Tout sur les médicaments הכל על תרופות كل شيئ عن الأدوية Все о наркотиках 关于药品的一切 డ్రగ్స్ గురించి అన్ని 마약에 관한 모든 것 Όλα για τα Ναρκωτικά Complete Tracking of Drugs Across the World by Dr Anthony Melvin Crasto, Worldpeacepeaker, worlddrugtracker, PH.D (ICT), MUMBAI, INDIA, Worlddrugtracker, Helping millions, 9 million hits on google on all websites, 2.5 lakh connections on all networks, “ALL FOR DRUGS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This is a compilation for educational purposes only. P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
Pramlintide (Symlin), a synthetic version of amylin, is a 37-amino acid peptide that is co-secreted with insulin by pancreatic β-cells. It was developed and approved in 2005 by the FDA for use in US patients with type I and II diabetes in conjunction with the administration of prandial insulin to improve postprandial glycemic control
Pramlintide
Pramlintide (Symlin) is a relatively new adjunct for diabetes (both type 1 and 2), developed by Amylin Pharmaceuticals (now a wholly owned subsidiary of Bristol Myers-Squibb). Pramlintide is delivered as an acetate salt.
Pramlintide is an analogue of amylin, a small peptide hormone that is released into the bloodstream by the β-cells of the pancreas along with insulin, after a meal.[1] Like insulin, amylin is completely absent in individuals with Type I diabetes.[2]
Reduction in glycated hemoglobin and weight loss have been shown in insulin-treated patients with type 2 diabetes taking pramlintide as an adjunctive therapy.[3]
By augmenting endogenous amylin, pramlintide aids in the absorption of glucose by slowing gastric emptying, promoting satiety via hypothalamic receptors (different receptors than for GLP-1), and inhibiting inappropriate secretion of glucagon, a catabolic hormone that opposes the effects of insulin and amylin. Pramlintide also has effects in raising the acute first-phase insulin response threshold following a meal.
Pramlintide has been approved by the FDA, for use by Type 1 and Type 2 Diabetics who use insulin.[4] Pramlintide allows patients to use less insulin, lowers average blood sugar levels, and substantially reduces what otherwise would be a large unhealthy rise in blood sugar that occurs in diabetics right after eating. Apart from insulin analogs, pramlintide is the only drug approved by the FDA to lower blood sugar in type 1 diabetics since insulin in the early 1920s.
Design and structure
Since native human amylin is highly amyloidogenic and potentially toxic, the strategy for designing pramlintide was to substitute residues from rat amylin, which is not amyloidogenic (but would presumably retain clinical activity). Proline residues are known to be structure-breaking residues, so these were directly grafted into the human sequence.
Amino acid sequences:
Pramlintide: KCNTATCATQRLANFLVHSSNNFGPILPPTNVGSNTY-(NH2)
Amylin: KCNTATCATQRLANFLVHSSNNFGAILSSTNVGSNTY-(NH2)
Rat amylin: KCNTATCATQRLANFLVRSSNNLGPVLPPTNVGSNTY-(NH2)
Ryan GJ, Jobe LJ, Martin R (2005). “Pramlintide in the treatment of type 1 and type 2 diabetes mellitus”. Clinical therapeutics27 (10): 1500–12. doi:10.1016/j.clinthera.2005.10.009. PMID16330288.
Molecular Formula: C26H29NO•C6H8O7 CAS Number: 54965-24-1 Brands: Nolvadex, TAMOXIFEN CITRATE
Chemically, NOLVADEX (tamoxifen citrate) is the trans-isomer of a triphenylethylene derivative. The chemical name is (Z)2-[4-(1,2-diphenyl-1-butenyl) phenoxy]-N, N-dimethylethanamine 2 hydroxy-1,2,3- propanetricarboxylate (1:1). The structural and empirical formulas are:
Tamoxifen citrate has a molecular weight of 563.62, the pKa’ is 8.85, the equilibrium solubility in water at 37°C is 0.5 mg/mL and in 0.02 N HCl at 37°C, it is 0.2 mg/mL.
Some breast cancer cells require estrogen to grow. Estrogen binds to and activates the estrogen receptor in these cells. Tamoxifen is metabolized into compounds that also bind to the estrogen receptor but do not activate it. Because of this competitive antagonism, tamoxifen acts like a key broken off in the lock that prevents any other key from being inserted, preventing estrogen from binding to its receptor. Hence breast cancer cell growth is blocked.
Tamoxifen was discovered by pharmaceutical companyImperial Chemical Industries (now AstraZeneca) and is sold under the trade names Nolvadex, Istubal, and Valodex. However, the drug, even before its patent expiration, was and still is widely referred to by its generic name “tamoxifen.
Breast cancer
Tamoxifen is currently used for the treatment of both early and advanced ER+ (estrogen receptor positive) breast cancer in pre- and post-menopausal women.Additionally, it is the most common hormone treatment for male breast cancer. It is also approved by the FDA for the prevention of breast cancer in women at high risk of developing the disease. It has been further approved for the reduction of contralateral (in the opposite breast) cancer.
In 2006, the large STAR clinical study concluded that raloxifene is equally effective in reducing the incidence of breast cancer, but after an average 4-year follow-up there were 36% fewer uterine cancers and 29% fewer blood clots in women taking raloxifene than in women taking tamoxifen, although the difference is not statistically significant.
Nolvadex (tamoxifen) 20 mg tablets
In 2005, the ATAC trial showed that after average 68 months following a 5 year adjuvant treatment, the group that received anastrozole (Arimidex) had significantly better results than the tamoxifen group in measures like disease free survival, but no overall mortality benefit. Data from the trial suggest that anastrozole should be the preferred medication for postmenopausal women with localized breast cancer that is estrogen receptor (ER) positive.Another study found that the risk of recurrence was reduced 40% (with some risk of bone fracture) and that ER negative patients also benefited from switching to anastrozole.
lTamoxifen was first developed in 1962 as a morning-after birth control pill that was successful in experiments with laboratory rats.
lTamoxifen (brand name Nolvadex) is the best-known hormonal treatment and the most prescribed anti-cancer drug in the world.
lUsed for over 20 years to treat women with advanced breast cancer, tamoxifen also is commonly prescribed to prevent recurrences among women with early breast cancer.
lIs a SERMs.
Anti-estrogens work by binding to estrogen receptors, blocking estrogen from binding to these receptors, stopping cell proliferation
lBreast-cancer prevention occurred in 1998 when the National Cancer Institute (NCI) announced results of a six-year study showing that tamoxifen reduced the incidence of breast cancer by 45 percent among healthy but high-risk women.
l13,388 healthy women considered at high risk for breast cancer were recruited
l85 developed breast cancer compared to 154 of those on the placebo or dummy pill.
lpotentially life-threatening side effects. There were 33 cases of endometrial cancer in the tamoxifen group
lThere were 30 cases of blood clots in major veins (deep-vein thrombosis)
lBecause these problems developed exclusively among postmenopausal women
–60-year-old, an age at which 17 out of every 1,000 women can be expected to develop breast cancer within five years
–ages of 35 and 59 were eligible to participate if their risks matched or exceeded those of a 60-year-old
lAlthough tamoxifen has been useful both in treating breast cancer patients and in decreasing the risk of getting breast cancer.
lSide effects arise from the fact that while tamoxifen acts as an antiestrogen that blocks the effects of estrogen on breast cells, it mimics the actions of estrogen in other tissues such as the uterus. Its estrogen-like effects on the uterus stimulate proliferation of the uterine endometrium and increase the risk of uterine cancer.
Adequate patent protection is required to develop an innovation in a timely manner. In 1962, ICI Pharmaceuticals Division filed a broad patent in the United Kingdom (UK) (Application number GB19620034989 19620913). The application stated, “The alkene derivatives of the invention are useful for the modification of the endocrine status in man and animals and they may be useful for the control of hormone-dependent tumours or for the management of the sexual cycle and aberrations thereof. They also have useful hypocholesterolaemic activity”.
This was published in 1965 as UK Patent GB1013907, which described the innovation that different geometric isomers of substituted triphenylethylenes had either oestrogenic or anti-oestrogenic properties. Indeed, this observation was significant, because when scientists at Merrell subsequently described the biological activity of the separated isomers of their drug clomiphene, they inadvertently reversed the naming. This was subsequently rectified.
Although tamoxifen was approved for the treatment of advanced breast cancer in post-menopausal women in 1977 in the United States (the year before ICI Pharmaceuticals Division received the Queen’s Award for Technological Achievement in the UK), the patent situation was unclear. ICI Pharmaceuticals Division was repeatedly denied patent protection in the US until the 1980s because of the perceived primacy of the earlier Merrell patents and because no advance (that is, a safer, more specific drug) was recognized by the patent office in the United States. In other words, the clinical development advanced steadily for more than a decade in the United States without the assurance of exclusivity. This situation also illustrates how unlikely the usefulness of tamoxifen was considered to be by the medical advisors to the pharmaceutical industry in general. Remarkably, when tamoxifen was hailed as the adjuvant endocrine treatment of choice for breast cancer by the National Cancer Institute in 1984, the patent application, initially denied in 1984, was awarded through the court of appeals in 1985. This was granted with precedence to the patent dating back to 1965! So, at a time when world-wide patent protection was being lost, the patent protecting tamoxifen started a 17 year life in the United States. The unique and unusual legal situation did not go uncontested by generic companies, but AstraZeneca (as the ICI Pharmaceuticals Division is now called) rightly retained patent protection for their pioneering product, most notably, from the Smalkin Decision in Baltimore, 1996. (Zeneca, Ltd. vs. Novopharm, Ltd. Civil Action No S95-163 United States District Court, D. Maryland, Northern Division, March 14, 1996.)
