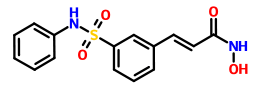
Dasantafil
Dasantafil

569351-91-3 CAS NO
405214-79-1 (racemate)
UNII-48P711MI2G, SCH 446132, D03657,
Molecular Formula: C22H28BrN5O5
Molecular Weight: 522.39222
Merck & Co. (Originator) IN PHASE 2
THERAPEUTIC CLAIM treatment of erectile dysfunction (phosphodiesterase (PDE) 5 isoenzyme inhibitor)
CHEMICAL NAMES
- 1H-purine-2,6-dione, 7-[(3-bromo-4-methoxyphenyl)methyl]-1-ethyl-3,7-dihydro-8-[[(1R,2R)-2-hydroxycyclopentyl]amino]-3-(2-hydroxyethyl)
- 7-(3-bromo-4-methoxybenzyl)-1-ethyl-8-[[(1R,2R)-2-hydroxycyclopentyl]amino]-3-(2-hydroxyethyl)-3,7-dihydro-1H-purine-2,6-dione
7-[(3-bromo-4-methoxyphenyl)methyl]-l-ethyl-8-[[(lR,2R)-2- hydroxycyclopentyl]amino]-3-(2-hydroxyethyl)purine-2,6-dione
Treatment of Erectile Dysfunction , Phosphodiesterase PDE5A Inhibitors
Dasantafil (SCH-446132) is a phosphodiesterase type 5 (PDE5) inhibitor which had been in early clinical development at Merck & Co. for the treatment of erectile dysfunction (ED); however, no recent development has been reported for this research. Phosphodiesterases regulate the tissue concentration of cyclic guanosine monophosphate (cGMP), which in turn triggers smooth muscle relaxation, allowing blood to flow into the penis and resulting in erection. PDE5 is the most abundant phosphodiesterase in the human corpus cavernosum, and as such its inhibition by dasantafil enhances erectile function by increasing the concentration of cGMP.
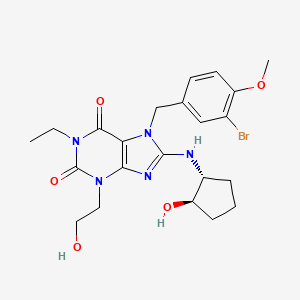 DASANTAFIL
DASANTAFIL
PDE V inhibitor compounds and their use in treating a variety of physiological conditions are described in a number of patents {e.g., U.S. Pat. Nos. 5,409,934, 5,470,579, 5,939,419 and 5,393,755) and foreign publications (e.g., WO 93/23401 , WO 92/05176, WO 92/05175, and WO 99/24433).
Specific PDE V inhibitors have been found useful for specific indications. For example, the use of PDE V inhibitors for treating impotence has met with commercial success with the introduction of sildenafil citrate, vardenafil, and tadalafil (i.e., Viagra®, Levitra®, and Cialis®, respectively). The chemistry and use of Viagra®, including its mechanism of action in treating erectile dysfunction, are taught in EP 0 702 555 B1. Accordingly, it is an object of this invention to provide a method of using a PDE V inhibitor to treat a patient who has, or is at risk of, congestive heart failure, and/or other cardiovascular conditions.
Processes for preparing PDE V inhibitor compounds can be found in US
6,207,829, US 6,066,735, US 5,955,611 , US 5,939,419, US 5,393,755, US 5,409,934, US 5,470,579, US 5,250,534, WO 02/24698, WO 99/24433, WO 93/23401 , WO 92/05176, WO 92/05175, EP 740,668 and EP 702,555. One type of PDE V inhibitor compound contains a xanthine functionality in its structure. Xanthines can be prepared as described by Peter K. Bridson and Xiaodong Wang in 1 -Substituted Xanthines, Synthesis, 855 (July, 1995), which is incorporated herein by reference in its entirety. WO 02/24698, which is incorporated herein by reference in its entirety, teaches a class of xanthine PDE V inhibitor compounds useful for the treatment of impotence. A general process disclosed therein for preparing xanthine PDE V inhibitor compounds having the formula (I) follows:

(III) (I) (i) reacting a compound having the formula (III) with an alkyl halide in the presence of a base (introduction of R11 or a protected form of R11); (ii) (a) debenzylating and then (b) alkylating the compound resulting from step (i) with an alkyl halide, XCH2R1“; (iii) (a) deprotonating and then (b) halogenating the compound resulting from step (ii);
(iv) reacting the compound resulting from step (iii) with an amine having the formula RlvNH2; and (v) removing a protecting portion of Rn, if present, on the compound resulting from step (iv) to form the compound having the formula (I). R1, R”, Rm and Rlv correspond to R1, R2, R3 and R4, respectively, in WO02/24698, and are defined therein. WO 02/24698 (pages 44 and 68-73) also teaches a synthesis for the following xanthine compound (identified therein as Compound 13 or Compound 114 of Table II): 1-ethyl-3,7-dihydro-8-[(1 R,2R)- (hydroxycyclopentyl) amino]-3-(2-hydroxyethyl)-7-[(3-bromo-4- methoxyphenyl)methyl]-1 H-purine-2,6-dione:
Compound 13. It would be beneficial to provide an improved process for preparing polycyclic xanthine PDE V inhibitor compounds
………………….
Patent description

