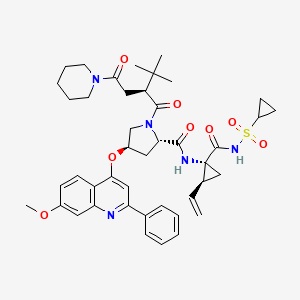
Clazosentan
IUPAC Name: N-[6-(2-hydroxyethoxy)-5-(2-methoxyphenoxy)-2-[2-(2H-tetrazol-5-yl)
pyridin-4-yl]pyrimidin-4-yl]-5-methylpyridine-2-sulfonamide
5-methyl-pyridine-2-sulphonic acid 6-(2-hydroxy-ethoxy)-5-(2- methoxy-phenoxy)-2-(2-1 H-tetrazol-5-yl-pyridin-4-yl)- pyrimidin-4-ylamide
5-methyl-pyridine-2-sulfonic acid [6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-2-[2-(1H-tetrazole-5-yl)-pyridine-4-yl]-pyrimidine-4-yl]-amide
VASODILATOR, Endothelin -1 – receptor antagonist
Clazosentan (Ro61-1790, AXV-034343)
- AXV 034
- AXV 034343
- AXV-034343
- AXV-343434
- Clazosentan
- Ro 61-1790
- Ro-61-1790
- UNII-3DRR0X4728
- VML 588
- VML-588
180384-56-9 cas no
CLINICAL TRIALS…http://clinicaltrials.gov/search/intervention=CLAZOSENTAN in phase 3
| Formula: | C25H23N9O6S |
|---|---|
| Molecular Weight: | 577.5718 |
Endothelin type-A receptor antagonist for the treatment of vasospasm in subarachnoid hemorrhage
Selective endothelin receptor antagonist (Pivlaz)
Acteliion…… innovator
Clazosentan is a drug with orphan drug status , which since 2007, currently in Phase III clinical trials CONSCIOUS-2 ( Clazosentan to O vercome N euro logical i SC Hemia and I nfarct O cc U rring after S ubarachnoid hemorrage) is located. It is for treatment of vasospasm after subarachnoid hemorrhage are used (SAH).
Clazosentan is used by the Swiss pharmaceutical company Actelion developed. Medicinally, the disodium salt is used.Clazosentan to come under the name Pivlaz on the market.
The endothelin -1 – receptor is one of the strongest known vasoconstrictors . Clazosentan is an E -1 – receptor antagonist , for the treatment of vasospasm after subarachnoid hemorrhage is under development. After subarachnoid hemorrhage , an irritation of theblood vessels to a vasospasm and the associated to a reduced supply of brain tissue with oxygen lead. A possible consequence may be a Ischemic stroke be. Clazosentan acts this vasoconstriction contrary.
The plasma half-life of 6-10 min .
Actelion has initiated a comprehensive global phase IIb/III development program for clazosentan sodium (formerly Ro-61-1790, VML-588, …

CLAZOSENTAN
 CLAZOSENTAN DI-NA SALT is discontinued
CLAZOSENTAN DI-NA SALT is discontinued
Clazosentan, shown below, is a well known endothelin receptor antagonist.

Since clazosentan is a known and useful pharmaceutical, it is desirable to discover novel derivatives thereof. Clazosentan is described in European Patent No. 0,799,209
IT IS DESCRIBED IN US6103902
- Paul LM van Giersbergen things Manse, J.: tolerability, pharmacokinetics, and pharmacodynamics of clazosentan, a parenteral endothelin receptor antagonist . In: European Journal of Clinical Pharmacology . 63, No. 2, February 2007, pp. 151-158. doi :10.1007/s00228-006-0117-z . PMID 16,636,870 .
- Paul LM van Giersbergen things Manse J., Gunawardena, KA: Influence of ethnic origin and sex on the pharmacokinetics of clazosentan . In: Journal of Clinical Pharmacology . 47, No. 11, November 2007, pp. 1374-1380. doi :10.1177/0091270007307337 . PMID 17,906,281
| US6004965 * | Dec 8, 1995 | Dec 21, 1999 | Hoffmann-La Roche Inc. | Sulfonamides |
| WO1996019459A1 * | Dec 8, 1995 | Jun 27, 1996 | Volker Breu | Novel sulfonamides |
|
8-13-1998
|
Pyrrolidine-3-carboxylic acids as endothelin antagonists. 3. Discovery of a potent, 2-nonaryl, highly selective ETA antagonist (A-216546).
|
Journal of medicinal chemistry
|
|
7-23-2009
|
In silico prediction of volume of distribution in human using linear and nonlinear models on a 669 compound data set.
|
Journal of medicinal chemistry
|
………………………………………………………..