Title: Tamoxifen
CAS Registry Number: 10540-29-1
CAS Name: (Z)-2-[4-(1,2-Diphenyl-1-butenyl)phenoxy]-N,N-dimethylethanamine
Percent Composition: C 84.06%, H 7.87%, N 3.77%, O 4.31%
Literature References: Nonsteroidal estrogen antagonist.
Prepn: BE637389 (1964 to ICI). Identification and separation of isomers: G. R. Bedford, D. N. Richardson, Nature212, 733 (1966); BE678807; M. J. K. Harper et al.,US4536516 (1966, 1985 both to ICI). Stereospecific synthesis: R. B. Miller, M. I. Al-Hassan, J. Org. Chem.50, 2121 (1985). Review of chemistry and pharmacology: B. J. A. Furr, V. C. Jordan, Pharmacol. Ther.25, 127-205 (1984). Reviews of clinical experience in treatment and prevention of breast cancer: I. A. Jaiyesimi et al.,J. Clin. Oncol.13, 513-529 (1995); C. K. Osborne, N. Engl. J. Med.339, 1609-1618 (1998).
Synthesis of the E and Z isomers of the antiestrogen Tamoxifen.
David W.Robertson and John A. Katzenellenbogen.
Journal of Organic Chemistry 1982 , 47, Pages 2387-2393.
An early synthesis of Tamoxifen : Production of non stereo specific products.
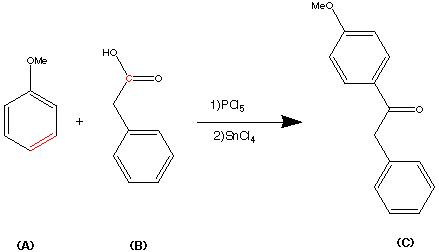
For easy of understanding the complete synthesis has been broken down into a number of steps.Step 1.
Step 1.
This step shows use of a simple friedel-craft acylation involving Anisole(A) and Phenylacetic acid (B). The acylating agent in this process was a mixture of PCl5 / SnCl4. The ketone C was formed in a 78% yield.
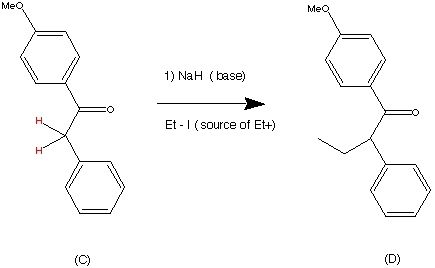
Step 2.
Step 2.
Alkylation was promoted by treating the ketone C with Sodium hydride (NaH). This removed the acidic protons (located on the position alpha to the carbonyl group) to produce the enolate ion. This could be isolated as the sodium enolate of the ketone treatment of this with ethyl iodide resulted in the formation of compound (D) in a 94% yield. The Ethyl iodide was chosen as the acylating agent probably as it contains the iodide ion , which is an excellent leaving group. It can therefore facilitate an SN2 substitution reaction with relative easy.
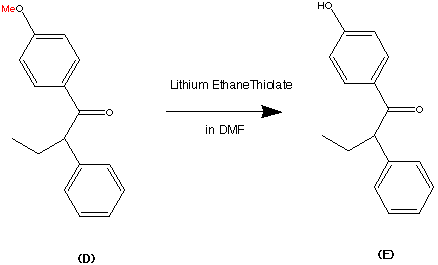
Step 3.
Step 3.
The phenol was deprotected using Lithium ethanthiolate in DMF ( Dimethyl This facilitated the removal of the methyl group and replaced it with a H to form a hydroxl group. Thus forming compound (E) in a 96% yield.
This is a key step as it has left a chink in the armour of the molecule. This can then be used to build up a characteristic part of the Tamoxifen molecule. (eg the (diemthylamino)ethyl group can be added easily from here)
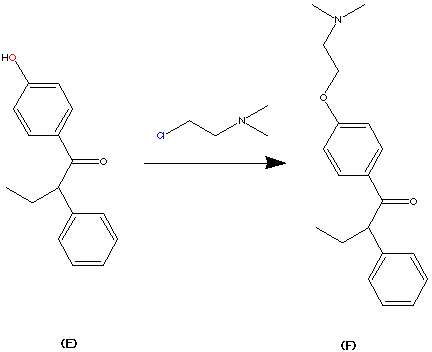
Step 4.
Step 4.
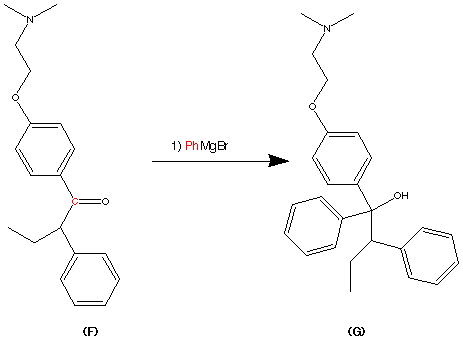
Then product E can be alkylated by treatment with 2-(dimethylamino) ethy chloride. The most facile site of alklation is the OH group on the phenyl ring. This can be interpreted roughly by using HSAB theory. e.g Hard and Soft acid/base theory. The carbon adjacent to the chloride ion of the reactant 2-(dimethylamino)ethyl chloride is made slightly harder due to the process of symbiosis. This can rationalise the formation between the hard oxygen atom to the normally soft carbon atom. In this case the carbon atom has become slightly harder due to the presence of the hard chorine atom. Hence the interaction is favourable by HSAB theory. The above reaction gives product F via a SN2 substitution reaction in 70% yield.
Step 5.
Step5.
F on treatment with PhMgBr forms the tertiary alcohol (G).
Formation of the Grignard reagent can be achieved via reaction of PhBr + Mg —–> PhMgBr. The Grignard reagent has effectively formed a carbanion species eg C delta negative (-ve). This is due to the presence of the C-Mg bond. the fact that Magnesium is a more electropositive element thus making the Carbon atom the more electronegative element and hence acquiring a negative charge. As a result of the negative nature of the carbon atom it can now attack the delta positive (+ve) Carbon atom of the carbonyl group.
step 6.
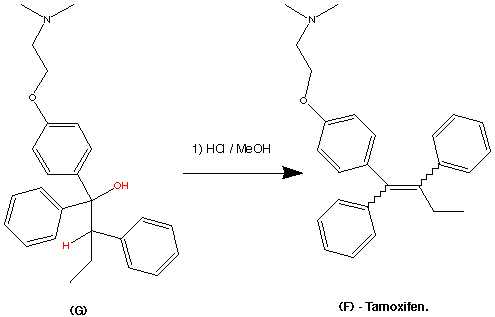
Step 6.
The dehydration of F was initiated by treatment of methanoic hydrogen chloride. this gives the required structure of Tamoxifen. However it gives a racemic mixture of both cis and trans isomers.
The ratio of the Cis / Trans isomers was (1.3 / 1). These isomers of Tamoxifen can be separated by Silica gel thin layer chromatography with benzene / triethylamine (9:1) as the developing solvent. Analysis of this technique revealed that the Z (Trans) isomer was more mobile than the E (Cis) isomer.
Synthetic Route 2: A Stereospecific Approach.
Stereospecific Synthesis of (Z) – Tamoxifen via carbometalation of Alkynylsilanes.
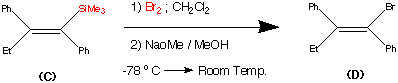
Studied for historical reasons rather than synthetic brilliance. This synthesis was the first stereo specific synthesis of (Z) Trans Tamoxifen. Comparison between this synthesis and the previous route I believe can illustrate the development of synthetic approaches to large molecules. In particular the quest for stereo specific reactions. So starting from an alkynylsilane (A) and through a series of reactions we can generate only the (Z) – Trans isomer of Tamoxifen.
Again for ease of understanding the complete synthesis has been broken down into a number of steps.
Step1.
Step1.
This step contains the vital stereo specific step. Namely the carbometalation of the alkynylsilane.It is this step which establishes the stereochemistry about the double bond. The phenyl (trimethyl silyl) – acetylene was carbometalated with diethylaluminium chloride – titanocene dichloride reactant to produce an organometallic intermediate. This organometallic intermediate was then cleaved with N bromosucciniamide to produce the alkene (B) in 85% yield.
The stereochemistry was assigned as E (Cis) mechanistic evidence suggests that this is linked to some steric reasons.
(Earlier work dedicated to this reaction see : Miller, R.B. Al-Hassan.M.I J.Org.Chem. 1984, 49, 725)
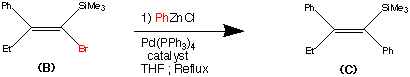
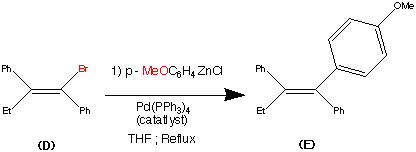
Step2.
Step 2.
The second step shows the stereo specific replacement of the Br group by a phenyl group. This was achieved by use of Palladium – catalysed coupling of compound (B) with phenyl zinc chloride to form (C) the vinylsilane in a 95% yield.
Step3.
Step3.
This step during the synthesis was reported to be tricky and several approaches were attempted before a successful technique was discovered.
The objective of this step was to replace the trimethyl Silyl group by a suitable halogen atom (e.g. Bromine or Iodine)
However a facile reaction was reported when (C) was treated with bromine – sodium methoxide at -78�C to produce the vinyl bromide (D) in a yield of 85%
Step 4.