WO2006055573A2
entry 129 is dasantafil

…………………
SYNTHESIS
WO2002024698A1

14X‘ CHs ‘ B” tX is Experimental Procedure: Compound 114 in Table II (13)
1 (20.0 g, 74.0 mmol) was dissolved in dimethylformamide (370 mL) under nitrogen and (2-bromoethoxy)-terf-butyldimethylsilane (31.8 mL, 148 mmol) was added dropwise. The reaction was stirred at room temperature for 115 hrs., then diluted with ethyl acetate and washed with water several times.
The organic mixture was dried over potassium carbonate, filtered and concentrated under vacuum. Purification via flash chromatography (30/70 ethyl acetate/hexanes) yielded 2 (28.1 g, 88%).
1H NMR (400 MHz, CDCI3): δ 7.52 (s, 1 H), 7.29-7.39 (m, 5H), 5.49 (s,
2H), 4.25 (t, 2H, J = 6.0 Hz), 4.07 (q, 2H, J = 7.2 Hz), 3.93 (t, 2H, J =
6.0 Hz), 1.24 (t, 3H, J = 7.2 Hz), 0.75 (s, 9H), 0.08 (s, 6H). HRMS: Calcd for C22H32N403Si (M+H): 429.2322. Found: 429.2329.
To a solution of 2 (2.10 g, 4.89 mmol) in methanol (375 mL) was added ammonium formate (4.64g, 73.6 mmol) and 20% palladium hydroxide on carbon (980 mg). The reaction was heated to reflux for 1.5 hrs., then cooled to room temperature, filtered and concentrated under vacuum. Purification via flash chromatography (50/50 ethyl acetate/hexanes) yielded 3 (1.26 g, 94%).
1H NMR (400 MHz, CDCI3): δ 7.82 (s, 1 H), 4.33 (t, 2H, J = 6.0 Hz), 4.16
(q, 2H, J = 7.2 Hz), 3.99 (t, 2H, J = 6.0 Hz), 1.29 (t, 3H, J = 7.2 Hz),
0.78 (s, 9H), 0.06 (s, 6H). HRMS: Calcd for Cι5H26N4O3Si (M+H): 339.1852. Found: 339.1864. To 3 (970 mg, 2.86 mmol) was added dimethylformamide (25 mL), 3- bromo-4-methoxybenzyl bromide 15 (1.62 g, 5.79 mmol), and potassium carbonate (800 mg, 5.79 mmol) under nitrogen. The reaction mixture was stirred at room temperature for 21 hrs., then diluted with ethyl acetate and washed with water several times. The organic mixture was dried over potassium carbonate, filtered and concentrated under vacuum. Purification by flash chromatography (30/70 ethyl acetate/hexanes) yielded 10 (1.55 g, 100%).
1H NMR (400 MHz, CDCI3): δ 7.52 (s, 1 H), 7.51 (d, 1 H, J = 2.4 Hz),
7.30 (dd 1 H, J = 2.0 Hz, J = 8.4 Hz), 6.87 (d, 1 H, J = 8.8 Hz), 5.40 (s,
2H), 4.25 (t, 2H, J = 6.0 Hz), 4.07 (q, 2H, J = 7.0 Hz), 3.93 (t, 2H, J =
6.0 Hz), 3.88 (s, 3H), 1.25 (t, 3H, J = 7.0 Hz), 0.75 (s, 9H), 0.08 (s, 6H).
HRMS: Calcd for C23H33BrN4O4Si (M+H): 537.1533. Found: 537.1540.
To solution of 10 (1.50 g, 2.80 mmol) in tetrahydrofuran (24 mL) under nitrogen at -78 °C (dry ice/acetone bath) was added lithium diisopropylamide (2M in THF/heptane, 2.2 mL, 4.33 mmol). After stirring for thirty minutes, 1 ,2- dibromotetrafluoroethane (0.69 mL, 5.77 mmol) was added dropwise over five minutes. The reaction was stirred for 1.25 hrs. at -78 °C then quenched with saturated aqueous sodium bicarbonate and warmed to room temperature.
The mixture was extracted with dichloromethane, dried over potassium carbonate, filtered and concentrated under vacuum. Purification via flash chromatography (30/70 ethyl acetate/hexanes) yielded 11 (600 mg, 34%). 1H NMR (400 MHz, CDCI3): δ 7.60 (d, 1 H, J = 2.4 Hz), 7.35 (dd, 1 H, J =
2.0 Hz, J = 8.4 Hz), 6.84 (d, 1 H, J = 8.4 Hz), 5.45 (s, 2H), 4.21 (t, 2H, J = 5.6 Hz), 4.07 (q, 2H, J = 6.8 Hz), 3.90 (t, 2H, J = 5.6 Hz), 3.87 (s, 3H), 1.24 (t, 3H, J = 6.8 Hz), 0.73 (s, 9H), 0.08 (s, 6H). HRMS: Calcd for C23H32Br2N4O4Si (M+H): 615.0638. Found: 615.0633.
