammatory response.14 The breakthrough discovery that IDO1 plays a crucial role in the maintenance of maternal immune tolerance ushered in a great deal of interest on the enzyme, by then considered a master regulatory hub of immunosuppressive pathways in pregnancy, autoimmune diseases, chronic inflammation, and cancer.15 In this framework, elevated levels of IDO1 expression found in several tumour cells were associated to the participation of the enzyme in the tumor immuno-editing process which sets up immune tolerance to tumor antigens.16,17 On this basis, academic groups and pharmaceutical companies have been engaged in the development of IDO1 inhibitors.18 Although part of these efforts has proved successful, with a large array of potent and selective inhibitors being disclosed in literature and patent applications, only few compounds have hitherto entered clinical trials (3-7, Figure 1).2,19-22 At this regard, some studies have highlighted challenges in the development of enzyme inhibitors mostly due to redox properties of the enzyme that may account for unspecific mechanism of inhibition of many compounds discovered in preclinical studies.23,24 Starting with an overview on the architecture of IDO1 and its structure-function relationships, in this article we discuss selected classes of inhibitors that have shaped advances in the medicinal chemistry of IDO1, providing outlooks on future trends in the design of next generation compounds.
Professor and Head of the Department E-mail:helen.p@rmp.srmuniv.ac.in Area: Chemistry Affiliation: Department of Chemistry, Ramapuram Campus, SRM University
Education
Ph.D.
Organic Synthesis
Bharathidasan University, Tiruchirapalli, 2000
M.Sc.
General Chemistry
Bharathidasan University, Tiruchirapalli, 1994
B.Sc.
General Chemistry
Bharathidasan University, 1992
Other Details:
Course
Chemistry
Principles of Environmental Science
Research Interests
Organic Synthesis
Medicinal Chemistry
Crystal Growth
Molecular Docking
Nano Synthesis
Selected PublicationS
A. Santhoshkumar, Helen P. Kavitha*, R. Suresh, Hydrothermal Synthesis, Characterization and Antibacterial Activity of NiO Nanoparticles, Journal of Advanced Chemical Sciences-Article in press
R. Kavipriya, Helen P. Kavitha, B. Karthikeyan, and A. Nataraj,” Molecular structure, spectroscopic (FT-IR, FT-Raman), NBO analysis of N,N0-diphenyl-6-piperidin-1-yl-[1,3,5]-triazine-2,4-diamine, Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 150 (2015) 476–487.
S. Sathishkumar, Helen P. Kavitha and S. Arulmurugan, In-silico anti-inflammatory evaluation of some novel tetrazolo and triazolodiazepine derivatives against COX-2 protien, International Journal of Advanced Chemical Science and Applications, 3(1), 2015
S. Arulmurugan and Helen P Kavitha, S. Sathishkumar and R. Arulmozhi. Review on biologically active benzimidazole, Miniriveviews in organic chemistry, 12(1), 178-195, 2015.
S. Sathishkumar and Helen P. Kavitha, Synthesis, Characterization and Anti-inflammatory Activity of Novel Triazolodiazepine Derivatives, IOSR Journal of Applied Chemistry, 8(1),47-52, 2015.
A. Silambarasan, Helen P. Kavitha, S. Ponnusamy, M. Navaneethan, Y. Hayakawa, Investigation of photocatalytic behavior of l-aspartic acid stabilized Zn(1−x)MnxS solid solutions on methylene blue Applied Catalysis A: General, 476, 22,1-8, 2014.
S. Sathish Kumar and Helen P. Kavitha, Synthesis and Biological Applications of Triazole Derivatives-A Review Mini-Reviews in Organic Chemistry, 10(1), 2013.
Helen P. Kavitha and S. Arulmurugan Synthesis, characterization and cytotoxic activity of benzoxazole, benzimidazole, imidazole and tetrazole Acta pharmaceutica 63(2), 253-264, 2013
Jasmine P. Vennila, Jhon Thiruvadigal, Helen P Kavitha, G. Chakkaravarthi and V. Manivannan, N-[2-(3,4-Dimeth-oxy¬phenyl)eth¬yl]-N-methyl-naphthalene-1-sulfonamide, Acta Crystallogr Sect E, 68(Pt 3): o890, 2012.