Step 4.
The vinyl bromide (D) coupled well with a Zinc organometallic species to produce (E) the ethyl triaryl olefin in a yield of 84%.
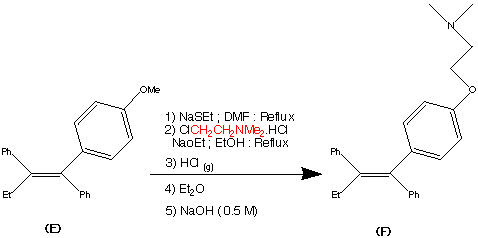
Step 5.
Step 5.
The formation of (F) Tamoxifen was achieved by demethylation with sodium ethylthoilate in refluxing dimethyl formamide. then reaction of the phenoxide ion with 2-( dimethylamino)ethyl chloride via a SN2 substitution.
Purification of the crude product was achieved via it’s hydrochloride salt ( via a reaction with HCl (g)) then F was regenerated by treatment with dilute base this produced the stereospecific (Z)- Trans isomer in an overall yield of 60%.
A solution of bromobenzene (3.92g, 25mmol) in ether (5ml) containing a crystal of iodine was added dropwise to a suspension of magnesium turnings (0.63g, 26mmol) in ether (5ml) at reflux, under nitrogen. After the addition was complete, the reaction mixture was cooled to room temperature and a solution of l- [ 4- ( 2- chloroethoxy)phenyl]-2-phenyl-l-butanone (3.75g, 12.4mmol) in ether (15ml) was added over 1 hour. The resulting mixture was refluxed for 16 hours, then poured into dilute hydrochloric acid (50ml) and extracted with ether (3x40ml) . The combined ether layers were concentrated, the residual oil was dissolved in ethanol (10ml) and refluxed with concentrated hydrochloric acid (5ml) for 4 hours. The organic phase was separated, dried (Na2S04) and evaporated to dryness to give a yellow oil. Η NMR (see Figures 1 to 4 and discussion below) showed this to be a 2:1 mixture of the Z and E isomers. The oil was then dissolved in warm methanol (about 40°C) and allowed to cool to room temperature. The colourless crystals formed proved to be pure Z isomer of 2-chloroethoxy tamoxifen (4.12g, 11.4mmol, 92% yield) . M.p. 107-109°C, m/z 362/364 (chlorine atom present), <SH 0.92 (3H, t, J = 7.33 Hz, CH3) , 2.46 (2H, q, J = 7.33 Hz, CH2CH3) , 3.72 (2H, t, J = 5.86 Hz, 0CH2CH2C1) , 4.09 (2H, t, J = 5.86 Hz, 0CH2CH2C1) , 6.55 (2H, d, J = 8.79 Hz, aromatic protons ortho to 0CH2CH2C1) , 6.79 (2H, d, J = 8.79 Hz, aromatic protons meta to 0CH2CH2C1) , 7.10-7.38 (10H, m, the two remaining C6H5,s) (see Figure 5) . The 2-chloroethoxy tamoxifen was reacted with dimethylamine in ethanol, under reflux, to produce the desired Z isomer of tamoxifen.
Analysis of Η NMR data
Figures 1 to 4 represent a mixture of the E- and Z- forms of compound XI described above.
The expansion of the region ό* 0.80 to 1.05 shows two overlapping triplets corresponding to the CH3 groups in the
Z- and E- derivatives respectively. The critical point is the ratio of the heights of the peaks at 0.92 (for the Z) and 0.94 (for the E) , which is approximately 2:1. The expansion of the 4.00 to 4.35 region reveals similar information where ratios are 10:6.4 and 5.56:3.43.
Similarly expansion of the region 3.6 to 3.9 shows the ratio to be 2.46:1. All of these measurements suggest an approximate 2:1 ratio.
Referring to Figure 5, this shows almost pure Z- isomer. It should be noted that there is 660 mg of this from an original mixture of a 2:1 ratio mixture of 780 mg which would contain only 520 mg of the Z-isomer.
Z isomer of tamoxifen and 4-hydroxytamoxi en include stereoselective syntheses (involving expensive catalysts) as described in J. Chem. Soc, Perkin Trans I 1987, 1101 and J. Org. Chem. 1990, 55, 6184 or chromatographic separation of an E/Z mixture of isomers as described in J. Chem. Res., 1985 (S) 116, (M) 1342, 1986 (S) 58, (M) 771.
(Z)-tamoxifen (1) and (E)-tamoxifen (2) in 52% yield. 1H-NMR (300 MHz, CDCl3) d 0.91 (Z isomer. 3H, t, J 7.3 Hz), 0.94 (E isomer. 3H, t, J 7.3 Hz), 2.28 (Z isomer. 6H, s), 2.34 (E isomer. 6H, s), 2.42-2.52 (Z and Eisomers. 4H, m), 2.63 (Z isomer. 2H, t, J 5.9 Hz), 2.74 (E isomer. 2H, t, J 5.9 Hz), 3.94 (Z isomer. 2H, t, J 5.9 Hz), 4.07 (E isomer. 2H, t, J 5.9 Hz), 6.68 (Z isomer. 2H, d, J 9.7 Hz), 6.76 (E isomer. 2H, d, J 9.3 Hz), 6.86-7.36 (Z and E isomers. 10H, m). IR (KBr film) nmax/cm-1: 3081, 3056, 2974, 2826, 2770, 1611, 1509, 1238, 1044. GC–MS (EI) m/z: Z isomer, 371(4%), 72 (24%), 58(100%); E isomer, 371(3%), 72 (24%), 58(100%). (the diastereoisomeric ratio was determined by capillary GC analysis and the configuration of the major diastereoisomer established by comparison of the NMR data of the synthetic mixture with an authentic sample of (Z)-tamoxifen (1).
nmr
ir
FTIR
shows the typical spectra’s of pure tamoxifen citrate, PCL, a physical mixture of tamoxifen citrate and PCL and drug-loaded implants. The spectrum of tamoxifen citrate shows characteristic absorption bands at 3027 cm−1 (=C-H stretching), 1507 and 1477 (C=C ring stretching) and 3180 cm -1 (-NH2). PCL displays a characteristic absorption band at strong bands such as the carbonyl stretching mode around 1727 cm−1 (C=O), asymmetric stretching 2949 cm−1 (CH 2 ) symmetric stretching 2865 cm−1 (CH 2 ). No changes in the spectrum of the physical mixture and drug-loaded microspheres were evident by FTIR spectroscopy. The strong bands such as the carbonyl peak were clear at all points.
Figure 2: Transmission FTIR spectra of (a) tamoxifen-loaded implant, (b) physical mixture of drug+PCL, (c) pure PCL, (d) pure tamoxifen citrate
enlarged view
FTIR spectra of A) tamoxifen citrate; B) PLGA; C) mixture of drug and excipients; D) freshly prepared nanoparticles in the formulation (BS-3HS).
Mentions: The pure drug tamoxifen citrate, PLGA-85:15, PVA, a mixture of PLGA and PVA, and a mixture of tamoxifen citrate, PLGA, and PVA; and a freshly prepared formulation were mixed separately with IR grade KBr in the ratio of 1:100 and corresponding pellets were prepared by applying 5.5 metric ton pressure with a hydraulic press. The pellets were scanned in an inert atmosphere over a wave number range of 4000–400 cm−1 in Magna IR 750 series II, FTIR instrument (Nicolet, Madison, WI, USA).
dsc
DSC thermograms of pure tamoxifen (a), pure PCL (b), physical mixture of drug+PCL (c) and (d) drug-loaded implant. The experiment was carried with crimped aluminum pans and a heating rate of 10ºC/min
xrd
X-ray diffraction studies of pure drug (a), pure PCL (b), physical mixture of drug+PCL (c) and (d) drug-loaded implant
synthesis
J.Chem. Research,1985(S) 116, (M) 1342 and 1986 (S) 58, (M) 0771.
TEVETEN® (eprosartan mesylate) is a non-biphenyl non-tetrazole angiotensin II receptor (AT1) antagonist. A selective non-peptide molecule, TEVETEN® is chemically described as the monomethanesulfonate of (E)-2-butyl-1 -(p-carboxybenzyl)-α-2-thienylmethylimid-azole-5 -acrylic acid.
Its empirical formula is C23H24N2O4S•CH4O3S and molecular weight is 520.625. Its structural formula is:
As with other angiotensin II receptor antagonists, eprosartan is generally better tolerated than enalapril (an ACE inhibitor), especially among the elderly.[1]
Ruilope L, Jäger B, Prichard B (2001). “Eprosartan versus enalapril in elderly patients with hypertension: a double-blind, randomized trial”. Blood Press.10 (4): 223–9. doi:10.1080/08037050152669747. PMID11800061.
