LM11A-31-BHS
(2S,3S)-2-amino-3-methyl-N-(2-morpholinoethyl)-pentanamide
2-Amino-3-methyl-N-[2-(4-morpholinyl)ethyl]-pentanamide dihydrochloride
- CAS Number 1214672-15-7
- Empirical Formula C12H25N3O2 · 2HCl
- Molecular Weight 316.27
LM11A-31 is a non-peptide ligand of the p75 neurotrophin receptor (p75NTR). LM11A-31 blocks pro-NGF induced cell death in neuronal cultures, and protects neuronal cells from the the cytotoxic effects of cisplatin or methotrexate. Oral administration of LM11A-31 promotes the survival of oligodendrocytes and myelinated axons in a mouse spinal cord injury model and improves function in both weight-bearing and non-weight bearing tests.Inhibits death of hippocampal neurons at 100–1,000 pM
http://amcrasto.wix.com/anthony-melvin-crasto/apps/blog/lm11a-31-new-drug-can-help-paralyzed
PharmatrophiX

LM11A-31, C12 H25 N3 O2, Pentanamide, 2-amino-3-methyl-N-[2-(4-morpholinyl)ethyl]- WO 2010102212 TO LONGO FRANK, PUB 10.09.2010 THE UNIVERSITY OF NORTH CAROLINA AT CHAPEL HILL
| PATENT LINK |
http://patentscope.wipo.int/search/en/WO2010102212
Scientists have developed a pill which they claim could help paralyzed people walk again.
The new drug allowed mice with no movement in their lower limbs to walk with ‘well-coordinated steps’ and even to replicate swimming motions, researchers said.
The experimental drug, called LM11A-31, was developed by Professor Frank Longo, of Stanford University, California.
The researchers gave three different oral doses of LM11A-31, as well as a placebo, to different groups of mice beginning four hours after injury and then twice daily for a 42 day experimental period, the ‘Daily Mail’ reported.
In tests, the experimental medication did not increase pain in the mice and showed no toxic effects on the animals.
It also efficiently crossed the blood brain barrier, which protects the central nervous system from potentially harmful chemicals carried around in the rest of the bloodstream.
An injury to the spinal cord stops the brain controlling the body and this is the first time an oral drug has been shown to provide an effective therapy.
“This is a first to have a drug that can be taken orally to produce functional improvement with no toxicity in a rodent model,” Professor Sung Ok Yoon, of Ohio State University, Columbus, said.
“So far, in the spinal cord injury field with rodent models, effective treatments have included more than one therapy, often involving invasive means. Here, with a single agent, we were able to obtain functional improvement,” Yoon said.
The small molecule in the study was tested for its ability to prevent the death of cells called oligodendrocytes.