SYNTHESIS
EXAMPLE 15
a) In analogy to Example 1a), from 5-methyl-pyridine-2-sulphonic acid 6-chloro-5-(2-methoxy-phenoxy)-2-(1-oxy-pyridin-4-yl)-pyrimidin-4-ylamide there is obtained 5-methyl-pyridine-2-sulphonic acid 6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-2-(1-oxy-pyridin-4-yl)-pyrimidin-4-ylamide, melting point 188-190
b) In analogy to Example 2, from 5-methyl-pyridine-2-sulphonic acid 6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-2-(1-oxy-pyridin-4-yl)-pyrimidin-4-ylamide there is obtained 5-methyl-pyridine-2-sulphonic acid 2-(2-cyano-pyridin-4-yl)-6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-pyrimidin-4-ylamide.
……………………………………………..
SYNTHESIS

4-Cyanopyridine (I) is reacted with ammonium chloride in methanolic NaOMe to afford the amidine (II), which is cyclized with diethyl (2-methoxyphenoxy)malonate (III) producing the dihydroxypyrimidine (IV). Chlorination of (IV) in hot POCl3, followed by oxidation of the obtained dichloropyrimidine (V) with peracetic acid leads to the pyridine N-oxide (VI). Subsequent condensation of the dichloropyrimidine derivative (VI) with the potassium salt of 5-methylpyridine-2-sulfonamide (VII) yields the sulfonamido pyrimidine (VIII). The remaining chloride group of (VIII) is then displaced with the sodium alkoxide of ethylene glycol in hot DME to furnish the hydroxyethyl ether (IX). Treatment of the pyridine N-oxide (IX) with cyanotrimethylsilane and Et3N in refluxing acetonitrile gives rise to the 2-cyanopyridine (X), which is finally converted to the title tetrazole derivative by treatment with sodium azide in the presence of NH4Cl in DMF (1).
………………………………………………………….
SYNTHESIS OF DISODIUM SALT OF CLAZOSENTAN

DESCRIBED IN WO 9619459.


- III is reacted with a compound of formula V

-
The reaction type is known in the art and may be performed under basic conditions for example in the presence of a coupling agent, e.g. 1,4-diazobicyclo[2.2.2]octane, together with potassium carbonate in acetone.
EXAMPLE 1
1360 ml of formamide were added to 136 g (437 mmol) of 5-(2-methoxy-phenoxy)-2-pyridine-4-yl-pyrimidine-4,6-diole. Then, at a temperature of 0 acid and thereafter 36.5 g (130 mmol) of iron(II)sulfate heptahydrate were added to the suspension. After that, 89 ml (874 mmol) of 30% hydrogen peroxide were added dropwise within 1 hr at a temperature of 0 to 5 0 sodium pyrosulfite in 680 ml of de-ionized water was added dropwise to the reaction mixture within 30 min. at 0 reaction mixture was stirred at 0 The suspension was then filtered under reduced pressure. The filtrate was first washed with 1750 ml of de-ionized water and thereafter with 700 ml of ethanol. Then the solid was dried at 80 There were obtained 132.4 g (91% of theory) of 4-[4,6-dihydroxy -5-(2-methoxy-phenoxy)-pyrimidine-2-yl]-pyridine-2-carboxylic acid amide with a HPLC purity of 91.4% (w/w).