Eprosartan mesylate was developed successfully by SmithKline Beecham Corporation in 1997, and marketed in Germany in 1998 under the trade-name Teveten and in the United States later in 1999. Eprosartan mesylate, as an angiotensin II receptor blocker, is an antihypertensive drug of the latest generation. Eprosartan mesylate is potent to lower systolic and diastolic pressures in mild, moderate and severe hypertensive patients, and is safe and tolerable. Eprosartan mesylate is rapidly absorbed when administrated orally, with a bioavailability of 13% and a protein binding rate of 98%. The blood peak concentration and AUC (Area Under Curve) can be elevated by about 50% in patients with liver and kidney dysfunction, or fullness after administration, and can be elevated by 2 to 3 folds in elderly patients. Eprosartan mesylate has a structure shown as follows:
U.S. Pat. No. 5,185,351 discloses a method for preparing eprosartan mesylate using Eprosartan and methanesulfonic acid in isopropanol (U.S. Pat. No. 5,185,351, Example 41 (ii)). However, it is found when following this method for preparing eprosartan mesylate in industry, an esterification reaction can occur between eprosartan and isopropanol and the following two impurities can be generated:
In addition to the above two esterification impurities, the salifying method provided by the above patent is prone to produce isopropyl mesylate. Considering currently known potential risk of gene toxicity of methylsulfonic acid ester on human as well as the stringent requirements of methylsulfonic acid ester from the Europe and the America authorities, it is important to produce eprosartan mesylate in a non-alcohol solvent during the process of producing eprosartan mesylate, since it avoids the formation of methylsulfonic acid ester and the residue thereof in the final product. Since the dosage of eprosartan mesylate is high, it is particularly important to strictly control methylsulfonic acid ester in eprosartan mesylate.
In addition, for the above salifying method, solid eprosartan is suspended in propanol at a low temperature, then methanesulfonic acid is added, about ten seconds later a great deal of eprosartan mesylate precipitate is obtained. Therefore, solid eprosartan may be embedded by the precipitated eprosartan mesylate. Since isopropyl alcohol has a high viscosity at low temperature, a heavy filtering operation burden is needed to obtain solid from isopropanol, and the obtained solid contains quite an amount of isopropanol.
Eprosartan has been obtained by several different ways: 1) The iodination of 2-butylimidazole (I) with I2 and Na2CO3 in dioxane/water gives 2-butyl-4,5-diiodoimidazole (II), which is treated with benzyl chloromethyl ether (III) and K2CO3 in DMF yielding the imidazole derivative (IV). The condensation of (IV) with N-methyl-N-(2-pyridyl)formamide (V) by means of butyllithium in THF affords 1-(benzyloxymethyl)-2-butyl-4-iodoimidazole-5-carbaldehyde (VI), which is deprotected with concentrated HCl ethanol to give 2-butyl-4-iodoimidazole-5-carbaldehyde (VII). The acylation of (VII) with methyl 4-(bromomethyl)benzoate (VIII) by means of K2CO3 in hot DMF yields 4-(2-butyl-5-formyl-4-iodoimidazol-1 ylmethyl)benzoic acid methyl ester (IX), which is deiodinated by hydrogenation with H2 over Pd/C in methanol affording compound (X). The condensation of (X) with methyl 3-(2-thienyl)propionate (XI) by means of lithium diisopropylamide (LDA) in THF gives (XII), which is acylated with acetic anhydride and dimethylaminopyridine (DMAP) in dichloromethane yielding the corresponding acetate (XIII). Elimination of acetic acid from (XIII) with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in hot toluene affords the expected propenoic ester (XIV), which is finally saponified with NaOH or KOH in ethanol/water.
Brevifoliol was first isolated from the leaves of the plant Taxus brevifolia (F. Balza et al Phytochemistry 30, p. 1613-1614 (1991)). The process of its isolation involved extracting the fresh leaves of Taxus wallichiana with ethyl alcohol to get an extract. The crude extract after concentration was diluted with water and partitioned between hexane, chloroform and ethyl acetate sequentially. The chloroform extract upon concentration yielded a dark brown residue. The resultant residue was subjected to column chromatography over silica gel and eluted with chloroform and chloroform-methanol gradient. Six fractions were collected and brevifoliol was isolated from fraction five by rechromatography over silica gel and eluting with hexane-ethyl acetate gradient.
Brevifoliol has been isolated from other species of Taxus including the Himalayan yew Taxus wallichiana which is available in India., Recently, the structure of brevifoliol has been revised and it was shown to belong to 11 (15→1) abeo taxoid bicyclic skeleton of formula (1). The isolation of brevifoliol from leaves of the plantTaxus wallichiana is also reported in S. K. Chattopadhyay et al Indian J. Chemistry 35B, 175-177(1996) as part of studies on the isolation of anticancer compounds. The process of this disclosure involved extracting the dried and crushed needles of Taxus wallichiana with methanol for 72 hours and the extract was concentrated in vacuo. The concentrate was diluted with water and extracted with hexane and chloroform respectively. Concentration of the chloroform phase under vacuum left a residue which was separated by column chromatography over silica gel. Fraction eluted with chloroform-methanol (98:5) contained brevifoliol which was further purified by re-chromatography over silica gel and eluted with chloroform-methanol (99:2). Fractions containing brevifoliol were combined and concentrated and recrystallized from pet-ether and ethyl acetate mixture to get brevifoliol as needles. In in vitro testing of brevifoliol, it was found to have significant anticancer activity against different cancer cell lines. The detection of anticancer activity in brevifoliol prompted the present investigators to develop an efficient processing technology for isolation of the compound in large quantities from the needles of the plant for further biological testing.
The prior art process of isolation of brevifoliol suffers from a number of disadvantages including partitioning of the aqueous extract with hexane and chloroform and repeated column chromatography to get the compound. Although the partitioning of the aqueous phase with organic solvents works on small scale, it forms thick emulsions on large scale partitioning process and creates hindrance in getting the fractions separated. Also, the use of repeated chromatography might be useful on small-scale isolation of brevifoliol, it is only cumbersome, tedious and not economical on large-scale process.
Assignee: Council of Scientific and Industrial Research, New Delhi, India
Title or Subject: Process for Preparing Brevifoliol
Brevifoliol is found in the leaves of the plants of the genus Taxus and is useful as an anticancer agent. This patent describes a method of extracting from the leaves of the plantTaxus wallichiana (Tw) Alternative methods are known for extracting from Tw but they are said to suffer from several disadvantages. These include partitioning of an aqueous extract with hexane and CHCl3 and the repeated use of chromatography to obtain the purified compound. The partitioning is said to work well on a small scale, but on a large scale thick emulsions are formed. Hence, the objective of the work in this patent is to provide a process that can be operated on a large scale. The process developed comprises the following steps:
(i) Dry and pulverize the leaves of the plant.
(ii) Extract the dried leaves with an alcohol such as MeOH or EtOH at 20−40 °C over 3 days.
(iii) Concentrate the alcoholic solution and adsorb the extract onto Celite.
(iv) Dry the Celite adsorbate at 20−50 °C for up to 48 h.
(v) Extract the dried adsorbate with 60−80 petroleum ether then CHCl3 and concentrate the CHCl3 extract.
(vi) Subject the concentrated mixture to gross fractionation over a column of silica gel using CHCl3 add 2% MeOH in CHCl3.
(vii) Subject the later eluate to further chromatography over alumina in petroleum ether using 10% EtOAc in petroleum ether.
(viii) Recrystallise from EtOAc/petroleum ether as needles.
Brevifoliol
Advantages
The patent claims that the solvents can be recycled so that the process is cost-effective. Certainly it is true that avoiding water removes the emulsification problem, but two chromatographic steps are involved, and the use of so many solvents would seem to create handling problems on a commercial plant.
MONTELUKAST (Singulair® Oral Granules) helps to reduce asthma symptoms (coughing, wheezing, shortness of breath, or chest tightness) and control your asthma. It does not provide instant relief and cannot be used to treat a sudden asthma attack. It works only when used on a regular basis to help reduce inflammation and prevent asthma attacks. This drug is also helpful in improving seasonal allergies, like hay fever. Montelukast is effective in adults and children
Amongst the US approvals, tentative FDA approvals have been identified for generic Montelukast sodium, awarded to Endo, Glenmark, Mylan, Roxane, Sandoz, Teva and Torrent. The large number of generic authorisations awaiting launch in the UK is indicative of the likely competition the Singulair product will face across Europe upon SPC expiry
EP Pat. No. 480,717 discloses Montelukast sodium along with other related compounds and the methods for their preparation. The reported method of synthesis proceeds through corresponding methyl ester namely, Methyl 2-[(3S)-[3-[(2E)-(7-chloroquinolin – 2yl) ethenyl] phenyl] – 3 – hydroxypropyl] benzoate and involves coupling methyl 1- (mercaptomethyi) cyclopropaneacetate with a mesylate generated in-situ. The methyl ester of Montelukast is hydrolyzed to free acids and the latter converted directly to Montelukast Sodium salt (Scheme -1). The process is not particularly suitable for large – scale production because it requires tedious chromatographic purification of the methyl ester intermediate and / or the final product and the product yield is low.