These cells surround and protect axons, long projections of a nerve cell, by wrapping them in a myelin sheath that protect the fibres.
In addition to functioning as axon insulation, myelin allows for the rapid transmission of signals between nerve cells.
The drug preserved oligodendrocytes by inhibiting the activation of a protein called p75. Yoon’s lab previously found p75 is linked to the death of these specialised cells after a spinal cord injury. When they die, axons that are supported by them degenerate.
“Because we know oligodendrocytes continue to die for a long period of time after an injury, we took the approach that if we could put a brake on that cell death, we could prevent continued degeneration of axons,” she said.
FULL TEXT – JOURNAL OF NEUROSCIENCE
Small, Nonpeptide p75NTR Ligands Induce Survival Signaling and Inhibit proNGF-Induced Death in Journal of neuroscience, 26(20): 5288-5300; doi: 10.1523/JNEUROSCI.3547-05.2006 by SM Massa – 2006 – Cited by 51 – Related articles
17 May 2006 – At 5 nm, LM11A-24 and -31 inhibit TUNEL staining to a degree … We further prioritized LM11A–31, because preliminary studies
Small, Nonpeptide p75NTR Ligands Induce Survival Signaling and Inhibit proNGF-Induced Death
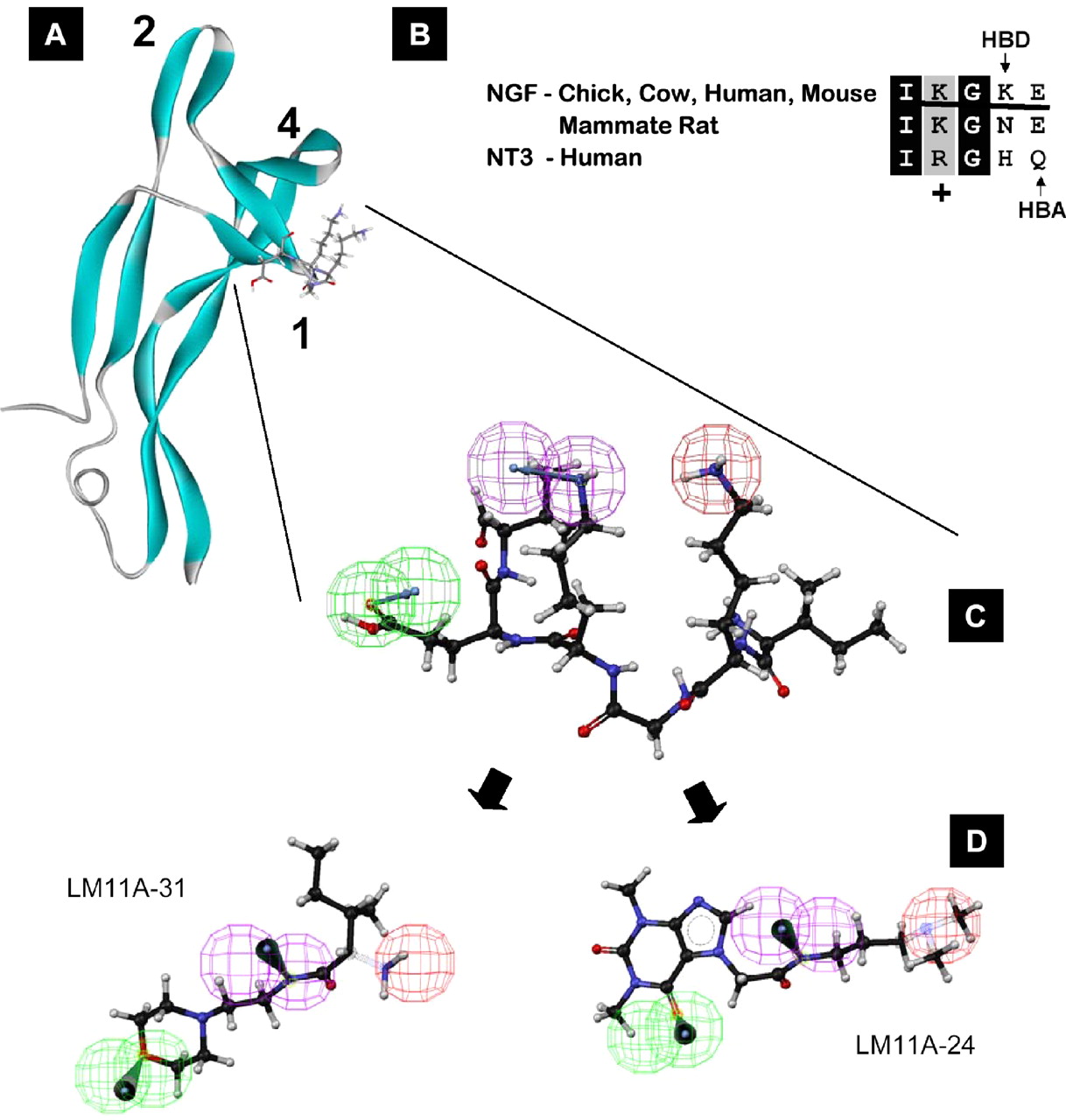
2010 SLIDE PRESENTATION RE P75 (E.G. LM11A–31) BY PHARMATROPHIX’S …
3 Nov 2010 – 2010 slide presentation re p75 (e.g. LM11A–31) by PharmatrophiX’s founder. Longo is PharmatrophiX’s founder.
 The experimental drug was developed by Prof Frank Longo from Stanford University
The experimental drug was developed by Prof Frank Longo from Stanford UniversityProf Frank Longo from Stanford University publications
http://med.stanford.edu/profiles/cancer/frdActionServlet?choiceId=showFacPublications&fid=7249&
Patents
1 US2013005731 (A1) ― 2013-01-03
2 WO2011150347 (A2) ― 2011-12-01
3 US2011230479 (A1) ― 2011-09-22
<a href=”http://www.bloglovin.com/blog/4674983/?claim=hj3e8pdf2nd”>Follow my blog with Bloglovin</a>
………………..
http://www.google.com.mx/patents/US7723328
| TABLE I | |
| Structures of Compounds 1-6 | |
| Compound | Name |
 |
Compound 1 (also referred to herein as “LM11A-28”) |
 |
Compound 2 (also referred to herein as “LM11A-7”) |
 |
Compound 3 (also referred to herein as “LM11A-24”, “24”, and “C24”) |
 |
Compound 4 (also referred to herein as “LM11A-31” and “31”) |
 |
Compound 5 (also referred to herein as “LM11A-36”, “36”, and “C36”) |
 |
Compound 6 (also referred to herein as “LM11A-38” and “C38”) |
 |
Compound 7 |
…………………….
http://www.google.co.in/patents/WO2010102212A2?cl=en
Table I. Structures of Compounds i-vii