Jasmine P. Vennila, D. Jhon Thiruvadigal, and Helen P. Kavitha Antibacterial evaluation of some organic compounds as potential inhibitors for glucosamine-6-phospate synthase Journal of Pharmacy Research, 5(4), 1963-1966, 2012.
Helen P. Kavitha and R. Arulmozhi , Synthesis, Characterization and Anti inflammatory Activity of Some New Tetrazoles Derived from Quinazoline-4-one , International Journal of Chemistry, 1-6, 2012.
S. Arulmurugan, Helen P. Kavitha and S. Sathish Kumar. Synthesis, characterization and molecular docking studies of some new benzoxazole, benzimidazole, imidazole and tetrazole compounds as potential inhibitors for thymidylate synthase, International Journal of Science and Technology, 1, 1-11 2012.
Jasmine P. Vennila, Jhon Thiruvadigal, G. E. Theboral Sugi Kamala, Helen P Kavitha, Chakkaravarthi and V. Manivannan N-[2-(3,4-Dimeth-oxy¬phen¬yl)eth¬yl]-N-methyl¬benzene-sulfonamide” Acta Crystallogr Sect E Struct Rep Online. 68(Pt 3), o882, 2012.
Helen P Kavitha, A. Silambarasan, S. Ponnusamy, M. Navaneethan and Y, Hayakawa, Monodispersed synthesis of hierarchical wurtzite ZnS nanostructures and its functional properties” Materials Letters 81, 209-211, 2012.
Jasmine P. Vennila, Jhon Thiruvadigal, Helen P Kavitha, G. Chakkaravarthi and V. Manivannan, 2-(4-Bromophenyl)-3-(4-hydroxyphenyl)-1,3-thiazolidin-4-one” Acta Cryst., E67, o1902, 2011.
Jasmine P. Vennila, D Jhon Thiruvadigal, Helen P Kavitha, G. Chakkaravarthi and V. Manivannan 2,4-Bis(morpholin-4-yl)-6-phenoxy-1,3,5-triazine” Acta Cryst. E67, o2451, 2011.
Jasmine P. Vennila, Jhon Thiruvadigal, Helen P Kavitha, G. Chakkaravarthi and V. Manivannan, 2-Chloro-4,6-bis(piperidin-1-yl)-1,3,5-triazine” Acta Cryst. E67, o312, 2011.
Helen P. Kavitha, Samiappan Sathish kumar and Ramachandran Balajee Antimicrobial Activity and Molecular Docking Studies of Some Novel Tetrazolo Diazepine Derivatives, Journal of Pharmacy Research,4(9), 2946-2949, 2011
Helen P. Kavitha and R. Arulmozhi Study of Antimicrobial and Analgesic Activities of Novel Tetrazoles Derived from Quinazolin-4-one, Journal of Pharmacy Research , 4(12), 4696-4698, 2011.
R.Thilagavathy, Helen.P.Kavitha, R.Amrutha and Bathey.R.Venkatraman Structural parameters, charge distribution and vibrational frequency analysis using theoretical SCF methods, Elixir Comp. Chem. 40, 5514-5516, 2011.
S. Sathish Kumar, Helen P. Kavitha, S. Arulmurugan and B. R. Venkatraman, Review on Synthesis of Biologically Active Diazepam Derivatives Mini-Reviews in Organic Chemistry, 8, 1-17, 2011.
Jasmine P. Vennila, Jhon Thiruvadigal, Helen P Kavitha, B. Gunasekaran and V. Manivannan, (E)-4-{(4-Bromopenzylidene) amino} phenol, Acta Cryst, E66, O316, 2010.
Subramaniyan Arulmurugan and Helen P. Kavitha, 2-Methyl-3-{4-[-(1H-tetrazol-5-yl)ethylamino]phenyl}-3H-quinazolin-4-one”, Molbank, M695,1-5, 2010.
S. Arulmurugan, Helen P. Kavitha and B. R. Venkatraman Biological Activities of Schiff Base and its Complexes”: A Review, Rasayan Journal of Chemistry, 3(3), 385-410, 2010.
R. Thilagavathy, Helen P Kavitha and B. R. venkatraman Isolation, Characterization and Anti-Inflammatory Property of Thevetia Peruviana E-journal of Chemistry,7(4), 1584-1590, 2010.