Scheme -1
U.S. Pat. No. 5,614632 disclosed a process for the preparation of crystalline Montelukast sodium, which comprises of the following steps (Scheme – 2):
■ Reaction of methyl 2-[3(S)-[3-[2-(7-chloroquinolin -2-yl) ethenyl] phenyl] -3- hydroxypropyl benzoate (I) with Grignard reagent, methyl magnesium chloride in presence of cerium chloride to give Diol (II) ■ Reaction of Diol (II) with methane sulfonyl chloride to afford 2-[2-[3 (s)-[3- (2-(7-chloro quinolin-2yl) ethenyl] phenyl]- 3 – methane sulfonyloxy propyl] phenyl] -2-propanol (III)
■ Condensation of 2-[2-[3(s)-[3-(2-(7-chloro quinolin – 2-yl) ethenyl] phenyl] –
3 – methane sulfonyloxypropyl] phenyl] – 2- propanol (III) with dilithium anion of 1-mercaptomethyl) cyclopropaneacetic acid, which has been generated by the reaction of l-(mercaptomethyl)cyclopropaneacetic acid (IV)with n-Butyl lithium
■ Isolation of the condensed product, Montelukast as solid Montelukast dicyclohexylamine salt
■ Purification and conversion of Montelukast dicyclohexylamine salt into Montelukast sodium
■ Crystallization of Montelukast sodium from a mixture of toluene and acetonitrile
The process disclosed in U.S Pat. No. 5,614,632 further involved the reaction of Diol (II) with methane sulfonyl chloride involves the reaction temperature of about – 25°C and the storage condition of the intermediate, 2-[2-[3(s)-[3-(2-(7-chloro quinolin – 2-yl) ethenyl] pheny] -3 -methane sulfonyloxy propyl] phenyl] -2-propanol (III) at temperature below – 150C for having the stability. The process further involves the reaction, formation of dilithium anion of l-(mercaptomethyl) cyclopropaneacetic acid which requires the usage of n-Butyl lithium, a highly flammable and hazardous reagent and the reaction is at temperature below -5°C further requires anhydrous conditions. Scheme – 2
Montelukast (trade names Singulair, Montelo-10, and Monteflo and Lukotas in India) is a leukotriene receptor antagonist(LTRA) used for the maintenance treatment of asthma and to relieve symptoms of seasonal allergies.[1][2] It is usually administered orally once a day. Montelukast is a CysLT1antagonist; it blocks the action of leukotriene D4 (and secondary ligands LTC4 and LTE4) on the cysteinyl leukotriene receptor CysLT1 in the lungs and bronchial tubes by binding to it. This reduces the bronchoconstriction otherwise caused by the leukotriene and results in less inflammation.
Because of its method of operation, it is not useful for the treatment of acute asthma attacks. Again because of its very specificmechanism of action, it does not interact with other asthma medications such as theophylline.
Another leukotriene receptor antagonist is zafirlukast (Accolate), taken twice daily. Zileuton (Zyflo), an asthma drug taken four times per day, blocks leukotriene synthesis by inhibiting 5-lipoxygenase, an enzyme of the eicosanoid synthesis pathway.
The Mont in Montelukast stands for Montreal, the place where Merck developed the drug.[3]
Singulair was covered by U.S. Patent No. 5,565,473[9] which expired on August 3, 2012.[10] The same day, the FDA approved several generic versions of montelukast.[11]
On May 28, 2009, the United States Patent and Trademark Office announced their decision to launch a reexamination of the patent covering Singulair. The decision to reexamine was driven by the discovery of references that were not included in the original patent application process. The references were submitted through Article One Partners, an online research community focused on finding literature relating to existing patents. The references included a scientific article produced by a Merck employee around the key ingredient of Singulair, and a previously filed patent in the same technology area.[12]
On December 17, 2009, the U.S. Patent and Trademark Office determined that the patent in question was valid based on the initial reexamination and new information provided.[13]
Montelukast is currently available in film coated tablet and orodispersible tablet formulations for once-daily administration, and also available as an oral granule formulation which is specifically designed for administration to paediatric patients.
Patent family US17493193A claims crystalline Montelukast sodium and processes for its preparation . Patents within this family are not considered to be a constraint to generic competition because the protected technology may possibly be circumvented by the synthesis and use of different molecular forms and/or salts. Patent family US33954901P relates to the specific marketed oral granule formulation of Montelukast.
The chemical name of montelukast sodium is: Sodium 1-[[[(1R)-1-[3-[(1E)-2-(7-chloro-2-quinolinyl)ethenyl]phenyl]-3-[2-(1-hydroxy-1-methylethyl)phenyl]propyl]thio]methyl]cyclopropaneacetic acid and its structure is represented as follows:
Montelukast is apparently a selective, orally active leukotriene receptor antagonist that inhibits the cysteinyl leukotriene CysLT1 receptor.
The chemical name for montelukast sodium is [R-(E)]-1-[[[1-[3-[2-(7-chloro-2-quinolinyl)ethenyl]phenyl]-3-[2-(1-hydroxy-1-methylethyl)phenyl]propyl]thio]methyl] cyclopropaneacetic acid, monosodium salt. Montelukast sodium salt is understood to be represented by the following structural formula:
U.S. patent No. 5,565,473 (“‘473 patent”) is listed in the FDA’s Orange Book for montelukast sodium. The ‘473 patent recites a broad class of leulcotriene antagonists as “anti-asthmatic, anti-allergic, anti-inflammatory, and cycloprotective agents” represented by a generic chemical formula. ‘473 patent, col. 2,1. 3 to col. 4,1. 4. Montelukast is among the many compounds represented by that formula. The ‘473 patent also refers to pharmaceutical compositions of the class of leukotriene antagonists of that formula with pharmaceutically acceptable carriers. Id. at col. 10,11. 42-46.
Montelukast sodium is currently marketed by Merck in the form of film coated tablets and chewable tablets under the trade name Singular®. The film coated tablets reportedly contain montelukast sodium and the following inactive ingredients: microcrystalline cellulose, lactose monohydrate, croscarmellose sodium, hydroxypropylcellulose, magnesium stearate, titanium dioxide, red ferric oxide, yellow ferric oxide, and carnauba wax. The chewable tablets reportedly containmontelukast sodium and the following inactive ingredients: mannitol, microcrystalline cellulose, hydroxypropylcellulose, red ferric oxide, croscarmellose sodium, cherry flavor, aspartame, and magnesium stearate. Physicians’ Desk Reference, 59th ed. (2005), p. 2141.
However, there is a need in the art to improve the stability of compositions of montelukast and particularly those of the sodium salt.
Montelukast sodium is a leukotriene antagonist and inhibits the leukotriene biosynthesis. It is a white to off-white powder that is freely soluble in methanol, ethanol, and water and practically insoluble in acetonitrile.
A montelukast sodium salt is a substance which exhibits efficacy of Singulair (available from Korean MSD) generally used for the treatment of asthma as well as for the symptoms associated with allergic rhinitis, which is pharmaceutically known as a leukotriene receptor antagonist. Leukotrienes produced in vivo by metabolic action of arachidonic acid include LTB4, LTC4, LTD4 and LTE4. Of these, LTC4, LTD4 and LTE4 are cysteinyl leukotrienes (CysLTs), which are clinically essential in that they exhibit pharmaceutical effects such as contraction of airway muscles and smooth muscles and promotion of secretion of bronchial mucus.
Montelukast sodium salt is a white and off-white powder which has physical and chemical properties that it is well soluble in ethanol, methanol and water and is practically insoluble in acetonitrile.
A conventionally known method for preparing a montelukast sodium salt is disclosed in EP Patent No. 480,717. However, the method in accordance with the EP Patent requires processes for introducing and then removing a tetrahydropyranyl (THP) protecting group and purification by chromatography, thus being disadvantageously unsuitable for mass-production. In addition, the method disadvantageously requires investment in high-cost equipment, for example, to obtain amorphous final compounds by lyophilization.
Meanwhile, U.S. Pat. No. 5,614,632 discloses an improved method for preparing a montelukast sodium salt by directly reacting a methanesulfonyl compound (2) with 1-(lithium mercaptomethyl)cyclopropaneacetic acid lithium salt, without using the tetrahydropyranyl protecting group used in EP Patent No. 480,717, purifying in the form of a dicyclohexylamine salt by adding dicyclohexylamine to the reaction solution, and converting the salt into a montelukast sodium salt (1).
However, the method in accordance with the US patent should use n-butyl lithium as a base in the process of preparing the 1-(lithium mercaptomethyl)cyclopropaneacetic acid lithium salt and thus requires an improved process due to drawbacks that n-butyl lithium is dangerous upon handling and is an expensive reagent.
PCT International Patent Laid-open No. WO 2005/105751 discloses a method for preparing a montelukast sodium salt, comprising coupling methyl 1-(mercaptomethyl)cyclopropane acetate (3) used in step 10 shown in Example 146 of EP Patent 480,717 with a methanesulfonyl compound (2) in the presence of a solvent/cosolvent/base, performing hydrolysis, recrystallizing the resultingmontelukast acid (4) in the presence of a variety of solvents to obtain highly puremontelukast acid (4), and converting the same into a montelukast sodium salt (1).
In addition, WO 2005/105751 claims that, in the coupling reaction, one is selected from tetrahydrofurane and dimethylcarbonate as a solvent, a highly polar solvent is selected from dimethylformamide, dimethylacetamide and N-methylpyrrolidone as a cosolvent, and one is selected from sodium hydroxide, lithium hydroxide, sodium hydride, sodium methoxide, potassium tert-butoxide, lithium diisopropylamine and quaternary ammonium salts, as a base.
However, WO 2005/105751 discloses that, since the coupling reaction requires use of a mixed solvent and the mixed solvent is different from the solvent used for hydrolysis, a process for removing the cosolvent through distillation under reduced pressure or extraction is further required prior to hydrolysis.
Further, in accordance with the method of WO 2005/105751, recrystallization is performed in the presence of a variety of solvents in order to obtain a highly puremontelukast acid (4) and the resulting recrystallization yield is varied in a range of 30 to 80%, depending on the solvent. In the case where desired purity is not obtained, recrystallization is repeated until montelukast acid (4) with a desired purity can be obtained. Disadvantageously, the method causes deterioration in overall yield.
European Patent No. 480,717 discloses montelukast sodium and its preparation starting with the hydrolysis of its ester derivative to the crude sodium salt, acidification of the crude to montelukast acid, and purification of the crude acid by column chromatography to give montelukast acid as an oil. The resulting crude oil in ethanol was converted to montelukast sodiumby the treatment with an equal molar aqueous sodium hydroxide solution. After removal of the ethanol, the montelukastsodium was dissolved in water and then freeze dried. The montelukast sodium thus obtained is of a hydrated amorphous form as depicted in FIG. 2.