Example 32: Preparation of enantiomerically pure 2-amino-3-methyl-N-(2- morpholino-ethyϊ)-pentanamide
[00332] 2-amino-3-methyl-N-(2-morpholinoethyl)-pentanamide can be prepared by a method shown in Scheme 4 below. First, 2-aminoethanol (Compound IE) is transformed to its derivative with a leaving group (Compound 2E). Examples of the leaving group include halides and alkoxy or other activated hydroxyl group. Second, Compound 2E reacts with morpholine at a neutral or basic condition to yield 2-morpholinoethanamine (Compound 3E). The aforementioned two steps may also be performed continuously as one step with Compound 2E being generated in situ. For example, Compound 3 E can be prepared from Compound IE directly through a Mitsunobu reaction wherein the hydroxyl group of Compound IE is activated by diethyl azodicarboxylate (DEAD) before morpholine is added. The final product, 2-amino-3-methyl-N-(2-moipholinoethyl)-pentanamide (Compound 5E), can be obtained by coupling 2-morpholinoethanamine with 2-amino-3- methylpentanoic acid (Compound 4E) via a peptide coupling agent. Examples of the peptide coupling agent include l,r-carbonyldiimidazole (CDI), hydroxybenzotriazole (HOBT), 1,3-dicyclohexylcarbodiimide (DCC), 1- hydroxybenzo-7-azatriazole (HOAt), and the like. Scheme 4:
H2N^0H — H2N^ / LG , p , .
1 Ot= LG: a leaving group
1E zt

[00333] A chiral 2-amino-3-methyl-N-(2-moφholinoethyl)-pentanamide (Compound 5E) can be obtained by using the corresponding chiral 2-amino-3- methylpentanoic acid (Compound 4E) in the above coupling step. For example, (2S,3S)-2-amino-3-methyl-N-(2-moφholinoethyl)-pentanamide; (2R,3R)-2-amino- 3 -methyl-N-(2-morpholinoethyl)-pentanamide; (2R,3 S)-2-amino-3 -methyl-N-(2- moφholinoethyl)-pentanamide; and (2S,3R)-2-ammo-3-methyl-N-(2- morpholinoethyl)-pentanamide can be obtained by using (2S,3S)-2-amino-3- methylpentanoic acid, i.e., L-isoleucine; (2R,3R)-2-amino-3-methylpentanoic acid, i.e., D-isoleucine; (2R,3S)-2-amino-3-methylpentanoic acid, i.e., D-alloisoleucine; and (2S,3R)-2-amino-3-methylpentanoic acid, i.e., L-alloisoleucine, respectively. [00334] The chiral purity, also known as, enantiomeric excess or EE, of a chiral Compound 5E can be determined by any method known to one skilled in the art. For example, a chiral Compound 5E can be hydrolyzed to Compound 3E and the corresponding chiral Compound 4E. Then, the chiral Compound 4E obtained through hydrolysis can be compared with a standard chiral sample of Compound 4E to determine the chiral purity of the chiral Compound 5E. The determination can be conducted by using a chiral HPLC.
……………….
http://www.google.co.in/patents/EP2498782A1?cl=en
Scheme A shows the chemical structures of the present compounds.