To 11 (1.89 g, 3.07 mmol) was added the amino alcohol hydrochloride salt (1.31 g, 12.27 mmol), diisopropylethylamine (15.4 mL), and 1-methyl-2- pyrrolidinone (15.4 mL). The reaction mixture was heated to 160 °C in a sealed tube for 13 hrs., then cooled to room temperature. Water was added, then the mixture was extracted with ethyl acetate and washed with water several times. The organic mixture was dried over potassium carbonate, filtered and concentrated under vacuum. Purification via flash chromatography (3/97 methanol/dichloromethane) yielded 12 (1.77 g, 90%).
1H NMR (400 MHz, CDCI3): δ 7.45 (d, 1 H, J = 2.0 Hz), 7.17 (dd, 1 H, J =
2.4 Hz, J = 8.6 Hz), 6.86 (d, 1 H, J = 8.4 Hz), 5.18-4.34 (m, 3H), 4.00- 4.23 (m, 5H), 3.86-3.98 (m, 6H), 3.69-3.79 (m, 1 H), 2.10-2.21 (m, 1 H), 1.99-2.10 (m, 1 H), 1.60-1.84 (m, 3H), 1.32-1.43 (m, 1 H), 1.24 (t, 3H, J = 7.2 Hz), 0.75 (s, 9H), 0.07 (d, 6H, J = 4.0 Hz). HRMS: Calcd for C28H43BrN5θ5Si (M+H): 636.2217. Found: 636.2207.
12 (1.77 g, 2.78 mmol) was dissolved in tetrahydrofuran (28 mL) under nitrogen and tetrabutylammonium fluoride (1M in THF, 28 mL) was added dropwise. The reaction was stirred at room temperature for 15 hrs., then diluted with dichloromethane and washed with water several times. The organic mixture was dried over potassium carbonate, filtered and concentrated under vacuum. Purification via flash chromatography (3/97 methanol/dichloromethane) yielded 13 (compound no. 114 in Table II) (760 mg, 52%).
DASANTAFIL
1H NMR (400 MHz, CDCI3):
δ 7.47 (d, 1 H, J = 2.0 Hz), 7.19 (dd, 1 H, J =2.0 Hz, J = 8.4 Hz), 6.88 (d, 1 H, J = 8.4 Hz), 5.25 (s, 2H), 5.09 (s, 1H), 4.21-4.27 (m, 3H), 4.06 (q, 2H, J = 7.0 Hz), 3.90-3.97 (m, 3H), 3.89 (s, 1 H), 3.74-3.82 (m, 1 H), 3.08 (s, 1 H), 2.12-2.22 (m, 1 H), 1.98-2.08 (m, 1 H), 1.60-1.86 (m, 3H), 1.33-1.43 (m, 1 H), 1.25 (t, 3H, J = 7.0 Hz),1.06-1.22 (m, 3H). HRMS: Calcd for C22H28BrN5O5 (M+H): 522.1352. Found: 522.1346.
2-Bromo-4-methyl anisole 14 (2.2 mL, 14.9 mmol) was dissolved in dichlomethane (30 mL) and N-bromosuccinimide (3.75 g, 16.4 mmol) was added followed by AIBN (26.0 mg). The reaction was heated to reflux for 19 hrs., then cooled to room temperature and the precipitate was filtered off. The filtrate was diluted with dichloromethane and washed with 0.5 M aqueous sodium bicarbonate, followed by water. The organic mixture was dried over sodium sulfate, filtered and concentrated under vacuum to yield 15 (4.16 g,
100%). The benzyl bromide was used as the crude material without further purification.
1H NMR (400 MHz, CDCI3): δ 7.59 (d, 1 H, J = 2.0 Hz), 7.30 (dd, 1 H, J =
2.4 Hz, J = 8.4 Hz), 6.85 (d, 1 H, J = 8.4 Hz), 4.37 (s, 2H), 3.90 (s, 3H).
General Synthesis of Compound No. 114 in Table II (13) a) Reacting 1 with an alkyl halide and base to form 2; b) Debenzylation of 2 to form 3; c) Alkylation of 3 with a benzyl halide to form 10; d) Deprotonation of 10 followed. by addition of a brominating agent to form 11 ; e) Displacement of bromo 11 with an amine to form 12; and f) Silyl ether cleavage of 12 to form compound no. 114 in Table II (13).
114 IN TABLE II./(13)