Subramaniyan Arulmurugan, Helen P. Kavitha, B. R. Venkatraman, Synthesis, Characterization and Study of antibacterial activity of some novel tetrazole derivatives” , Orbital Elec. J. Chem, 2(3), 271-276, 2010.
With R. Thilagavathi “Synthesis of 3-{4-[4-(benzylideneamino) benzene sulfonyl]-phenyl}-2-phenylquinazoline-4(3H)-one” Molbank, M589, 2009.
With S.Sathish Kumar “Synthesis of 3-Methyl-1-Morpholin-4-ylmethyl-2,6-Diphenylpiperidin-4-One”, Molbank, M617, 2009.
With S. Sathish Kumar “6-Methyl-2,7-Diphenyl-1,4-Diazepan-5-One”, Acta Cryst., E65, (o3211), 2009.
With R. Thilagavathi “2-phenyl-4H-3,1-benzoxazian-4-one”, Acta., Cryst. (E), E65, (o127), 2009.
With Jasmine P. Vennila “4-nitrophenyl napthalene-1-sulfonate”, Acta Cryst. (E), (o1848), E64, 2008.
With R. Arulmozhi,”1- Naphthyl-9-HCarbazole-4-Sulphonate”, Acta Cryst., E66, 010208, 2008.
With T. Nithya “Antibacterial activity of Solanum Trilobatum”, Journal of Ecotoxicol.Environ. Monit., 14, (237-239), 2004.
“Synthesis and Antimicrobial activity of1-(9’Acridinyl)-5-substituted phenyl Tetrazoles”, Asian Journal of chemistry, 16, (1191-1192), 2004.
With S. V. Selva bala “Study of Hypoglysemic Activity of Solanum Xanthocarpum L. on Alloxanised Diabetic Rats”, Adv. Pharmacol Toxicol., 4, (19-24), 2003.
Helen P. Kavitha “Study of anajesic activity some novel 1-(9’Acridinyl)-5-substituted phenyl tetrazoles”, Indian Journal of Chemical Technology, 9, (361-362), 2002.
With S.Malliga, “Effect of Soaking the Wood of Emblica officinalis,on Some Water Parameters”, Journal of Swamy Bot. 15, (89-90), 1998.
Working Papers
With S. Arulmurugan, “Review on Biologically Active Benzimidazole derivatives”: Mini reviews in organic Chemistry.
Academic Experiences
Assistant Professor(S. G), SRM University, Ramapuram from Sep 2007 to Jun 2012
Lecturer, SRM University, Ramapuram from Aug 2004 to Aug 2007
Senior Lecturer, VRS College, Villupuram from Aug 2002 to May 2004
Lecturer, VRS College, Villupuram, from Aug 2000 to May 2002
Lecturer, ADM College for Women, Nagapattinam from July 94 to April 95
Other Professional Experiences
4 Scholars have been awarded Ph.D Degree
Guiding 3 Ph.D candidates
Guided 8 M. Phil and 20 M. Sc projects
Principal Investigator for a pilot project funded by SRM University (completed)
Co-investigator for a UGC major project (completed)
Reviewer for International Journals
Convenor for the National Conference on New Renaissance in Chemical Research, 2011 and 2015.
Member Board of Studies –Chemistry,SRM University.
Doctoral Committee member in Karunya University
Undertaking consultancy work in the department
Question paper setter for various universities
Convenor for many programmes conducted in the campus
Chief Superintendent for SRM University-Ramapuram campus
Member in various professional bodies such as MISTE, FICCE and CTA
Author of five books in chemistry
Executive council member in Association of Chemistry Teachers, Mumbai
Achievement and Award
Received Award and cash prize for Research from SRM University from the year 2006-15
SYNTHESIS, UncategorizedComments Off on Control of stereoselectivity of benzylic hydroxylation catalysed by wild-type cytochrome P450BM3 using decoy molecules
Control of stereoselectivity of benzylic hydroxylation catalysed by wild-type cytochrome P450BM3 using decoy molecules
Catal. Sci. Technol., 2017, Advance Article DOI: 10.1039/C7CY01130J, Paper
Kazuto Suzuki, Joshua Kyle Stanfield, Osami Shoji, Sota Yanagisawa, Hiroshi Sugimoto, Yoshitsugu Shiro, Yoshihito Watanabe
The benzylic hydroxylation of non-native substrates was catalysed by cytochrome P450BM3, wherein “decoy molecules” controlled the stereoselectivity of the reactions.