(2S,3S)-2-amino-3-methyl-/V-(2-mor holinoethyl)pentanamide

(2R,3R)-2-amin -3-methyl-A/-(2-morpholinoethyl)pentanamide

(2S,3R)-2-amino-3-meth l-A/-(2-morpholinoethyl)pentanamide

] Q (2R,3S)-2-amino-3-methyl-/ /-(2-morpholinoethyl)pentanamide
The free base compound of 2-amino-3-niethyl- -(2-morpholinoethyl)-pentanamide can be prepared from isoleucine by synthetic methods known to one skilled in the art.
Standard procedures and chemical transformation and related methods are well known to one skilled in the art, and such methods and procedures have been described, for example, in standard references such as Fiesers’ Reagents for Organic Synthesis, John Wiley and Sons, New York, NY, 2002: Organic Reactions, vols, 1-83, John Wiley and Sons, New York, NY, 2006; March J, and Smith M,, Advanced Organic Chemistry, 6th ed., John Wiley and Sons, New York, NY; and Larock R.C., Comprehensive Organic Transformations, Wiley-VCH Publishers, New York, 1999. All texts and references cited herein are incorporated by reference in their entirety. Other related synthetic methods can be found in U.S. Patent Application Publication Nos. 2006/024072 and 2007/0060526, the contents of which are herein incorporated by reference in their entirety for all purposes. The amorphous dihydrochloride (di-HCl) salt of 2-amino-3-methyl-N-(2-morpholinoethyl)-pentanamide can be prepared by mixing two molar ecjuivalents of HC1 with one molar equivalent of 2-amino- 3-methyl-N-(2-morpholinoethyl)~pentanamide in appropriate solvent(s) and then separating the di-HCl salt from the solvent(s) mixture.
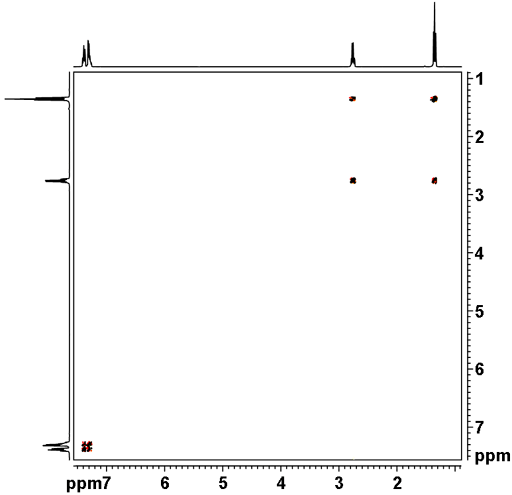
The amorphous di-HCl salt of 2-aniino-3-methyl-N-(2-moi holinoethyl)-pentariamide was analyzed via the methods as described above. The XRD analysis indicated it was amorphous/low ordered as shown in Figure 1 , The DSC thermogram exhibited a broad endotherm with onset temperature 37 °C and peak temperature 74 °C and an enthalpy value of ΔΗ = 80 J/g. The TGA thermogram indicated the di-HCl salt is anhydrous and starts to decompose after about 200°C. An overlay of DSC and TGA thermograms are shown in Figure 2. The moisture sorption-desorpiion isotherm of the di-HC! salt (Figures 3 A and 3 B ) was collected using dynamic vapor sorption (DVS) analysis. The material did not adsorb much moisture from 0% to 20% RH, then it showed steady sorption up to 140 wt% moisture at 95% RH (likely deliquescence). This sample showed rapid desorption from 95% to 70% RH and then continues desorbing at a relatively slower pace to a mass about 5 wt% greater than the original value at 0% RH. This sample shows a small hysteresis between the sorption and desorption phase. O verall this material is quite hygroscopic. The crude solubility of the di-HCl salt in water was >30 mg/niL. The proton N MR spectrum of the amorphous di-HCl salt is shown in Figure 4. Example 2. Preparation of 2-amino-3-methyl- -(2-morpholinoethy[)-pentanamide (free base):