Preparation of Starting Material
a) 53.1 g of 4-cyano-pyridine (98%) are added all at once to a solution of 1.15 g of sodium in 200 ml of abs. MeOH. After 6 hr 29.5 g of NH.sub.4 Cl are added while stirring vigorously. The mixture is stirred at room temperature overnight. 600 ml of ether are added thereto, whereupon the precipitate is filtered off under suction and thereafter dried at 50 4-amidino-pyridine hydrochloride (decomposition point 245-247
b) 112.9 g of diethyl (2-methoxyphenoxy)malonate are added dropwise within 30 min. to a solution of 27.60 g of sodium in 400 ml of MeOH. Thereafter, 74.86 g of the amidine hydrochloride obtained in a) are added all at once. The mixture is stirred at room temperature overnight and evaporated at 50 of ether and filtered off under suction. The filter cake is dissolved in 1000 ml of H.sub.2 O and treated little by little with 50 ml of CH.sub.3 COOH. The precipitate is filtered off under suction, washed with 400 ml of H.sub.2 O and dried at 80 obtained 5-(2-methoxy-phenoxy)-2-(pyridine-4-yl)-pyrimidine-4,6-diole (or tautomer), melting point above 250
EXAMPLE 2
Within 20 min. 61 ml (633 mmol) of POCl.sub.3 were added dropwise to 34 ml (200 mmol) of diisopropyl ethylamine at 5 followed by stirring at 5 23.5 g (66 mmol) of 4-[4,6-dihydroxy-5-(2-methoxy-phenoxy)-pyrimidine-2-yl]-pyridine-2-carboxylic acid amide were added in four portions under cooling followed by stirring at 90 to 20 dichloromethane. Volatile components (i.e. excess of POCl.sub.3) was removed by evaporation from 20 re-distillation with 100 ml of toluene. After adding 250 ml of dichloromethane to the residue (88 g of a black oil) the solution was heated to 35 were added dropwise within 30 min. whereby the pH was kept constant by the subsequent addition of 28% NaOH solution (60 ml) within 5 to 6 hr. The mixture was stirred at 35 by removal of dichloromethane by distillation. The resulting suspension was allowed to cool down to 20 hr. The solid was filtered off under suction, washed with 500 ml of water and dried at 70 (86% of theory) of 4-[4,6-dichloro-5-(2-methoxy-phenoxy)-pyrimidine-2-yl]-pyridine-2-carbonitrile with a HPLC purity of 94.3% (w/w).
EXAMPLE 3
12.5 g (33.5 mmol) of 4-[4,6-dichloro-5-(2-methoxy-phenoxy)-pyrimidine-2-yl]-pyridine-2-carbonitrile and 6.06 g (35 mmol) of 5-methyl-pyridine-2-sulfonamide were added to 130 ml of acetone. 15 g of potassium carbonate and 190 mg (1.6 mmol) of 1,4-diazobicyclo[2.2.2]octane were added and the suspension was stirred at 40 de-ionized water were added followed by dropwise addition of 50 ml of 3 N hydrochloric acid (pH of the solution=1). Acetone was removed by evaporation and the suspension was stirred for 1 hr. The solid was filtered and washed with 100 ml of water. The residue was heated (reflux) in 100 ml of methanol for 1 hr followed by cooling to 20 solid was filtered and dried at 80 were obtained 16.0 g (93% of theory) of 5-methyl-pyridine-2-sulfonic acid [6-chloro-2-(2-cyano-pyridine-4-yl)-5-(2-methoxy-phenoxy)-pyrimidine-4-yl]-amide with a HPLC purity of 90.3% (w/w).
EXAMPLE 5
20 g (39 mmol) of 5-methyl-pyridine-2-sulfonic acid [6-chloro-2-(2-cyano-pyridine-4-yl)-5-(2-methoxy-phenoxy)-pyrimidine-4-yl]-amide were suspended in 100 ml of N,N-dimethyl formamide and 7.6 ml (156 mmol) of hydrazine hydrate were added within 15 min. The reaction mixture was allowed to warm up slowly to 20 temperature of 15 followed by slow addition of 10.5 ml acetic acid (until pH=5.4). The resulting suspension was stirred for 2 hr at 20 additional 2 hr 0 firstly washed with 200 ml of de-ionized water and thereafter with 100 ml of t-butylmethylether. The residue was dried at 40 18 hr. There were obtained 21.7 g (102% of theory) of 5-methyl-pyridine-2-sulfonic acid [6-chloro-2-[2-(hydrazino-imino-methyl)-pyridine-4-yl]-5-(2-methoxy-phenoxy)-pyrimidine-4-yl]-amide with a HPLC purity of 81.4% (w/w).