The hydroxylation of non-native substrates catalysed by wild-type P450BM3 is reported, wherein “decoy molecules”, i.e., native substrate mimics, controlled the stereoselectivity of hydroxylation reactions. We employed decoy molecules with diverse structures, resulting in either a significant improvement in enantioselectivity or clear inversion of stereoselectivity in the benzylic hydroxylation of alkylbenzenes and cycloalkylbenzenes. For example, supplementation of wild-type P450BM3 with 5-cyclohexylvaleric acid-L-phenylalanine (5CHVA-Phe) and Z-proline-L-phenylalanine yielded 53% (R) ee and 56% (S) ee for indane hydroxylation, respectively, although 16% (S) ee was still observed in the absence of any additives. Moreover, we performed a successful crystal structure analysis of 5CHVA-L-tryptophan-bound P450BM3 at 2.00 Å, which suggests that the changes in selectivity observed were caused by conformational changes in the enzyme induced by binding of the decoy molecules.
UncategorizedComments Off on Guest blogger, Dr Pravin Patil, Synthesis of Extended Oxazoles III: Reactions of 2-(Phenylsulfonyl)methyl-4,5-Diaryloxazoles
Professor, Organic Chemistry: Organic and Medicinal Chemistry
Typical Procedure for Aluminum/HgCl2-Mediated Desulfonylation for Synthesis of 4 (Eq. 1) and 18 (Table 2). To a solution of the alkylated 2-(sulfonylethyl)-4,5-diphenyloxazole 5 (0.12 mmol, 1.0 equiv) and crystals of mercuric chloride (0.034 mmol, 0.3 equiv), in methanol (15 mL), was added an excess of food-grade aluminum foil (2.32 mmol, 20 equiv) with vigorous stirring under a nitrogen atmosphere. The resulting heterogeneous mixture was heated at reflux until the metal disappeared. The reaction mixture was then allowed to cool to room temperature and filtered through a Celite bed followed by washing with methanol (2 x 15mL). The filtrate was concentrated to a crude residue which was submitted to gravity-column chromatography on silica gel to provide 2-methyl-4,5-diphenyloxazole 4 (96%) or 2-ethyl-4,5-diphenyloxazole 18 (97%).
General procedure for Magnesium/HgCl2-Mediated Desulfonylation of Alkylated Sulfones 5-17. To a stirred solution of an alkylated 2-(phenylsulfonyl)methyl-4,5-diphenyloxazole (0.12 mmol, 1.0 equiv. from Table 1) in methanol (5 mL) was added magnesium turnings (1.73 mmol, 15 equiv) and crystals of mercuric chloride (0.012 mmol, 0.1 equiv) at room temperature. The reaction mixture was stirred at room temperature (2 h) while monitoring the reaction progress by TLC. After the reaction was complete, the reaction mixture was filtered through a Celite bed followed by washing with methanol (2 x 10 mL). The filtrate was concentrated and the resultant crude residue was submitted to gravity-column chromatography on silica gel (hexane/ethyl acetate) to afford the pure products 18–27 listed in Table 2.
Typical Procedure for Aluminum/HgCl2-Mediated Desulfonylation for Synthesis of 4 (Eq. 1) and 18 (Table 2). To a solution of the alkylated 2-(sulfonylethyl)-4,5-diphenyloxazole 5 (0.12 mmol, 1.0 equiv) and crystals of mercuric chloride (0.034 mmol, 0.3 equiv), in methanol (15 mL), was added an excess of food-grade aluminum foil (2.32 mmol, 20 equiv) with vigorous stirring under a nitrogen atmosphere. The resulting heterogeneous mixture was heated at reflux until the metal disappeared. The reaction mixture was then allowed to cool to room temperature and filtered through a Celite bed followed by washing with methanol (2 x 15mL). The filtrate was concentrated to a crude residue which was submitted to gravity-column chromatography on silica gel to provide 2-methyl-4,5-diphenyloxazole 4 (96%) or 2-ethyl-4,5-diphenyloxazole 18 (97%).