Five grams of 2-amino-3-methyl-N-(2-morpholinoethyl)-pentanamide di-HCl salt was dissolved in 150 mL of ethanol. Sodium bicarbonate (5.3 g), dissolved in 100 mL of HPLC water, was added to this solution. The mixed solution was sonicated for ~10 minutes. This solution was concentrated using a rotovap, and the residue was dissolved in 300 mL of methylene chloride. This solution was passed through a short plug of carbonate bonded silica gel. This solution was concentrated using rotovap and the residue was lyophilized to dry, resulting in 3.6 g of the free base as a white solid. Proton NMR, C-13 NMR and LC/MS confirmed the structure of this material as the free base of 2-amino-3-methyl-N-(2- morpholmoethyl)-pentanamide.
In the process of converting the di-HCl salt to free base, the sample was lyophilized to avoid formation of oil. XRD analysis of the lyophilized free base surprisingly re vealed it was crystalline, as shown in Figure 5. The DSC thermogram exhibited an endotherm with extrapolated onset temperature 51 °C and peak temperature 53 °C and an enthalpy value of Δ¾= 104 J/g. The TGA thermogram shows less than 0.6 wt% loss at 105 °C, suggesting it was solvent free. An overlay of the DSC and TGA thermograms can be seen in Figure 6. The crude solubility of free base in water was >30 mg/mL. The proton NMR was consistent with the free base. The NMR and Raman spectra are shown in Figures 7 and 8A and 8B, respectively. The moisture sorption-desorption isotherm (Figures 9 A and 9B) was collected using dynamic vapor sorption (DVS) analysis. The sample did not adsorb much moisture content from 0% to 45% RH under the experimental conditions. Above 45 %RH the sample appears to adsorb moisture of – 10 wt% from 45% to 50% RH followed by rapid sorption up to 96 wt% moisture at 95% RH. In the desorption phase, the free base shows a rapid desorption from 95% to 80°/» RH, then the sample desorbs at a relatively slow pace to the original weight at 0% RH. The sample may form a hydrate near 45 %>RH, The putative hydrate appears to deliquesce resulting in an amorphous glass by the end of the scan.
……………
new patent
Crystalline forms of neurotrophin mimetic compounds and their salts
Type II TNF receptor agonist; NGF receptor modulator
Crystalline forms of (2S,3S)-2-amino-3-methyl-N-(2-morpholinoethyl)-pentanamide (LM11A-31-BHS), useful for the treatment of neurodegenerative disorders such as Alzheimer’s disease (AD), Parkinson’s disease and multiple sclerosis. See WO2011066544 claiming deuterated compounds of LM11A-31-BHS, useful for treating neurodegenerative diseases. PharmatrophiX is investigating the p75 neutrophin receptor ligand, LM11A-31-BHS, for the oral treatment of AD. By March 2013, a phase I trial was planned. The drug was formerly being investigated in collaboration with Elan Corp and the deal was terminated by the fourth quarter of 2010.