EXAMPLE 7
20 g (37 mmol) of 5-methyl-pyridine-2-sulfonic acid [6-chloro-2-[2-(hydrazino-imino-methyl)-pyridine-4-yl]-5-(2-methoxy-phenoxy)-pyrimidine-4-yl]-amide were added to 160 ml of N,N-dimethyl formamide. To this solution was added dropwise 23 ml of 6 N aqueous hydrochloric acid at a temperature of 15 mmol) of sodium nitrite in 20 ml de-ionized water was added slowly. The reaction mixture was allowed to warm up to 20 for 1.5 hr. Then 160 ml of de-ionized water were added and the suspension was stirred for 1 hr. The solid was filtered off under suction, washed with 100 ml of de-ionized water and dried at 50 hr. There were obtained 18.9 g (92% of theory) of 5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2-methoxy-phenoxy)-2-[2-(1H-tetrazole-5-yl)-pyridine-4-yl]-pyrimidine-4-yl]-amide with a HPLC purity of 89.6% (w/w).
EXAMPLE 9
15 g (27 mmol) of 5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2-methoxy-phenoxy)-2-[2-(1H-tetrazole-5-yl)-pyridine-4-yl]-pyrimidine-4-yl]-amide were suspended in 75 ml of ethylene glycol and 6.5 g (163 mmol) of sodium hydroxide were added. The reaction mixture was heated to 85 thereafter 55 ml of 3 N aqueous hydrochloric acid were added dropwise. The suspension was stirred at 20 off under suction, washed with 150 ml of de-ionized water and dried at 70 in 50 ml of N,N-dimethyl formamide and 40 ml of dioxane at 70 Gaseous ammonia was introduced into this solution until pH=9. The resulting suspension was allowed to cool down slowly. The suspension was stirred at 0 washed with 25 ml of dioxane and thereafter with 25 ml of ethanol. Then the solid was dried at 50 ammonium salt (10.4 g, 17.5 mmol) was suspended in 50 ml of methanol and thereafter 6.5 ml (35 mmol) of a 5.4 N sodium methylate solution were added. The solution was heated (reflux) for 3 hr, cooled down slowly to 20 filtering, washed with 10 ml of ice-cold methanol and dried at 70 C., 2000 Pa for 17 hr. There were obtained 6.9 g (41% of theory) of 5-methyl-pyridine-2-sulfonic acid [6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-2-[2-(1H-tetrazole-5-yl)-pyridine-4-yl]-pyrimidine-4-yl]-amidesodium salt (1:2) with a HPLC purity of 98.2% (w/w).
This application claims benefit to EP 98114978.4 filed Aug. 10, 1998.