General procedure for Magnesium/HgCl2-Mediated Desulfonylation of Alkylated Sulfones 5-17. To a stirred solution of an alkylated 2-(phenylsulfonyl)methyl-4,5-diphenyloxazole (0.12 mmol, 1.0 equiv. from Table 1) in methanol (5 mL) was added magnesium turnings (1.73 mmol, 15 equiv) and crystals of mercuric chloride (0.012 mmol, 0.1 equiv) at room temperature. The reaction mixture was stirred at room temperature (2 h) while monitoring the reaction progress by TLC. After the reaction was complete, the reaction mixture was filtered through a Celite bed followed by washing with methanol (2 x 10 mL). The filtrate was concentrated and the resultant crude residue was submitted to gravity-column chromatography on silica gel (hexane/ethyl acetate) to afford the pure products 18–27 listed in Table 2.
Typical procedure: Synthesis of Oxaprozin
Ethyl 3-(4,5-diphenyloxazol-2-yl)-3-(phenylsulfonyl)propanoate(28). To a prechilled solution of 2-(phenylsulfonyl)methyl-4,5-diphenyloxazole 3 (100 mg, 0.27 mmol) in dry THF (15 mL) was added potassium tert-butoxide (33 mg, 0.29 mmol) under a nitrogen atmosphere. The resulting yellow solution was stirred (5°C) for 30 min. To the reaction mixture was slowly added ethyl bromoacetate (49 mg, 32.4 μL, 0.29 mmol) and stirring was continued (16 h) at room temperature. Upon completion of reaction as indicated by TLC, the reaction mixture was quenched with cold water (20 mL) and extracted with dichloromethane (2 x 20 mL). The organic layers were combined, dried over anhydrous sodium sulfate and concentrated to obtain a crude oily residue. The residue was submitted to gravity-column chromatography on silica gel (hexane/ethyl acetate, 4:1) afford pure ethyl 3-(4,5-diphenyloxazol-2-yl)-3-(phenylsulfonyl)propanoate 28 as off-white solid ( 88 mg, 72%).
Ethyl 3-(4,5-diphenyloxazol-2-yl)acrylate (29). To a cooled (5°C) solution of sulfonyloxazole ester 28 (225 mg, 0.49 mmol) in dry THF was added potassium tert-butoxide (60.2 mg, 0.54 mmol) under nitrogen and the reaction mixture was then stirred at 5-10°C (2 h) while monitoring by TLC. After completion of the reaction, the reaction mixture was extracted with dichloromethane (2 x 25 mL) followed by washing the extracts with water and brine then drying over anhydrous Na2SO4. Removal of the drying agent and concentration of the filtrate gave a crude residue which was submitted to gravity-column chromatography (hexane/ethylacetate, 4:1) to provide unsaturated oxazole ester 29 as a colorless oil (100 mg, 65%).
Ethyl 3-(4,5-diphenyloxazol-2-yl)propanoate (30).17 The unsaturated oxazole ester 30 (160 mg, 0.50 mmol) was dissolved in methanol (25 mL) then 10% Pd/C (16 mg, 10% wt/wt) was added at room temperature. The reaction mixture was purged with nitrogen while stirring followed by the addition of hydrogen gas (balloon) and then stirring was continued (16 h) under an atmosphere of hydrogen. Upon completion of reaction, the reaction mixture was filtered through a bed of Celite while washing with methanol (2 x 30 mL). The combined filtrates were concentrated and the crude residue was submitted to gravity-column chromatography (hexane/ethyl acetate, 4:1) to afford 30 as an off-white solid (129 mg, 80%).
Methyl 3-(4,5-diphenyloxazol-2-yl)propanoate (31).13To a clear solution of sulfonyloxazole ester 28 (80 mg, 0.173 mmol) in methanol (10 mL) was added magnesium turnings (63 mg, 2.60 mmol) followed by solid mercuric chloride (4.7 mg, 0.017 mmol) at room temperature. The resulting reaction mixture was stirred (2 h) while monitoring the reaction progress by TLC. After completion of the reaction, the heterogeneous mixture was then filtered through a Celite bed followed by washing with methanol (2 x 15 mL). The methanolic filtrates were combined and concentrated to afford a crude residue. The residue was submitted to gravity-column chromatography (hexane/ethylacetate, 4:1) to provide ester 31 as an off-white solid (52 mg, 97%).