……………
WO2003101992A1
GENERAL SCHEME

SYNTHESIOS
1A

9A 13A DASANTAFIL
SYNTHESIS
Compound 1A:
glycine-A/-r(4-methoxyphenyl)methyl1 ethyl ester
To a mixture of glycine ethyl ester hydrochloride (about 1.4 equiv) and potassium carbonate (about 1.0 equiv) was added anhydrous ethanol. The mixture
was stirred at about 40-45 °C for about 3 hours. Then, p-anisaldehyde (about 1.0
equiv.) was added, and the reaction mixture was stirred for a minimum of about 3 hours to provide an imine (not shown). Upon reaction completion (about <5.0 % p- anisaldehyde remaining by GC analysis), the reaction mixture was cooled to about 0-
10 °C. Then, an aqueous solution of sodium borohydride (about 0.50 equiv) was
added to the reaction mixture at a temperature of between about 0 °C and about 20
°C, and stirred for about 1 hour to provide Compound 1 A. Upon completion of the
reduction reaction, the reaction mixture was quenched with the slow addition of an aqueous solution of aqueous glacial acetic acid. After quenching, the reaction mixture was warmed to room temperature and filtered to remove solids. The filtrate was then concentrated under vacuum, followed by the addition of toluene and water to facilitate layer separation. Aqueous potassium carbonate solution was added to adjust the pH of the mixture to about 8-9. The organic layer was separated and the aqueous layer was extracted with toluene. The combined toluene extracts were concentrated to provide the product in about a 80-85% yield (based on GC and HPLC in solution assay). 1H NMR 400 MHz (CDCI3): δ 7.23 (d, J = 8.5 Hz, 2H), 6.85 (d, J = 8.5 Hz, 2H),
4.17 (q, J = 7.1 Hz, 2H), 3.78 (s, 3H), 3.73 (s, 2H), 3.38 (s, 2H), 1.88 (s, br, 1 H), 1.26
(t, J = 7.1 Hz, 3H); 13C NMR 100 MHz (CDCI3): δ 172.8, 159.2, 132.0, 129.9, 114.2,
61.1, 55.6, 53.1 , 50.4, 14.6.
Compound 2:
/V-cvanomethanimidic acid ethyl ester
To cyanamide (about 1.2 mole) was added triethylorthoformate (about 1.33 mole), and the reaction mixture was heated to about 85-95 °C for approximately 2 hours to form Compound 2. Estimated in-solution yield was about 95-100%. The product was optionally purified by vacuum distillation.
1H NMR 400 MHz (CDCI3): δ 8.38 (s, 1H), 4.28 (t, J = 6.7 Hz, 2H), 1.29 (t, J =
6.8 Hz, 3H); 13C NMR 100 MHz (CDCI3): δ 171.5, 113.4, 65.5, 13.1.
Compound 3A:

cis- and frans-glvcine Λ/-r(cvanoimino,methyl1-Λ/-r(4- methoxyphenvDmethvπ ethyl ester
A solution of Compound 1A (about 1.0 mole) in toluene was concentrated under vacuum to distill off toluene. Anhydrous tetrahydrofuran (“THF”) was added to the concentrate, then Compound 2 (about 1.2 moles, obtained above) was added to that, and the solution was heated at reflux for about 1 hour. At this stage, the formation of Compound 3A was complete. Estimated in-solution yield was about
95% (about 2:1 mixture of cis and trans isomers). Compound 4A: 1H-imidazole-5-carboxylic acid, 4-amino-1-[(4- methoxyphenvDmethvn ethyl ester
Compound 3A (obtained above) was concentrated by distilling off THF. Then, anhydrous ethanol was added to afford a reaction mixture solution. Separately, potassium t-butoxide (about 0.15 mole) was dissolved in anhydrous ethanol to afford a solution. The potassium t-butoxide solution was added to the reaction mixture solution and heated to about 75-85 °C for about 1 hour. The overall in-solution yield of Compound 4A was about 85-90%.

1H NMR 400 MHz (CDCI3): δ 7.16 (s, 1H), 7.08 (d, J = 8.6 Hz, 2H), 6.82 (d, J
=8.7 Hz, 2H), 5.23 (s, 2H), 4.93 (s, br, 2H), 4.23 (q, J = 7.1 , 2H), 3.76 (s, 3H), 1.26 (t,
J = 7.1 Hz, 3H); 13C NMR 400 MHz (CDCI3): δ 160.9, 159.2, 139.0, 128.6, 128.5,
114.0, 101.8, 59.5, 55.2, 50.1 , 14.4.
Compound 5AK:

4A 5AK
1 -ethyl-3,7-dihydro-7-F(4-methoxyphenyl)methvπ-1 H-Purine-2.6- dione potassium salt
The reaction mixture containing Compound 4A in ethanol (obtained above) was added to diglyme and distilled under vacuum to remove the ethanol. After being cooled to room temperature, Λ/-ethylurethane (about 1.2 equiv.) was added and the
reaction mixture was heated to about 110-120 °C. A solution of potassium t-butoxide
(2.2 equiv.) in diglyme was added to the hot solution. The reaction mixture was cooled to room temperature. THF was added to precipitate additional product, which was filtered and washed to provide Compound Salt 5AK in 55-65% overall yield. The wet cake can be used as such for conversion to Compound 6A.
1H NMR (DMSO-de, 400 MHz): δ 7.73 (s, 1H) 7.31 (d, J = 8.6 Hz , 2H) 6.86 (d,
J = 8.6 Hz, 2H) 5.24 (s, 1 H) 3.88 (q, J = 6.8 Hz, 2H) 3.71 (s, 3H) 1.07 (t, J = 6.8 Hz, 3H); 13C NMR (DMSO-d6, 100 MHz): δ 161.1 , 159.0, 158.4, 157.2, 141.4, 131.0,
129.5, 114.1 , 105.6, 55.4, 48.2, 34.4, 14.3.
Optional Neutralization of Compound Salt 5AK to Compound 5A: Compound 5A: 1-ethyl-3,7-dihvdro-7-r(4-methoχyphenyl,methvπ-1 H-Purine-2,6- dione
The wet cake filtered solid of Compound Salt 5AK (obtained above) was suspended in water and then acidified to a pH of about 5 using glacial acetic acid. The resulting slurry was filtered to obtain the neutralized product, which was then washed with water and dried. The overall isolated yield of neutralized Compound 5A from Compound 1 A was about 45-55%. Spectroscopic data for neutralized Compound 5A was identical to that of Compound Salt 5AK.
Compound 6A:
3-r2-(acetyloxy,ethvn-1-ethyl-3,7-dihvdro-7-r(4- methoxyphenyl,methvπ-1H-purine-2,6-dione
To the wet cake filtered solid of Compound Salt 5AK (obtained above) were added tetrabutylammonium bromide (about 0.05 mole) and 2-bromoethyl acetate
(about 1.2 moles) in THF. After being heated to reflux for about 2 hours, part of the THF was distilled off, and isopropyl alcohol was added to the reaction mixture. The reaction mixture was then concentrated under reduced pressure and cooled to around room temperature. Water was added to precipitate the product. After being cooled to about 0-5 °C for about a few hours, the product was isolated by filtration. The wet cake was washed with aqueous isopropyl alcohol (about 30% in water), and dried under vacuum to afford Compound 6A as a pale yellow solid in about a 45- 55% overall yield (based on Compound 1A). The crude product may be purified further by decolorizing with Darco in methanol, followed by filtration and concentration to afford crystalline Compound 6A.
1H NMR (CDCI3 , 400 MHz): δ 7.54 (s, 1 H) 7.32 (d, J = 8.6 Hz, 2H) 6.90 (d, J =
8.6 Hz, 2H) 5.43 (s, 2H) 4.41 (m, 2H) 4.38 (m, 2H) 4.10 (q, J = 7.2 Hz, 2H) 3.79 (s,
3H) 1.96 (s, 3H) 1.25 (t, J = 7.2 Hz, 3H); 13C NMR (CDCI3 , 100 MHz): δ 171.1 ,
160.2, 155.3, 151.4, 148.9, 140.9, 130.1 , 127.7, 114.8, 107.5, 61.7, 55.6, 50.2, 42.4, 36.9, 21.2, 13.6.
After Optional Neutralization of Compound Salt 5AK to Compound 5A:
Compound 6A:
3-r2-(acetyloxy.ethvπ-1-ethyl-3,7-dihvdro-7-r.4- methoxyphenyl)methyn-1H-purine-2,6-dione
Acetonitrile was added to a mixture of Compound 5A (about 1.0 mole), anhydrous potassium carbonate (about 1.5 moles) and tetrabutylammonium hydrogen sulfate (about 0.05 mole). 2-bromoethyl acetate (about 1.5 moles) was added in three separate portions (0.72 mole in the beginning, another 0.45 mole after about 2 hours of reaction, and then the remaining 0.33 mole after about another
1 hour of reaction) during the course of the reaction at about 80-85 °C. The total reaction time was about 7 hours. The reaction mixture was cooled to about room temperature and filtered. The filtrate was concentrated. Aqueous isopropanol was added to crystallize the product. The product was filtered, washed with aqueous isopropanol, and dried to provide Compound 6A in about a 75-80% yield. Compound 7A: 8-bromo-1 -ethyl-3-r2-(acetyloxy)ethvπ-3,7-dihvdro-7-r(3-bromo-4- methoxyphenyl)methvπ-1 – -Purine-2,6-dione
Compound 6A (about 1 mole) and NBS (about 2.8 moles) were dissolved in
dry acetonitrile and agitated at about 15-20 °C. To this reaction mixture, a solution of
sulfuric acid (about 0.03 mol) in acetonitrile was added, while maintaining the
reaction temperature below about 25 °C. The reaction mixture was agitated at about
20-25 °C for about 12-15 hours until complete consumption of the starting material
was indicated. The reaction mixture was cooled to about 0-5 °C and a cold (about 5-
10 °C) aqueous solution of sodium sulfite was added, keeping the temperature below
about 10 °C. The reaction was agitated for about 2 hours at about 0-10 °C, and then
filtered. The isolated cake was washed with water, followed by methanol, then dried under a vacuum to obtain Compound 7A in about an 85% yield.

1H NMR (CDCIs, 400 MHz): D 7.60 (d, J=2.0 Hz, 1H), 7.35 (dd, J=8.4 Hz, 2.0 Hz, 1 H), 6.83 (d, J=8.4 Hz, 1 H), 5.43 (s, 2H), 4.35 (m, 4H), 4.05 (q, J=7.0 Hz, 2H), 3.85 (s, 3H), 1.96 (s, 3H), 1.23 (t, J=7.0 Hz, 3H); 13C NMR (CDCI3, 100 MHz): D 171.0, 156.2, 154.2, 150.8, 148.2, 138.3, 128.9, 128.7, 127.5, 112.1 , 112.0, 109.1 , 61.5, 56.5, 49.3, 42.5, 37.0, 21.0, 13.3. MS (ES) m/e 545.2 (M+H)+.
Compound 13A:
1-ethyl-3.7-dihvdro-8-r(1f?,2 )-(hvdroxycvclopentyl)amino1-3-(2- hvdroxyethvπ-7-r(3-bromo-4-methoxyphenvhmethvπ-1/–purine-2.6-dione
Compound 7A (about 1 mole) was combined with (R,R)-2-amino-1- cyclopentanol hydrochloride (Compound 8A, about 1.2 moles) and sodium bicarbonate (about 3 moles). To this reaction mixture was added N,N- dimethylacetamide (“DMA”), and the reaction mixture was agitated at about 135-140 °C for about 15-17 hours until complete consumption of the starting material was
indicated.