SIMILAR SYNTHESIS OF TEZOSENTAN AND INTERMEDIATES… AN EXPERT WILL PICK UP NAMES AND INTERMEDIATES… just change the isopropyl gp in vii to methyl
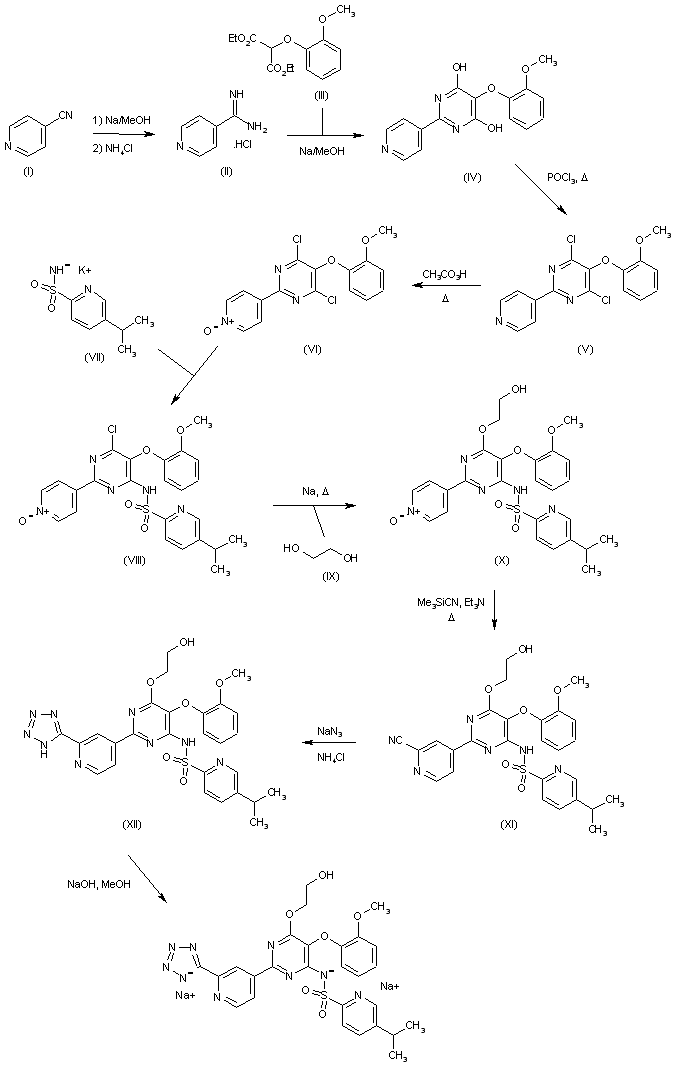
Reaction of 4-cyano-pyridine (I) with Na in methanol followed by treatment with ammonium chloride provides 4-amidino-pyridine hydrochloride (II), which is then converted into 5-(2-methoxyphenoxy)-2-(pyridin-4-yl)-pyrimidine-4,6-diol (IV) by condensation with diethyl malonate derivative (III) by means of Na in MeOH. By heating compound (IV) with phosphorus oxychloride (POCl3), 4,6-dichloro-5-(2-methoxyphenoxy)-2-pyridin-4-yl)pyrimidine (V) is obtained, which in turn is oxidized with peracetic acid in refluxing acetonitrile to afford N-oxide derivative (VI). Condensation of (VI) with 5-isopropylpyridine-2-sulfonamide potassium (VII) furnishes 5-isopropylpyridine-2-sulfonic acid 6-chloro-5-(2-methoxyphenoxy)-2-(1-oxy-pyridin-4-yl)-pyrimidin-4-yl amide (VIII), which is then dissolved in dimethoxyethane and subjected to reaction with Na in hot ethylene glycol (IX) to provide N-[6-(2-hydroxyethoxy)-5-(2-methoxyphenoxy)-2-(1-oxy-pyridin-4-yl)-pyrimidin-4-yl]-5-isopropylpyridine-2-sulfonamide (X). Refluxing of (X) with trimethylsilylcyanide and Et3N in acetonitrile yields cyano derivative (XI), which is then converted into the tetrazole derivative (XII) by reaction with sodium azide and NH4Cl in DMF at 70 C. Finally, the disodium salt of tezosentan is obtained by treatment of (XII) with Na/MeOH in THF.
………………………………..
SYNTHESIS
Example 29
In analogy to Example 3, from 5-methyl-pyridine-2- sulphonic acid 2-(2-cyano-pyridin-4-yl)-6-(2-hydroxy-ethoxy)- 5-(2-methoxy-phenoxy)-pyrimidin-4-ylamide there is obtained 5-methyl-pyridine-2-sulphonic acid 6-(2-hydroxy-ethoxy)-5-(2- methoxy-phenoxy)-2-(2-1 H-tetrazol-5-yl-pyridin-4-yl)- pyrimidin-4-ylamide as a white substance of melting point 239- 241 °C from CH3CN
Exgmple, 1 5
a) In analogy to Example l a), from 5-methyl-pyridine-2- sulphonic acid 6-chloro-5-(2-methoxy-phenoxy)-2-(1 -oxy- pyridin-4-yl)-pyrimidin-4-ylamide there is obtained 5-methyl- pyridine-2-sulphonic acid 6-(2-hydroxy-ethoxy)-5-(2-methoxy- phenoxy)-2-(l -oxy-pyridin-4-yl)-pyrimidin-4-ylamide, melting point 188-190°C (from acetonitrile).