![12,14-ditbutylbenzo[g]chrysene](http://chem.ch.huji.ac.il/nmr/techniques/2d/cosy/cosy_files/dtbbgc.gif)
![COSY of 12,14-ditbutylbenzo[g]chrysene](http://chem.ch.huji.ac.il/nmr/techniques/2d/cosy/cosy_files/cosydtbbgc.gif)
![Aromatic region of COSY of 12,14-ditbutylbenzo[g]chrysene](http://chem.ch.huji.ac.il/nmr/techniques/2d/cosy/cosy_files/aromaticdtbbgc.gif)
![Aromatic region of COSY of 12,14-ditbutylbenzo[g]chrysene](http://chem.ch.huji.ac.il/nmr/techniques/2d/cosy/cosy_files/aromaticdtbbgc2.gif)
![Aromatic region of COSY of 12,14-ditbutylbenzo[g]chrysene in color](http://chem.ch.huji.ac.il/nmr/techniques/2d/cosy/cosy_files/coloraromatic.gif)
























































































 1,5-Cyclooctadiene-iridium(I) chloride dimer, Chloro(1,5-cyclooctadiene)iridium(I) dimer, Di-μ-chlorobis[(1,2,5,6-η)-1,5-cyclooctadiene]diiridium, Iridium(I) chloride 1,5-cyclooctadiene complex dimer, [Ir(1,5-cod)Cl]2, [Ir(1,5-cod)Cl]2, [Ir(cod)Cl]2
1,5-Cyclooctadiene-iridium(I) chloride dimer, Chloro(1,5-cyclooctadiene)iridium(I) dimer, Di-μ-chlorobis[(1,2,5,6-η)-1,5-cyclooctadiene]diiridium, Iridium(I) chloride 1,5-cyclooctadiene complex dimer, [Ir(1,5-cod)Cl]2, [Ir(1,5-cod)Cl]2, [Ir(cod)Cl]2
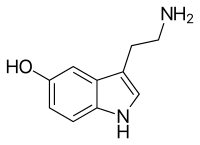




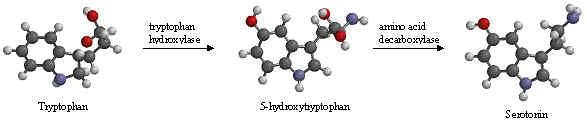
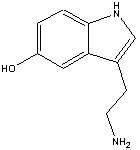 The chemical name for serotonin is 5-hydoxytryptamine which is often abbreviated to 5-HT.
The chemical name for serotonin is 5-hydoxytryptamine which is often abbreviated to 5-HT.

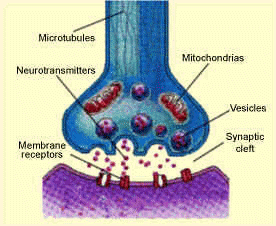 into the synaptic cleft. The serotonin molecules can then bind to receptor proteins within the postsynaptic cell, which causes a change in the electrical state of the cell. This change in electrical state can either excite the cell, passing along the chemical message, or inhibit it. Excess serotonin molecules are taken back up by the presynaptic cell and reprocessed.
into the synaptic cleft. The serotonin molecules can then bind to receptor proteins within the postsynaptic cell, which causes a change in the electrical state of the cell. This change in electrical state can either excite the cell, passing along the chemical message, or inhibit it. Excess serotonin molecules are taken back up by the presynaptic cell and reprocessed. worthlessness, insomnia and fatigue. The most concrete evidence for the connection between serotonin and depression is the decreased concentrations of serotonin metabolites in the cerebrospinal fluid and brain tissues of depressed people.
worthlessness, insomnia and fatigue. The most concrete evidence for the connection between serotonin and depression is the decreased concentrations of serotonin metabolites in the cerebrospinal fluid and brain tissues of depressed people. serotonin from sending neural messages in the brain. Once the LSD molecule is bound to the receptor proteins the message is not carried any further. Instead the impulse is redirected to the older parts of the brain, where the bloodstream then takes it to the sense interpretive centres and the motor areas.
serotonin from sending neural messages in the brain. Once the LSD molecule is bound to the receptor proteins the message is not carried any further. Instead the impulse is redirected to the older parts of the brain, where the bloodstream then takes it to the sense interpretive centres and the motor areas.