3-(4,5-Diphenyloxazol-2-yl)propanoic acid(Oxaprozin)(32).13 Ethyl ester 30 (128 mg, 0.39 mmol) or methyl ester 31 (65 mg, 0.21 mmol) and 20% aquous NaOH solution (3 mL) was stirred overnight at room temperature. Upon completion of reaction as indicated by TLC, the reaction mixture was slowly acidified to pH 3-4 using conc. HCl (3 mL) at room temperature and stirring was continued (3 h). After the neutralization was complete the reaction mixture was diluted with cold water (15 mL) and extracted with dichloromethane (2 x 15 mL). The organic extracts were combined, dried over anhydrous Na2SO4 and concentrated to give a white solid residue. The residue was submitted to gravity-column chromatography (chloroform/methanol, 9:1) to afford pure Oxaprozin 32 as white solid (80 mg, 68%, from the ethyl ester 30) or (60 mg, 97%, from the methyl ester 31).
ABOUT GUEST BLOGGER
Dr. Pravin C. Patil
Postdoctoral Research Associate at University of Louisville
Dr. Pravin C Patil completed his B.Sc. (Chemistry) at ASC College Chopda (Jalgaon, Maharashtra, India) in 2001 and M.Sc. (Organic Chemistry) at SSVPS’S Science College Dhule in North Maharashtra University (Jalgaon, Maharashtra, India) in year 2003. After M.Sc. degree he was accepted for summer internship training program at Bhabha Atomic Research Center (BARC, Mumbai) in the laboratory of Prof. Subrata Chattopadhyay in Bio-organic Division. In 2003, Dr. Pravin joined to API Pharmaceutical bulk drug company, RPG Life Science (Navi Mumbai, Maharashtra, India) and worked there for two years. In 2005, he enrolled into Ph.D. (Chemistry) program at Institute of Chemical Technology (ICT), Matunga, Mumbai, aharashtra, under the supervision of Prof. K. G. Akamanchi in the department of Pharmaceutical Sciences and Technology.
After finishing Ph.D. in 2010, he joined to Pune (Maharashtra, India) based pharmaceutical industry, Lupin Research Park (LRP) in the department of process development. After spending two years at Lupin as a Research Scientist, he got an opportunity in June 2012 to pursue Postdoctoral studies at Hope College, Holland, MI, USA under the supervision of Prof. Moses Lee. During year 2012-13 he worked on total synthesis of achiral anticancer molecules Duocarmycin and its analogs. In 2014, he joined to Prof. Frederick Luzzio at the Department for Chemistry, University of Louisville, Louisville, KY, USA to pursue postdoctoral studies on NIH sponsored project “ Structure based design and synthesis of Peptidomimetics targeting P. gingivalis.
During his research experience, he has authored 23 international publications in peer reviewed journals and inventor for 4 patents.
Prof K. G. Akamanchi
Prof. K. G. Akamanchi
Professor, Pharmaceutical Sciences and Technology
Email ID : kg.akamanchi@ictmumbai.edu.in
Reseach Area : Development of Novel Methodologies, Hypervalent Iodine Chemistry, Synthesis of drug and drug intermediates, Design and Synthesis of Potential bioactive molecules, Protein Isolation and Bioassay
SEE…………
Frederick A. Luzzio
Professor, Organic Chemistry: Organic and Medicinal Chemistry
502-852-7323
502-852-6066
faluzz01@louisville.edu
About
The long term goals of our research are focused at the interface of chemistry and biology. We are interested in solving problems in biomedicine using the techniques and application of synthetic organic, medicinal and natural products chemistry. Toward our goals in biomedicine we concentrate our efforts in the following three areas of organic chemistry: (1) the development of new methods and strategy which are applicable to the synthesis of biologically active compounds; (2) the total synthesis of a wide range of complex molecules including natural products, pharmaceutical leads and their analogues; and (3) the isolation and discovery of biologically active compounds from natural sources. Within our objectives in item 1 (above), we have had a long-term collaboration with the Clinical Pharmacology Section of the National Cancer Institute in which we have synthesized metabolites and analogues of thalidomide, a small-molecule immunomodulator and angiogenesis inhibitor. The derivatives and analogues of thalidomide were stereospecifically synthesized in order to ascertain the mode of action and the molecular target of this small molecule. Ultimately, the synthetic studies are leading to analogues of thalidomide which are more potent, but which have less undesirable side effects than the parent compound. In the neurosciences area we have completed an enantioselective synthesis of both optical isomers of a key intermediate in preparing the histrionicotoxins, a group of compounds which are isolated for the neurotoxic Amazon “poison dart” frogs. One of our present natural products projects (under item 3,above) entails the isolation, neurotoxicity assays and synthesis of a series of naturally-occurring compounds called acetogenins from the North American paw paw tree Asimina triloba. The isolation, purification and structural confirmation of the natural products has been conducted in collaboration with the Neurosciences Department within the University of Louisville School of Medicine. In the area of anti-infectives (under 1), we are designing and synthesizing an array of nitrogen and nitrogen/oxygen heterocyclic scaffolds bearing acetylenic and azido groups for use in the so-called “click reaction.” The multiply-connected scaffolds have proven to be effective for inhibiting micro-organisms working in tandem to produce biofilms necessary for their establishment and survival.