The reported syntheses of montelukast sodium, as pointed out by the inventor in EP 737,186, are not suitable for large-scale production, and the product yields are low. Furthermore, the final products, as the sodium salts, were obtained as amorphous solid which are often not ideal for pharmaceutical formulation. Therefore, they discloses an efficient synthesis of montelukastsodium by the use of 2-(2-(3-(S)-(3-(7-chloro-2-quinolinyl)ethenyl)phenyl)-3-methanesulfonyloxypropyl)phenyl)-2-propanol to couple with the dilithium salt of 1-(mercaptomethyl)cyclopropaneacetic acid. The montelukast acid thus obtained is converted to the corresponding dicyclohexylamine salt and recrystallized from a mixture of toluene and acetonitrile to obtain crystallinemontelukast sodium. This process provides improved overall product yield, ease of scale-up, and the product sodium salt in crystalline form.
According to the process described in EP 737,186, the chemical as well as optical purities of montelukast sodium depends very much on the reaction conditions for the mesylation of the quinolinyl diol with methanesulfonyl chloride. For instance, the reaction temperature determinates the chemical purity of the resulting coupling product montelukast lithium, due to the fact that an increase in the reaction temperature resulted in decreased selectivity of mesylation toward the secondary alcohol. Mesylation of the tertiary alcohol occurred at higher temperature will produce, especially under acidic condition, the undesired elimination product, the styrene derivative. This styrene impurity is difficult to remove by the purification procedure using DCHA salt formation; while excess base, butyl lithium in this case, present in the reaction mixture causes the formation of a cyclization by-product, which will eventually reduce the product yield.
PCT WO 2005/105751 discloses an alternative process for preparing montelukast sodium by the coupling of the same mesylate as disclosed in ‘186 patent with 1-(mercaptomethyl)cyclopropane alkyl ester in the presence of a base. In this patent, the base butyl lithium, a dangerous and expensive reagent, is replaced with other milder organic or inorganic base. However, the problem concerning the formation of the styrene impurity is still not resolved.
Process for the manufacture of 1-[[[(1R)-1-[3-[(1E)-2-(7-chloro-2-quinolinyl)ethenyl]phenyl]-3-[2-(1-hydroxy-1-methylethyl)phenyl]propyl]thio]methyl]cyclopropane acetic acid, sodium salt [montelukast sodium (I)] consisting of: i. Converting methyl 1-(mercaptomethyl)-cyclopropaneacetate to a metal salt (X) using a metal hydroxide, ii. Subjecting the metal salt (X) to monometallation to provide a dimetallide (XI). iii. Converting a diol of formula (II) to a mesylate of formula (III) and reacting (III) in situ with (XI) affordin the metal salt of 1-[[[(1R)-1-[3-[(1E)-2-(7-chloro-2-quinolinyl)ethenyl]phenyl]-3-[2-(1-hydroxy-1-methylethyl)phenyl]propyl]thio]methyl]cyclopropane acetic acid. iv. Reacting the metal salt in-situ with a base and purifying to afford an amine salt (XII). v. Treating (XII) with a sodium base and precipitating out montelukast sodium (I).
more info
European Patent No. 480,717 disclose the montelukast and its preparation method first be hydrolyzed to the crude ester derivatives sodium, then this crude product was acidified to montelukast acid (montelukastacid), Finally, column chromatography purification of this crude acid into oily montelukast acid. This oilMontelukast acid in ethanol, by equimolar amounts of sodium hydroxide solution and converted to montelukast sodium. The ethanol was removed aftermontelukast sodium dissolved in water, followed by freeze-drying. Finally obtainedmontelukast shown in Figure 2 is amorphous hydrated.
The invention, in European Patent No. 737,186 points out, thismontelukast synthesis method is not suitable for mass production, and the low yield. Moreover, the resulting amorphous solid salt, are generally not used in pharmaceutical formulations. Therefore, they disclose the synthesis of an effective method of montelukast sodium, which uses 2 – (2 – (3 – (S) – (3 – (7 – chloro-2 – quinolinyl) ethenyl) phenyl) -3 – methylsulfonyl) phenyl) -2 – propanol and 1 – (methylthio alcohol) cyclopropane coupling the lithium salt of acetic acid, the resulting Montelukast acid is converted into a corresponding bicyclic hexyl amine salt, and from a mixture of toluene and acetonitrile recrystallization to prepare crystalline montelukast. This method greatly improves the productivity, ease of mass production, and the product is crystalline sodium salt.
According to European Patent No. 737,186 described method for preparingmontelukast chemical purity and optical purity depends largely quinoline diol with methanesulfonyl chloride in the reaction between the mesylated condition. For example, the reaction temperature resulted in an increase of the secondary alcohols methanesulfonyl selective reduction, the reaction temperature determines the coupling product (montelukast lithium) chemical purity. Occurs at a higher temperature mesylation tertiary alcohols, in particular under acidic conditions, will produce impurities, such as styrene derivatives. This impurity is difficult styrene generated by using the DCHA salt (DCHA salt formation) in the purification process to remove; present in the reaction mixture and excess base, butyl lithium cyclized by-products resulting in the formation will eventually reduce the yield of the product.
W02005/105751 disclose another preparation method of montelukast sodium, which is the methanesulfonic acid (European Patent No. 737,186 is the same) in an alkaline state where 1_ (methyl mercaptan yl) cyclopropyl alkyl ester and coupling thereof. In this patent, the dangerous and expensive alkaline-butyl lithium reagent, is replaced by other more moderate organic or inorganic base. However, the formation of styrene impurity problem is still not resolved
Lipkowitz, Myron A. and Navarra, Tova (2001) The Encyclopedia of Allergies (2nd ed.) Facts on File, New York, p. 178, ISBN 0-8160-4404-X
…………………….
Improved Process for the Preparation of Montelukast: Development of an Efficient Synthesis, Identification of Critical Impurities and Degradants
HPLC (isocratic mode) chromatograms were measured with the EliteLachrom device made by the Hitachi Company. Stationary phase: RP-18e was used for the analyses; column temperature was 20 °C. Mobile phase: Acetonitrile (80%) and a 0.1 M aqueous solution of ammonium formate adjusted to pH 3.6 with formic acid (20%) were used. The flow rate of the mobile phase was 1.5 mL/min. Detection at the wavelength of 234 nm was used. Methanol was used as the solvent for preparation of samples; 10−20 μL of the solution was used for the injection. The isocratic HPLC method was used for checking the compositions of the reaction mixtures. HPLC (gradient mode) chromatograms were measured with the Alliance HPLC device with PDA detector. Stationary phase: STAR RP-8e, 250 mm × 4 mm, 5 μm was used for the analyses; column temperature was 15 °C. Mobile phase: Acetonitrile (A) and 0.01 M aqueous solution of KH2PO4 adjusted to pH 2.2 with phosphoric acid (B) were used. Gradient mode with the flow rate of mobile phase 0.8 mL/min was used. Composition on the start was 60% of A and 40% of B, then changed to 15% of A and 85% of B over 20 min; this composition was held for 5 min, then changed to 60% of A and 40% of B over 5 min, and this composition was held to the end (overall time 35 min.). Detection at the wavelength of 234 nm was used. Methanol was used as the solvent for the preparation of the samples; 10−20 μL of the solution was used for the injection. The gradient HPLC method was used for checking the quality of the target substance including its salts with amines and of isolated standards of impurities. HPLC (determination of (S)-enantiomer by HPLC) chromatograms were measured with the Alliance HPLC device with PDA detector. Stationary phase: Chiralpak IA (5 μm), size 0.25 m, internal diameter 4.6 mm (manufactured by Daicel) was used for the analyses, column temperature 10 °C. Mobile phase: hexane/ethanol/1,4-dioxan/trifluoroacetic acid (77:3:20:0,1 v/v/v) was used. The flow rate of the mobile phase was 1.0 mL/min. Detection at the wavelength of 285 nm was used. Methanol was used as the solvent for preparation of samples; 10 μL of the solution was used for the injection. The isocratic elution was used for checking the optical purity of target montelukast. Typical retention times: montelukast: 9.3 min, (S)-montelukast: 12.9 min.
KEY REFERENCES
(a) Ray, U. K.;Boju, S.; Pathuri, S. R.; Meenakshisunderam, S. (Aurobindo Pharma Limited, India). PCT Patent Application WO/2008/001213, 2008.
(b) Wang, Y.; Wang, Y.; Brand, M.; Kaspi, J. (Chemagis Ltd., Israel). PCT Patent Application WO/2007/088545, 2007.
(c) Turchetta, S.;Tuozzi, A.; Ullucci, E.; de Ferra, L. (Chemi S.P.A.; Italy). European Patent Application EA 1,693,368, 2008.
(d) Srinivas, P. L.; Rao, D. R.; Kankan, R. N.; Relekar, J. P. (Cipla Limited, India). PCT Patent Application WO/2006/064269, 2006.
(e) Reguri, B. R.; Bollikonda, S.;Bulusu, V. V. N. C. S.; Kasturi, R. K.; Aavula, S. K. (Dr. Reddy’s Laboratory, India). U.S. Patent Application U.S.2005/0107612, 2005.
(f) Coppi, L.; Bartra Sanmarti, M.; Gasanz Guillen, Y.; Monsalvatje Llagostera, M.; Talavera Escasany, P. (Esteve Quimica, S.A., Spain). PCT Patent Application WO/2007/051828, 2007.
(g) Hung, J. T.; Wei, C. P. (Formosa Laboratories, Inc., Taiwan). U.S. Patent Application U.S.2008/0097104, 2008.