b) In analogy to Example 2, from 5-methyl-pyridine-2- sulphonic acid 6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-2- (1 -oxy-pyridin-4-yl)-pyrimidin-4-ylamide there is obtained 5- methyl-pyridine-2-sulphonic acid 2-(2-cyano-pyridin-4-yl)-6- (2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-pyrimidin-4- ylamide
Example 1
a) 200 ml of dimethoxyethane and 1 10.9 g of 4-[4-(4-tert- butyl-phenyl-sulphonylamino)-6-chloro-5-(2-methoxy-phenoxy)- pyrimidin-2-yl]-pyridine 1 -oxide are added all at once to a solution of 23.80 g of sodium in 660 ml of ethylene glycol. The solution is heated at 90°C for 20 hours while stirring, thereafter cooled, poured into 2500 ml of H2O and thereafter treated with CH3COOH to pH 5. The mixture is extracted three times with EtOAc, the organic phase is washed with H2O, dried with Na2Sθ4 and evaporated under reduced pressure. The residue is recrystall- ized from CH3CN and thereafter twice from a mixture of acetone and CH3CN. There is thus obtained 4-[4-(4-tert-butyl-phenyl- sulphonylamino)-6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)- pyrimidin-2-yl]-pyridine 1 -oxide.
Preparation of the starting material:
b) 53.1 g of 4-cyano-pyridine (98%) are added all at once to a solution of 1.15 g of sodium in 200 ml of abs. MeOH. After
6 hours 29.5 g of NH4CI are added while stirring vigorously. The mixture is stirred at room temperature overnight. 600 ml of ether are added thereto, whereupon the precipitate is filtered off under suction and thereafter dried at 50°C under reduced pressure. There is thus obtained 4-amidino-pyridine hydro- chloride (decomposition point 245-247°C).
c) 1 12.9 g of diethyl (2-methoxyphenoxy)malonate are added dropwise within 30 minutes to a solution of 27.60 g of sodium in 400 ml of MeOH. Thereafter, 74.86 g of the amidine hydro- chloride obtained in b) are added all at once. The mixture is stirred at room temperature overnight and evaporated at 50°C under reduced pressure. The residue is treated with 500 ml of ether and filtered off under suction. The filter cake is dissolved in 1000 ml of H2O and treated little by little with 50 ml of CH3COOH. The precipitate is filtered off under suction, washed with 400 ml of H2O and dried at 80°C under reduced pressure. There is thus obtained 5-(2-methoxy-phenoxy)-2-(pyridin-4-yl)- pyrimidine-4,6-diol (or tautomer), melting point above 250°C.
d) A suspension of 1 54.6 g of 5-(2-methoxy-phenoxy)-2- (pyridin-4-yl)-pyrimidine-4,6-diol (or tautomer) in 280 ml of POCI3 is heated at 120°C in an oil bath for 24 hours while stirring vigorously. The reaction mixture changes gradually into a dark brown liquid which is evaporated under reduced pressure and thereafter taken up three times with 500 ml of toluene and evaporated. The residue is dissolved in 1000 ml of CH2CI2, treated with ice and H2O and thereafter adjusted with 3N NaOH until the aqueous phase has pH 8. The organic phase is separated and the aqueous phase is extracted twice with CH2CI2. The combined CH2CI2 extracts are dried with MgSθ4, evaporated to half of the volume, treated with 1000 ml of acetone and the CH2CI2 remaining is distilled off at normal pressure. After standing in a refrigerator for 2 hours the crystals are filtered off under suction and dried at 50°C overnight. There is thus obtained 4,6-dichloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl)- pyrimidine, melting point 1 78-1 80°C.
e) A solution of 1 7.4 g of 4,6-dichloro-5-(2-methoxy- phenoxy)-2-pyridin-4-yl)-pyrimidine in 100 ml of CH3CN is boiled at reflux for 3 hours with 1 5 ml of a 32% peracetic acid solution, thereafter cooled and stored in a refrigerator overnight. The crystals are filtered off under suction and dried at 50°C under reduced pressure. There is thus obtained 4-[4,6-dichloro- 5-(2-methoxy-phenoxy)-pyrimidin-2-yl]-pyridine 1 -oxide, melting point 189-1 90°C.