Education
1976 B.S. Vanderbilt University
1979 M.S. Tufts University
1982 Ph.D. Tufts University
1982-1985 Postdoctoral Fellow, Harvard University
Current Service
Executive Committee/Treasurer, International Society of Heterocyclic Chemistry HETCHEM@louisville.edu












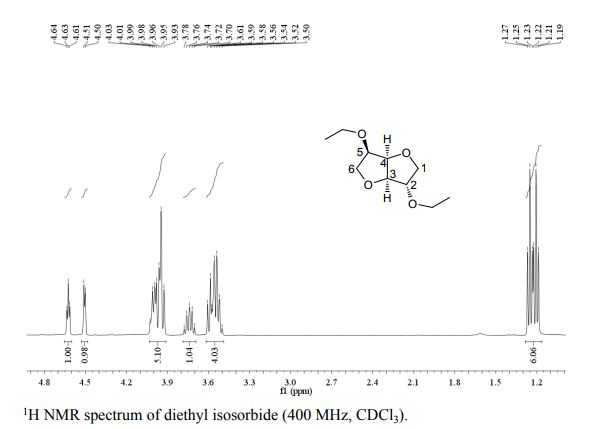
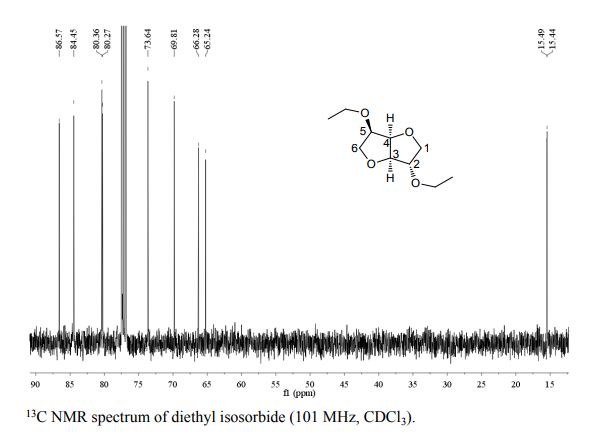
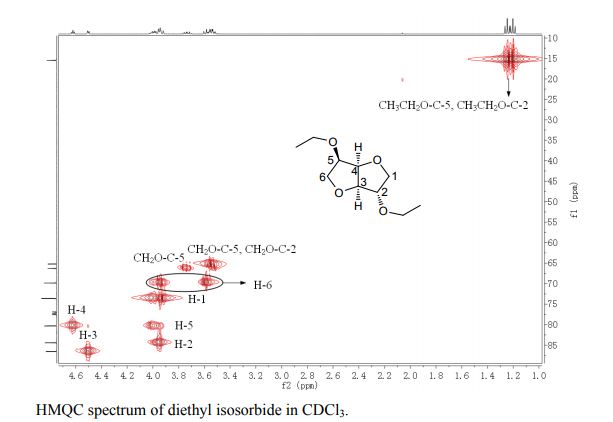
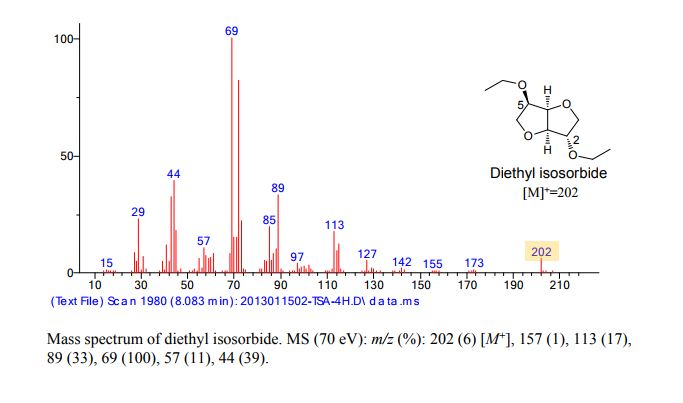


![[1860-5397-13-51-1]](/bjoc/content/figures/1860-5397-13-51-1.png?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i1]](/bjoc/content/inline/1860-5397-13-51-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i2]](/bjoc/content/inline/1860-5397-13-51-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i3]](/bjoc/content/inline/1860-5397-13-51-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i4]](/bjoc/content/inline/1860-5397-13-51-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i5]](/bjoc/content/inline/1860-5397-13-51-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i6]](/bjoc/content/inline/1860-5397-13-51-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i7]](/bjoc/content/inline/1860-5397-13-51-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-2]](/bjoc/content/figures/1860-5397-13-51-2.png?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i8]](/bjoc/content/inline/1860-5397-13-51-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i9]](/bjoc/content/inline/1860-5397-13-51-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i10]](/bjoc/content/inline/1860-5397-13-51-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