J. Liang*, J. Lalonde, B. Borup, V. Mitchell, E. Mundorff, N. Trinh, D. A. Kochrekar, R. N. Cherat, G. G. Pai
Codexis, Inc., Redwood City, USA and Arch PharmaLabs Limited, Mumbai, India
Development of a Biocatalytic Process as an Alternative to the (-)-DIP-Cl-Mediated Asymmetric Reduction of a Key Intermediate of Montelukast
Org. Process Res. Dev. 2010, 14: 193-198
Montelukast sodium (Singulair®) is a leukotriene receptor antagonist prescribed for the treatment of asthma and allergies. Workers at Codexis used directed evolution and high-throughput screening to engineer a robust and efficient ketoreductase enzyme (CDX-026) that accomplished the asymmetric reduction of ketone A, which is essentially water insoluble, at a loading of 100 g/L in the presence of ca. 70% organic solvents at 45 ˚C. The (S)-alcohol B was obtained in >95% yield in >99.9% ee and in >98.5% purity on a >500 mol scale.
The enzymatic reduction entails the reversible transfer of a hydride from isopropanol to the ketone A with concomitant formation of acetone. The reaction is driven to completion by the fortuitous crystallization of the monohydrate B. The four-step conversion of B into montelukast sodium is described in the Merck process patent (M. Bhupathy, D. R. Sidler, J. M. McNamara, R. P. Volante, J. J. Bergan US 6320052, 2001). This biocatalytic reduction is superior to the reduction of A with (-)-DIPCl previously used in the manufacture of montelukast
Auraptene has shown a remarkable effect in the prevention of degenerative diseases. Many studies have reported the effect of auraptene as a chemopreventative agent against cancers of liver, skin, tongue, esophagus, and colon in rodent models.[1] The effect in humans is not yet known.
Curini, M., Carvotto, G., Epifano, F. and Giannone, G. “Chemistry and Biological Activity of Natural and Synthetic Prenyloxycoumarins”(2006). Current Medicinal Chemistry, 13, 199-222.
Fever is one of the most common problems and there is not a single person who does not suffer from this in his life at least once. But in some people symptoms such as tingling sensation, temporary paralysis of legs and arms may appear after an attack of fever. These are the symptoms of Guillian Barre syndrome.
Brain and backbone are very important for the movement of the body. Nerves that control the movements are also very important as they carry the signals from brain to the muscles. Muscles move according to the signals brought by the muscles. If the nerves get damaged due to some problems, symptoms such as paralysis of legs and arms, tingling sensation, losing muscles movements etc are seen. Nerve disorders are usually found in diabetic patients and alcoholics. But these may sometime appear in people after a viral infection.
When symptoms of Guillian Barre syndrome appear, the patient should be taken to the Neurologist immediately. NCS test is prescribed for the patient and depending on the test results, Guillian Barre syndrome can be confirmed. Some other test may also be required to rule out other diseases that have the same symptoms.
Treatment for Guillian Barre syndrome
If nerve damage due to Guillian Barre is only minor, the condition can be improved with few injections and physiotherapy. Improvement can be observed in two to three months. But if the severity is more and if the patient finds breathing also difficult, he should be admitted into the ICU. The patient is given treatment with medicines for five days. Treatment for Guillian Barre syndrome is expensive. The dosage of the medicine is decided depending on the body weight of the patient. Medicines may cost between Rs. 2 lakhs to 3 lakhs.
There is alternative treatment for Guillian Barre syndrome for those who cannot bear the expense of medicines. It is called the Plasmapheresis procedure. This helps in preventing the condition of the patient from deteriorating further. In this procedure, patient’s blood is taken out and plasma is separated. Blood without plasma is again transfused into the patient’s body. If necessary, healthy plasma taken from donors is also given to the patient. But this treatment should begin immediately after Guillian Barre syndrome is confirmed for effective results. This treatment costs nearly Rs. 1 lakh and the patient should be given physiotherapy treatment for muscle movement.
With 11 treatments in Phase I trials, 8 in Phase II, and 13 in Phase III, Bayer has a strong pipeline.
By far the most interest currently, given that the latest reports came out October 21st, is riociguat (BAY 63-2521),
which has had good news from its ongoing Phase III clinical trials of the treatment for pulmonary arterial hypertension, also known as PAH. PAH is a progressive condition that overburdens the heart.
Trials indicate subjects had improved heart function and could better tolerate physical exercise. Patients on riociguat improved their walking distance by 36 meters on average, while those on placebo showed no improvement.
Professor Hossein Ardeschir Ghofrani of University Hospital Giessen, the principal investigator, was quite pleased with the results and explained the value of the measurement. “The six-minute walk distance test is a well-validated clinical measure in patients with PAH, and therefore, the results of the PATENT-1 trial are encouraging. . .These data from the PATENT study suggest that riociguat may be a potential treatment option both for patients who have never been treated for PAH as well as for those who have received prior treatment.”
Hossein A. Ghofrani Associate Professor of Internal Medicine, MD (University of Giessen) 1995 Research interests: pulmonary hypertension, ischaemia-reperfusion, experimental therapeutics, clinical trials
Bayer Accelerates Clinical Development of Promising New Drug Candidates
Five new molecular entities projected to enter Phase III by 2015 / Addressing unmet medical needs in the areas of oncology, cardiology, and women’s health / Initiation of further studies with recently launched products planned to add new treatment options
Leverkusen, October 8, 2013 – Following the recent commercial introduction of five new drugs to address the medical needs of patients with various diseases, Bayer is now accelerating the development of further five promising drug candidates which are currently undergoing phase I and II clinical studies. The company today announced that it plans to progress these five new highly innovative drug candidates in the areas of oncology, cardiology, and women’s health into phase III clinical studies by 2015.
“Our Pharma research and development has done a tremendous job of bringing five new products to the market offering physicians and patients new treatment alternatives for serious diseases”, said Bayer CEO Dr. Marijn Dekkers. “Following our mission statement ‘Science For A Better Life’, the five chosen further drug candidates all have the potential to impact the way diseases are treated for the benefit of patients.”
Bayer CEO Dr. Marijn Dekkers “Our research and development activities are strongly focused on areas where treatment options are not available today or where true breakthrough innovations are missing”, said Prof. Andreas Busch, member of the Bayer HealthCare Executive Committee and Head of Global Drug Discovery at Bayer HealthCare. “Our drug development pipeline holds a number of promising candidates which we want to bring to patients who need them urgently”, said Kemal Malik, member of the Bayer HealthCare Executive Committee, Chief Medical Officer and Head of Pharmaceutical Development at Bayer HealthCare. “Furthermore we are continuing to expand the range of indications for all our recently launched products Xarelto, Stivarga, Xofigo, Riociguat as well as Eylea and further refine the profile of these drugs in specific patient populations.”
The five mid-stage candidates have been selected for accelerated development based on positive “proof-of-concept” data from early clinical studies. Three of them are development compounds in the area of cardiology or the cardio-renal syndrome: Finerenone (BAY 94-8862) is a next generation oral, non-steroidal Mineralocorticoid Receptor antagonist which blocks the deleterious effects of aldosterone. Currently available steroidal MR antagonists have proven to be effective in reducing cardiovascular mortality in patients with heart failure but have significant side effects that limit their utilization. Finerenone is currently in clinical Phase IIb development for the treatment of worsening chronic heart failure, as well as diabetic nephropathy.
The second drug candidate in the area of cardiology is an oral soluble guanylate cyclase (sGC) stimulator (BAY 1021189). The start of a Phase IIb study in patients with worsening chronic heart failure is expected later this year.
For the cardio-renal syndrome, a Phase IIb program with the investigational new drug Molidustat (BAY 85-3934) is under initiation in patients with anemia associated with chronic kidney disease and/or end-stage renal disease. Molidustat is a novel inhibitor of hypoxia-inducible factor (HIF) prolyl hydroxylase (PH) which stimulates erythropoietin (EPO) production and the formation of red blood cells. Phase I data have shown that inhibition of HIF-PH by Molidustat results in an increase in endogenous production of EPO.
In oncology, Copanlisib (BAY 80-6946), a novel, oral phosphatidylinositol-3 kinases (PI3K) inhibitor, was selected for accelerated development. Copanlisib demonstrated a broad anti-tumor spectrum in preclinical tumor models and promising early clinical signals in a Phase I study in patients with follicular lymphoma. A Phase II study in patients with Non-Hodgkin’s lymphoma is currently ongoing.
Bayer has also made good progress in the development of new treatment options for patients with gynecological diseases: sPRM (BAY 1002670) is a novel oral progesterone receptor modulator that holds the promises of long-term treatment of patients with symptomatic uterine fibroids. Based on promising early clinical data the initiation of a Phase III study is planned for mid-2014.
Initiation of further studies with recently launched products Bayer has successfully launched five new pharmaceutical products, namely Xarelto™, Stivarga™, Xofigo™, Eylea™, and Riociguat, which has very recently been approved in Canada under the trade name Adempas™.