for analogy
f) A solution of 36.4 g of 4-[4,6-dichloro-5-(2-methoxy- phenoxy)-pyrimidin-2-yl]-pyridine 1 -oxide and 52.8 g of p-tert- butylphenyl-sulphonamide potassium in 1 50 ml of abs. DMF is stirred at room temperature for 24 hours. Thereafter, it is poured into a mixture of 1 500 ml of H2O and 1000 ml of ether while stirring mechanically, whereby a precipitate forms. The suspension is adjusted to pH 5 with CH3COOH, suction filtered, the crystals are washed with cold water and thereafter with ether and dried at 50°C. There is thus obtained 4-[4-(4-tert- butyl-phenylsulphonylamino)-6-chloro-5-(2-methoxy-phenoxy)- pyrimidin-2-yl]-pyridine 1 -oxide as a colourless material of melting point 247-249°C.
Example 2
A solution of 78.45 g of 4-[4-(4-tert-butyl-phenyl- sulphonylamino)-6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)- pyrimidin-2-yl]-pyridine 1 -oxide, 122.5 g of trimethylsilyl cyanide, 127.8 g of triethylamine and 1200 ml of CH3CN is boiled at reflux for 20 hours and thereafter evaporated under reduced pressure. The oily residue is taken up in 1000 ml of EtOAc and the solution is washed with CH3COOH:H2θ 9:1 and then with H2O. The EtOAc extracts are dried with Na2SO4. After evaporation of the solvent the residue is taken up in a mixture of CH3CN and CF3COOH (20:1 ), whereby a crystalline precipitate separates. There is thus obtained 4-tert-butyl-N-[2-(2-cyano-pyridin-4- yl)-6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-pyrimidin-4- yl]-benzenesulphonamide of melting point 176-1 79°C.
Example 3
A suspension of 50.0 g of 4-tert-butyl-N-[2-(2-cyano- pyridin-4-yl)-6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)- pyrimidin-4-yl]-benzenesulphonamide, 46.33 g of NH4CI and 56.47 g of NaN3 in 1600 ml of DMF is heated to 70°C for 24 hours while stirring vigorously. The majority of the solvent is distilled off under reduced pressure, the residue is dissolved in H2O, the solution is extracted four times at pH 6.5 with ether, thereafter treated with CH3COOH to pH = 4.5 and extracted with EtOAc. After working up there is obtained a residue which is treated with ether and filtered off under suction therefrom. There is thus obtained 4-tert-butyl-N-[6-(2-hydroxy-ethoxy)-5-(2- methoxy-phenoxy)-2-(2-1 H-tetrazol-5-yl-pyridin-4-yl)- pyrimidin-4-yl]-benzenesulphonamide, melting point 225-227°C.
////////////////////////////////
EXTRA INFO
Bosentan (Ro-470203), Atransentan (ABT627), Tezosentan (Ro-610612), Sitaxsentan (TBC-11251), Darusentan (LU-135252), Clazosentan (Ro61-1790, AXV-034343), ZD-4054, Ambrisentan (LU-208075), TAK-044, Avosentan (SPP301), and BQ-123 (Ihara et al Life Sci 1992, 50(4):247-55).