i11]](/bjoc/content/inline/1860-5397-13-51-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i12]](/bjoc/content/inline/1860-5397-13-51-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i13]](/bjoc/content/inline/1860-5397-13-51-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i14]](/bjoc/content/inline/1860-5397-13-51-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i15]](/bjoc/content/inline/1860-5397-13-51-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i16]](/bjoc/content/inline/1860-5397-13-51-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i17]](/bjoc/content/inline/1860-5397-13-51-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i18]](/bjoc/content/inline/1860-5397-13-51-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i19]](/bjoc/content/inline/1860-5397-13-51-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i20]](/bjoc/content/inline/1860-5397-13-51-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i21]](/bjoc/content/inline/1860-5397-13-51-i21.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i22]](/bjoc/content/inline/1860-5397-13-51-i22.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i23]](/bjoc/content/inline/1860-5397-13-51-i23.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-3]](/bjoc/content/figures/1860-5397-13-51-3.png?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-4]](/bjoc/content/figures/1860-5397-13-51-4.jpg?scale=2.0&max-width=1024&background=FFFFFF)




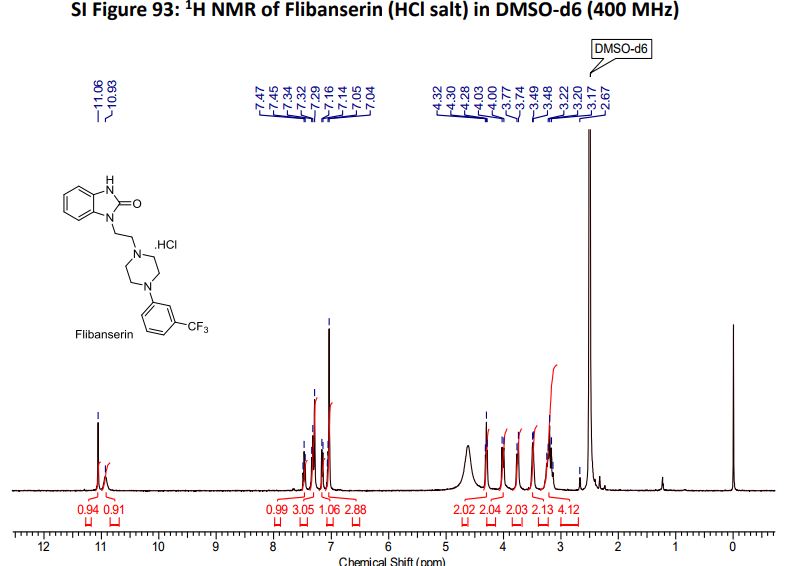
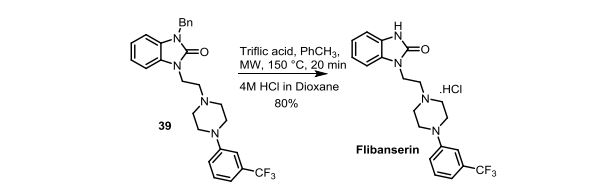







 Open Access
Open Access