Xarelto has been approved globally for five indications across seven distinct areas of use, allowing doctors to treat patients in a greater variety of venous and arterial thromboembolic conditions than any other novel oral anticoagulant. The company continues to study the use of Xarelto for the treatment of further cardiovascular diseases. Ongoing clinical Phase III studies include COMPASS and COMMANDER-HF. The COMPASS study will assess the potential use of Xarelto in combination with aspirin, or as a single treatment to prevent major adverse cardiac events (MACE) in nearly 20,000 patients with atherosclerosis related to coronary or peripheral artery disease. The COMMANDER-HF study will evaluate the potential added benefit of Xarelto in combination with single or dual-antiplatelet therapy to help reduce the risk of death, heart attack and stroke in approximately 5,000 patients with chronic heart failure and coronary artery disease, following hospitalization for exacerbation of their heart failure. In order to answer medically relevant questions for specific patient populations Bayer has initiated a range of additional Xarelto studies in patients with atrial fibrillation (AF) undergoing percutaneous coronary intervention with stent placement (PIONEER-AF-PCI), cardioversion (X-VERT) or an AF ablation procedure (VENTURE-AF). As an extension to the Xarelto clinical trial programme, a number of real-world studies are designed to observe and further evaluate Xarelto in everyday clinical practice. These include the XAMOS study of more than 17,000 orthopaedic surgery patients, which confirmed the clinical value of oral, once-daily Xarelto in routine clinical practice in adults following orthopaedic surgery of the hip or knee. XANTUS is designed to collate data on real-world protection with Xarelto in over 6,000 adult patients in Europe with non-valvular AF at risk of stroke while XANAP is designed to collate data on real-world protection with Xarelto in over 5,000 adult patients in Europe and Asia with non-valvular AF at risk of stroke. XALIA will generate information from over 4,800 patients treated for an acute DVT with either Xarelto or standard of care.
In the area of oncology, Stivarga has been approved in 42 countries for use against metastatic colorectal cancer that is refractory to standard therapies, and additionally for gastrointestinal stromal tumor (GIST) in the US and Japan. Bayer is now planning to assess Stivarga in earlier stages of colorectal cancer as well as other cancer types. A Phase III trial in patients with colorectal cancer after resection of liver metastases is currently under initiation. Based on early clinical data Bayer has also initiated a Phase III study in liver cancer in patients who have progressed on sorafenib treatment.
Furthermore, the anti-cancer drug Xofigo (radium 223 dichloride) is a first-in-class alpha-pharmaceutical which is designed for use in prostate cancer patients with ‘bone metastases’ (secondary cancers in the bone) to treat the cancer in the bone and to help extend their lives. Xofigo is approved in the US for the treatment of patients with advanced castrate-resistant prostate cancer with symptomatic bone metastases. In addition, the European CHMP recently gave a positive opinion for radium 223 dichloride for the same use. The decision of the European Commission on the approval is expected in the fourth quarter of 2013. Based on the excellent Phase III results for Xofigo in patients with castration resistant prostate cancer and symptomatic bone metastases Bayer is looking to expand the use of Xofigo to earlier stages of the disease, and plans to initiate a Phase III study in combination with the novel anti-hormonal agent abiraterone. In addition, early stage signal-generating studies in other cancer forms where bone metastases are important causes of morbidity and mortality are planned.
In the area of pulmonary hypertension Adempas (Riociguat) is the first member of a novel class of compounds – so-called ‘soluble guanylate cyclase (sGC) stimulators’ – being investigated as a new and specific approach to treating different types of pulmonary hypertension (PH). Adempas has the potential to overcome a number of limitations of currently approved treatments for pulmonary arterial hypertension (PAH) and addresses the unmet medical need in patients with chronic thromboembolic pulmonary hypertension (CTEPH). It was approved for the treatment of CTEPH in Canada in September 2013, making it the world’s first drug approved in this deadly disease. Riociguat has already shown promise as a potential treatment option beyond these two PH indications. An early clinical study was conducted in PH-ILD (interstitial lung disease), a disease characterized by lung tissue scarring (fibrosis) or lung inflammation which can lead to pulmonary hypertension, and, based on positive data, the decision was taken to initiate Phase IIb studies in PH-IIP (idiopathic pulmonary fibrosis), a subgroup of PH-ILD. Moreover, scientific evidence was demonstrated in preclinical models that the activity may even go beyond vascular relaxation. To prove the hypothesis Bayer is initiating clinical studies in the indication of systemic sclerosis (SSc), an orphan chronic autoimmune disease of the connective tissue affecting several organs and associated with high morbidity and mortality. If successful, Riociguat has the potential to become the first approved treatment for this devastating disease.
In the area of ophthalmology, Eylea (aflibercept solution for injection) is already approved in Europe and several additional countries for the treatment of neovascular (wet) age-related macular degeneration and for macular edema following central retinal vein occlusion. In September, Bayer HealthCare and Regeneron Pharmaceuticals presented data of the two phase III clinical trials VIVID-DME and VISTA-DME of VEGF Trap-Eye for the treatment of diabetic macular edema (DME) at the annual meeting of the Retina Society in Los Angeles and at the EURetina Congress in Hamburg, Germany. Both trials achieved the primary endpoint of significantly greater improvements in best-corrected visual acuity from baseline compared to laser photocoagulation at 52 weeks. Bayer plans to submit an application for marketing approval for the treatment of DME in Europe in 2013.
About Bayer HealthCare The Bayer Group is a global enterprise with core competencies in the fields of health care, agriculture and high-tech materials. Bayer HealthCare, a subgroup of Bayer AG with annual sales of EUR 18.6 billion (2012), is one of the world’s leading, innovative companies in the healthcare and medical products industry and is based in Leverkusen, Germany. The company combines the global activities of the Animal Health, Consumer Care, Medical Care and Pharmaceuticals divisions. Bayer HealthCare’s aim is to discover, develop, manufacture and market products that will improve human and animal health worldwide. Bayer HealthCare has a global workforce of 54,900 employees (Dec 31, 2012) and is represented in more than 100 countries. More information at www.healthcare.bayer.com.
AltheRx Pharmaceuticals has received a notice of allowance for its patent application from the US Patent and Trademark Office (USPTO) for the use of solabegron, a beta 3-adrenergic receptor agonist, in combination with antimuscarinics at both therapeutic and sub-therapeutic doses, for the treatment of overactive bladder (OAB).
Solabegron was discovered by GlaxoSmithKline and acquired by AltheRx in March 2011. Solabegron relaxes the bladder smooth muscle by stimulating beta-3 adrenoceptors, a novel mechanism compared to older established drug treatments for overactive bladder syndrome such as the anticholinergic agents. Astellas Pharma have developed the first commercially available β3 adrenergic receptor, mirabegron, which is now licensed in Japan[6] and the US[7] for overactive bladder. Mirabegron is not licensed for irritable bowel syndrome.
A Phase II study of Solabegron for overactive bladder (OAB) looked at 258 patients with moderate to severe incontinence experiencing an average of 4.5 wet episodes per day. Results demonstrated a statistically significant improvement with Solabegron as compared to placebo, as measured by the percent reduction of the number of wet episodes and the absolute number of daily voids.
A Phase II study for irritable bowel syndrome (IBS) evaluated 102 patients with IBS. Solabegron demonstrated significant reduction in pain associated with the disorder and a trend for greater improvement in the quality of life, compared to placebo.
Both Phase II studies indicated a tolerability profile for Solabegron that was similar to placebo. The OAB patients did not suffer from dry mouth, constipation, increase in heart rate or cognitive issues.
AltheRx is currently preparing to advance Solabegron into a large clinical study in OAB.
Synthesis
Hicks A, McCafferty GP, Riedel E, Aiyar N, Pullen M, Evans C, Luce TD, Coatney RW, Rivera GC, Westfall TD, Hieble JP. GW427353 (solabegron), a novel, selective beta3-adrenergic receptor agonist, evokes bladder relaxation and increases micturition reflex threshold in the dog. Journal of Pharmacology and Experimental Therapeutics. 2007 Oct;323(1):202-9.doi:10.1124/jpet.107.125757PMID 17626794
Grudell AB, Camilleri M, Jensen KL, Foxx-Orenstein AE, Burton DD, Ryks MD, Baxter KL, Cox DS, Dukes GE, Kelleher DL, Zinsmeister AR. Dose-response effect of a beta3-adrenergic receptor agonist, solabegron, on gastrointestinal transit, bowel function, and somatostatin levels in health.American Journal of Physiology. Gastrointestinal and Liver Physiology. 2008 May;294(5):G1114-9. PMID 18372395
Kelleher DL, Hicks KJ, Cox DS, et al. Randomized, double-blind, placebo (PLA)-controlled, crossover study to evaluate efficacy and safety of the beta 3-adrenergic receptor agonist solabegron (SOL) in patients with irritable bowel syndrome (IBS). Neurogastroenterol Motil 2008;20 (Suppl 2):131.
Cellek S, Thangiah R, Bassil AK, Campbell CA, Gray KM, Stretton JL, Lalude O, Vivekanandan S, Wheeldon A, Winchester WJ, Sanger GJ, Schemann M, Lee K. Demonstration of functional neuronal beta3-adrenoceptors within the enteric nervous system. Gastroenterology. 2007 Jul;133(1):175-83.
Schemann M, Hafsi N, Michel K, Kober OI, Wollmann J, Li Q, Zeller F, Langer R, Lee K, Cellek S. The beta3-adrenoceptor agonist GW427353 (solabegron) decreases excitability of human enteric neurons via release of somatostatin.Gastroenterology 2009 Sep 25. [Epub ahead of print]























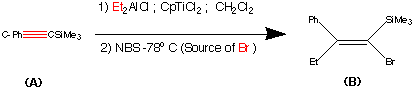







































 Copper-based radiopharmaceuticals for diagnostic imaging can target the amyloid-β plaques implicated in Alzheimer’s disease
Copper-based radiopharmaceuticals for diagnostic imaging can target the amyloid-β plaques implicated in Alzheimer’s disease