| Antagonists of Endothelin type A receptor ETA | |
| Name | Structure |
| BQ-123 |
 |
| Bosentan |
 |
| Atrasentan |
 |
| Tezosentan |
 |
| Sitaxsentan |
 |
| Darusentan |
 |
| Clazosentan |
|
| ZD-4054 (Zibotentan) |
 |
| Ambrisentan |
 |
| Tak-044 |
 |
| Avosentan |
 |






 rapamycin
rapamycin SIROLIMUS
SIROLIMUS











 SIROLIMUS
SIROLIMUS



 MIDOSTAURIN
MIDOSTAURIN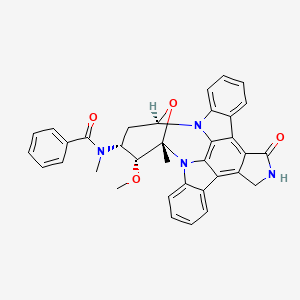 MIDOSTAURIN
MIDOSTAURIN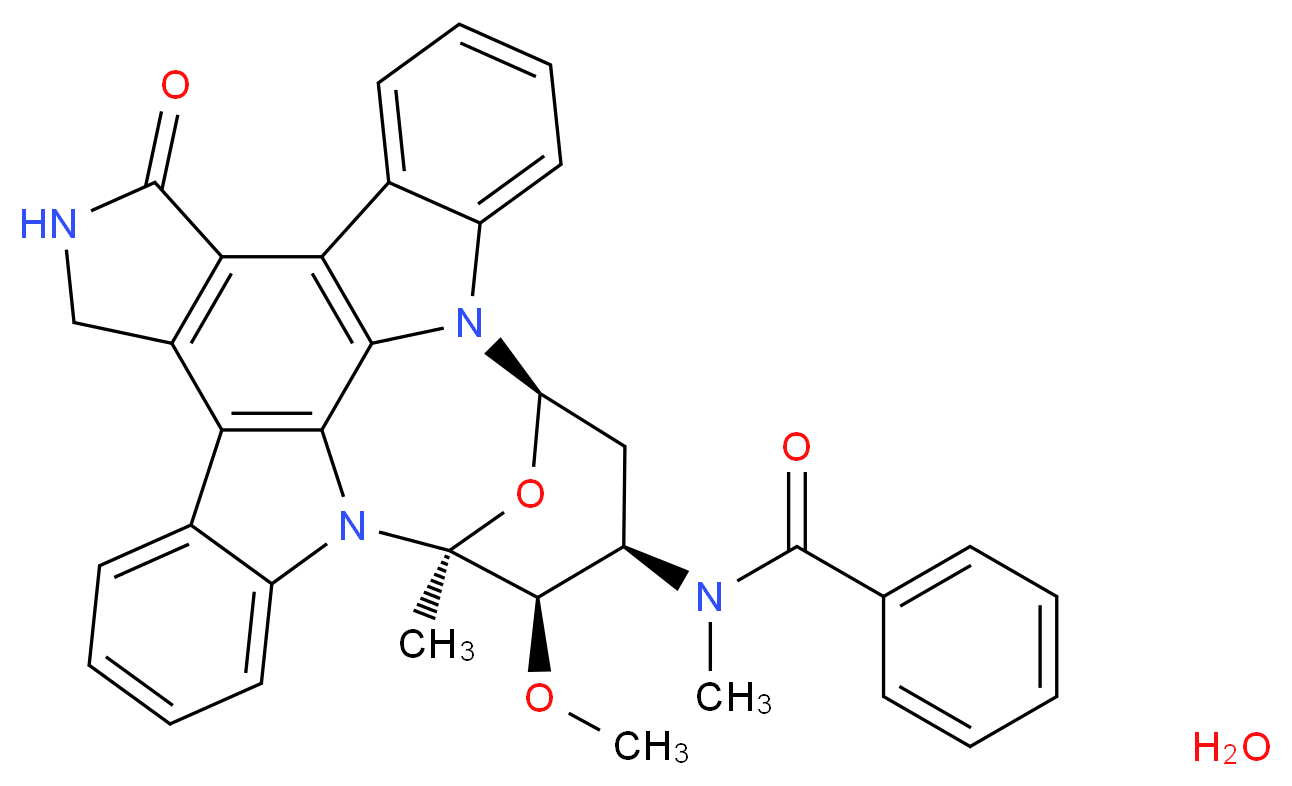






 cicaprost
cicaprost cicaprost
cicaprost



















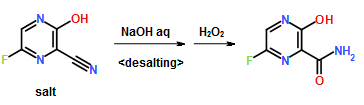













 11.25 M in NH3)], Na2CO3 [15% w/w solution (4 L)]. More EtOAc (4 L) was added, and the organic layer was washed with water (4 L). The organic phase was then concentrated to 2.5 L; again fresh EtOAc (4 L) was added, and the solution was concentrated to 2.5 L to give a solution of casopitant 2.
11.25 M in NH3)], Na2CO3 [15% w/w solution (4 L)]. More EtOAc (4 L) was added, and the organic layer was washed with water (4 L). The organic phase was then concentrated to 2.5 L; again fresh EtOAc (4 L) was added, and the solution was concentrated to 2.5 L to give a solution of casopitant 2.