Compound 9A is an intermediate that is formed, but not isolated, from the
reaction mixture. The reaction mixture was then cooled to about 45-50 °C, and
tetrabutylammonium hydroxide (about 0.05 moles of about a 40% solution in water) was charged therein, followed by methanol. The reaction mixture was refluxed at
about 80-85 °C for about 8-9 hours until complete deprotection of the acetate group
was indicated. The reaction mixture was cooled to about 40-45 °C and concentrated
under vacuum. The pH of the reaction mixture was adjusted to about 5-6 with dilute
acetic acid, and the reaction mixture was heated to about 55-65 °C, and seeded with
a small amount of Compound 13A. The reaction mixture was then cooled to about
30-35 °C over a period of about 2 hours, and water was added over a period of
about 1 hour. The reaction mixture was further cooled to about 0-5 °C over a period
of about 1 hour, and agitated at that temperature for about 4 hours. The Compound 13A product was isolated by filtration, washed with water and dried to provide about an 85-90% yield.

9A 13A DASANTAFIL
1H NMR (CDCI3, 400 MHz): D 7.47 (d, J=2.1 Hz, 1 H), 7.18 (dd, J=8.4 Hz, 2.0 Hz, 1 H), 6.87 (d, J=8.4 Hz, 1H), 5.23 (s, 2H), 5.01 (s, 1 H), 4.22 (m, 2H), 4.15 (m, 1H), 4.05 (q, J=7.0 Hz, 2H), 3.93 (m, 3H), 3.88 (s, 3H), 3.77 (m, 1H), 2.95 (m, 1H), 2.15 (m, 1H), 2.05 (m, 1 H), 1.60-1.80 (m, 4H), 1.35 (m, 1 H), 1.23 (t, J=7.0 Hz, 3H); 13C NMR (CDCI3, 100 MHz): D 156.2, 154.0, 153.5, 151.8, 148.3, 132.6, 129.1 , 127.9, 112.5, 103.2, 79.5, 77.8, 63.2, 61.3, 56.7, 46.5, 45.9, 36.8, 32.9, 31.5, 21.4, 13.8. MS (ES) m/e 523.4 (M+H)+. Micronization
INTERPRETATION
1H NMR (CDCI3, 400 MHz): DELTA
7.47 (d, J=2.1 Hz, 1 H), SANDWICHED AROM H BETWEEN BROMO AND -CH2-PY RING
7.18 (dd, J=8.4 Hz, 2.0 Hz, 1 H), AROM H ORTHO TO -CH2-PH RING AND PARA TO BROMO
6.87 (d, J=8.4 Hz, 1H), AROM H ORTHO TO O ATOM OF PH RING
5.23 (s, 2H), CH2 OF N-CH2-PH RING
5.01 (s, 1 H), OH OR NH 1H OUT OF 3 NOS
4.22 (m, 2H), OH OR NH 2H OUT OF 3 NOS
4.15 (m, 1H), –NCH2CH2OH 1H OUT OF 4 NOS
4.05 (q, J=7.0 Hz, 2H), CH2 OF NCH2 CH3
3.93 (m, 3H), —NCH2CH2OH 3H OUT OF 4 NOS
3.88 (s, 3H), -OCH3
3.77 (m, 1H), OH-CH OF CYCLOPENTANE RING
2.95 (m, 1H),NH-CH OF CYCLOPENTANE RING
2.15 (m, 1H),
2.05 (m, 1 H), 1H ON CYCLOPENTANE RING
1.60-1.80 (m, 4H), 4H ON CYCLOPENTANE RING
1.35 (m, 1 H), 1 H PARA TO SUBS IN CYCLOPENTANE RING
1.23 (t, J=7.0 Hz, 3H) –NCH2 CH3
………………………..

 DASANTAFIL
DASANTAFIL
REFERENCES
1 WANG Y ET AL: “DESIGN AND SYNTHESIS OF XANTHINE ANALOGUES AS POTENT AND SELECTIVE PDE5 INHIBITORS” BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, OXFORD, GB, vol. 12, no. 21, 2002, pages 3149-3152, XP009014973 ISSN: 0960-894X
2. Peter K. Bridson and Xiaodong Wang in 1 -Substituted Xanthines, Synthesis, 855 (July, 1995)

PATENTS
1. WO 2002024698..
2. WO 2003101992..
3.WO 2010062366
4. WO 2007002125
5. WO 2006104870
6. WO 2005051368
7. US 2004167137
8. WO 2003101991
9.WO2006104870A2 , Schering Corp Methods of treating benign prostatic hyperplasia or lower urinary track symptoms by using pde 5 inhibitors
| WO2005012303A1 * |
Jul 29, 2004 |
Feb 10, 2005 |
Kevin B Alton |
Metabolite of xanthine phosphodiesterase 5 inhibitor and derivatives thereof useful for treatment of erectile dysfunction |
| US7312223 |
Jul 29, 2004 |
Dec 25, 2007 |
Schering Corporation |
Metabolite of xanthine Phosphodiesterase 5 inhibitor and derivatives thereof useful for treatment of erectile dysfunction |
| WO2002024698A1 * |
Sep 17, 2001 |
Mar 28, 2002 |
Schering Corp |
Xanthine phosphodiesterase v inhibitors |
| WO2003020724A1 * |
Aug 26, 2002 |
Mar 13, 2003 |
Schering Corp |
Polycyclic guanine phosphodiesterase v inhibitors |
| WO2003042216A1 * |
Nov 7, 2002 |
May 22, 2003 |
Schering Corp |
Polycyclic guanine derivative phosphodiesterase v inhibitors |
| DE4411660A1 * |
Apr 5, 1994 |
Oct 12, 1995 |
Hoechst Ag |
Verwendung von Xanthinderivaten zur Reduktion der pathologischen Hyperreagibilität eosinophiler Granulozyten, neue Xanthinverbindungen und Verfahren zu deren Herstellung |
| EP0430025A2 * |
Nov 20, 1990 |
Jun 5, 1991 |
Hokuriku Pharmaceutical Co., Ltd. |
Xanthine compound, method for preparing thereof, and a pharmaceutical composition comprising the same |
| JP3072480A * |
|
|
|
Title not available |
| JP4279586A * |
|
|
|
Title not available |
| JP5001065A * |
|
|
|
Title not available |
| US5728686 * |
Oct 31, 1996 |
Mar 17, 1998 |
Hoechst Aktiengesellschaft |
Alkylxanthine phosphonates and alkylxanthine phosphine oxides and their use as pharmaceuticals |
| US6214992 * |
Jun 4, 1997 |
Apr 10, 2001 |
Hoechst Aktiengesellschaft |
Use of theophylline derivatives for the treatment and prophylaxis of states of shock, novel xanthine compounds and processes for their preparation |
| WO2003057200A2 * |
13 jan 2003 |
17 juli 2003 |
Richard David Carr |
Compositions comprising inhibitors of dpp-iv and nep enzymes for the treatment of diabetes |
| WO2003101991A1 * |
30 mei 2003 |
11 dec 2003 |
Schering Corp |
Xanthine phosphodiesterase v inhibitor polymorphs |
| WO2006026395A1 * |
26 aug 2005 |
9 maart 2006 |
Richard Dixon |
Endothelin a receptor (eta) antagonists in combination with phosphodiesterase 5 inhibitors (pde5) and uses thereof |
| US20020169174 * |
28 aug 2001 |
14 nov 2002 |
Samuel Chackalamannil |
Xanthine phosphodiesterase V inhibitors |
| US20030060627 * |
25 jan 2002 |
27 maart 2003 |
Jordi Gracia Ferrer |
8-Phenyl-6, 9-dihydro-[1,2,4] triazolo[3,4-i]purin-5-one derivatives |
| US20040063731 * |
14 jan 2002 |
1 april 2004 |
Hans-Michael Eggenweiler |
Pharmaceutical formulation comprising pyrazolo[4,3-d]pyrimidines and endothelin receptor antagonists or thienopyrimidines and endothelin receptor antagonists |
|
|
6-17-2011
|
USE OF A PDE 5 INHIBITOR FOR TREATING AND PREVENTING HYPOPIGMENTARY DISORDERS
|
|
|
8-32-2010
|
Process for preparing xanthine phosphodiesterase V inhibitors and precursors thereof
|
|
|
6-12-2009
|
METHODS OF USING PDE V INHIBITORS FOR THE TREATMENT OF CONGESTIVE HEART FAILURE
|
|
|
5-13-2009
|
Xanthine Phosphodiesterase V Inhibitors
|
|
|
4-24-2009
|
METHODS OF TREATING BENIGN PROSTATIC HYPERPLASIA OR LOWER URINARY TRACT SYMPTOMS BY USING PDE 5 INHIBITORS
|
|
|
3-20-2009
|
PHARMACEUTICAL FORMULATIONS
|
|
|
2-29-2008
|
Use of a Pde 5 Inhibitor for Treating and Preventing Hypopigmentary Disorders
|
|
|
12-26-2007
|
Metabolite of xanthine phosphodiesterase 5 inhibitor and derivatives thereof useful for treatment of erectile dysfunction
|
|
|
9-12-2007
|
Xanthine phosphodiesterase V inhibitors
|
|
|
5-32-2007
|
RAPIDLY ABSORBING ORAL FORMULATIONS OF PDE 5 INHIBITORS
|
|
|
|
|
|
3-21-2007
|
Xanthine phosphodiesterase V inhibitor polymorphs
|
|
|
2-16-2007
|
Methods of using PDE 5 inhibitors for the treatment of congestive heart failure
|
|
|
2-9-2007
|
Rapidly absorbing oral formulations of PDE 5 inhibitors
|
|
|
1-5-2007
|
Methods of treating benign prostatic hyperplasia or lower urinary tract symptoms by using PDE 5 inhibitors
|
|
|
7-12-2006
|
Process for preparing xanthine phosphodiesterase V inhibitors and precursors thereof
|
|
|
2-24-2006
|
Pharmaceutical formulations
|
|
|
8-19-2005
|
Xanthine phosphodiesterase V inhibitors
|
|
|
4-8-2005
|
Xanthine phosphodiesterase V inhibitors
|
|
|
11-24-2004
|
Xanthine phosphodiesterase V inhibitors
|
|
|
11-19-2004
|
Xanthine phosphodiesterase V inhibitors
|






































 DOXOFYLLINE
DOXOFYLLINE




















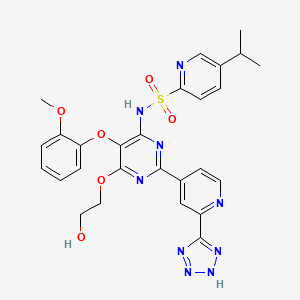 TEZOSENTAN
TEZOSENTAN
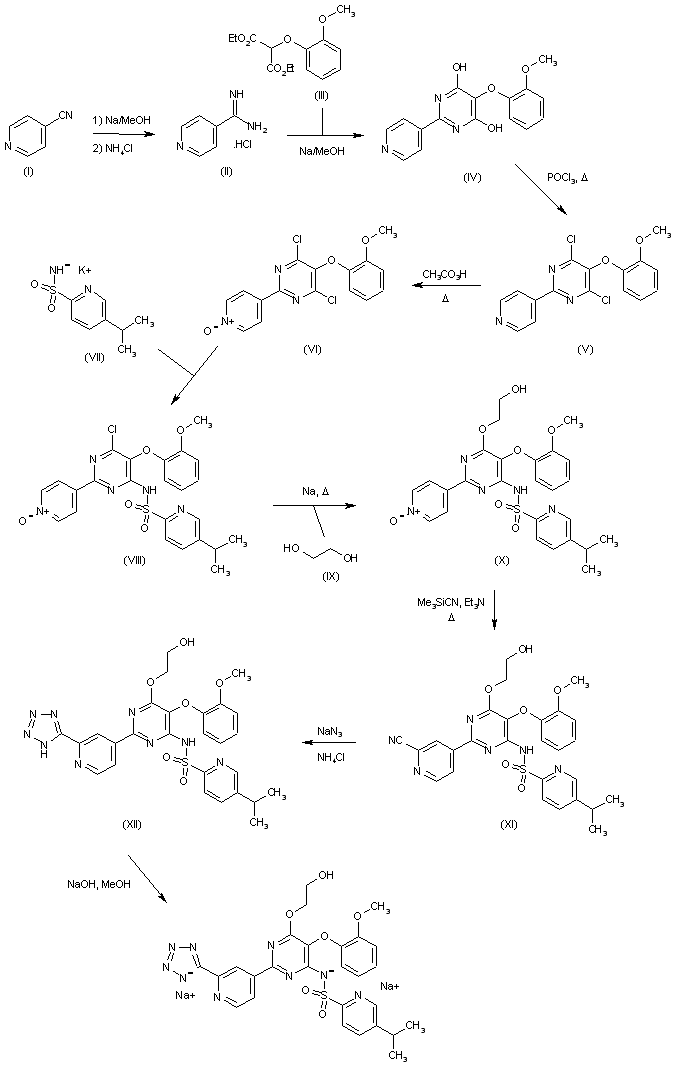
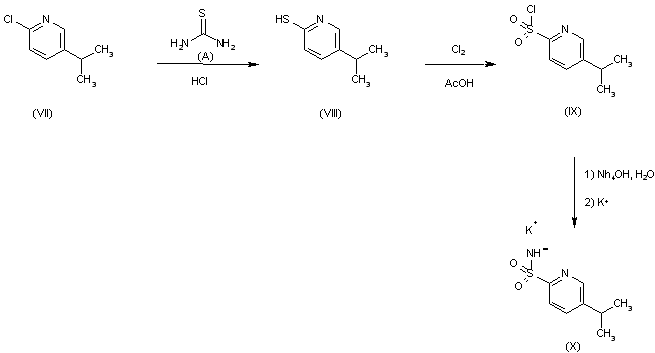

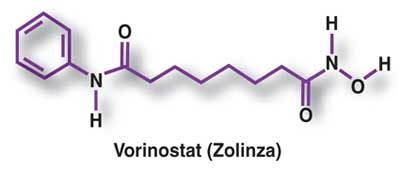
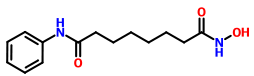
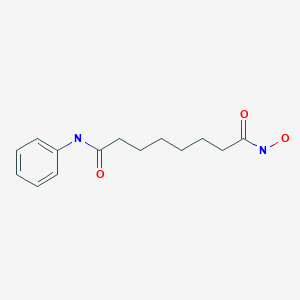 VORINOSTAT
VORINOSTAT






















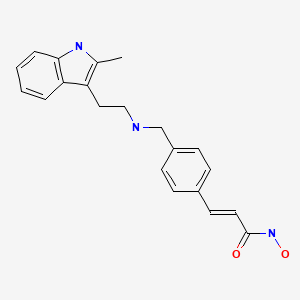 panobinostat
panobinostat













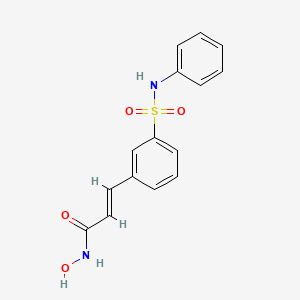 BELINOSTAT
BELINOSTAT