Tout sur les médicaments הכל על תרופות كل شيئ عن الأدوية Все о наркотиках 关于药品的一切 డ్రగ్స్ గురించి అన్ని 마약에 관한 모든 것 Όλα για τα Ναρκωτικά Complete Tracking of Drugs Across the World by Dr Anthony Melvin Crasto, Worldpeacepeaker, worlddrugtracker, PH.D (ICT), MUMBAI, INDIA, Worlddrugtracker, Helping millions, 9 million hits on google on all websites, 2.5 lakh connections on all networks, “ALL FOR DRUGS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This is a compilation for educational purposes only. P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
Celltrion announced that the company, on August 8, 2014, completed the filing procedure to obtain US FDA approval for its infliximab biosimilar. This marks the first 351(k) biosimilar mAb application to be filed in the U.S.A. and the second application for a biosimilar to be filed through the US BPCIA.
2-(8-oxo-7-((5-trifluromethyl)-1H-benzo[d]imidazol-2-yl)methyl)7,8-dihydropyrazin[2,3-d]pyridazin-5-yl)acetic acid and [4-oxo-(5-trifluoromethyl-benzothaiazol-2-ylmethyl)-3,4-dihydro-phthalazin-1-yl]-acetic acid (also known as zopolrestat), pharmaceutical compositions thereof and methods of treating diabetic complications in mammals comprising administering to mammals these salt and compositions. 2-(8-oxo-7-((5-trifluromethyl)-1H-benzo[d]imidazol-2-yl)methyl)8-dihydropyrazin[2,3-d]pyridazin-5-yl) acetic acid (formula II), is disclosed in WO 2012/009553 A1. Zopolrestat (formula III) is disclosed in U.S. Pat. No. 4,939,140.
Each of the patents, applications, and other references referred to herein are incorporated by reference. The diabetic complications include neuropathy, nephropathy, retinopathy, cataracts and cardiovascular complications, including myocardial infarction and cardiomyopathy. This invention is also directed to combinations of these salts and antihypertensive agents. These combinations are also useful in treating diabetic complications in mammals.
2-(8-oxo-7-((5-trifluoromethyl)-1H-benzo[d]imidazol-2-yl)methyl)8-dihydropyrazin[2,3-d]pyridazin-5-yl)acetic acid is prepared as disclosed in WO 2012/009553 A1, which is incorporated herein by reference. Zopolrestat is prepared as disclosed in U.S. Pat. No. 4,939,140.
…………………………
Zopolrestat can be obtained by several different ways: 1) The reaction of 2- (4-oxo-3,4-dihydrophthalazin-1-yl) acetic acid ethyl ester (I) with 2-chloroacetonitrile by means of potassium tert-butoxide in DMF gives 2- [3- (cyanomethyl) -4-oxo-3,4-dihydrophthalazin-1-yl] acetic acid ethyl ester (II), which is cyclized with 2-amino-4- (trifluoromethyl) thiophenol (III) in refluxing ethanol yielding zopolrestat ethyl ester (IV). Finally, this compound is hydrolyzed with KOH in methanol / water / THF. 2) Compound (IV) can also be obtained by cyclization of (II) with 4-chloro-3-nitrobenzotrifluoride . (V) in hot DMF saturated with H2S 3) Compound (II) can also be obtained as follows: The reaction of phthalazine (I) with aqueous formaldehyde gives 2- [3- (hydroxymethyl) -4-oxo-3,4 -dihydrophthalazin-1-yl] acetic acid ethyl ester (VI), which is treated with PBr3 in ethyl ether yielding the bromomethyl derivative (VII). Finally, this compound is treated with potassium cyanide and KI in acetone / water.
EXAMPLE 18 Sodium 3-(5-trifluoromethylbenzothiazol-2-ylmethyl)-4-oxo-3H-phthalazin-1-ylacetateSodium methoxide (54 mg) was added to 3-(5-trifluoromethylbenzothiazol-2-ylmethyl)-4-oxo-phthalazin-1-ylacetic acid (0.4 g) in methanol 10 ml) at room temperature. After the addition was complete, a clear solution was obtained which was stirred for 15 minutes at room temperature. The excess methanol was evaporated. The residue was triturated with ether (20 ml) and filtered to obtain the product (0.43 g; m.p. 300° C.).EXAMPLE 19 3-(5-Trifluoromethylbenzothiazol-2-ylmethyl)-4-oxo-3H-phthalazin-1-ylacetate, dicyclohexylamine saltTo a mixture of 3-(5-trifluromethylbenzothiazol-2ylmethyl)-4-oxo-phthalazin-1-ylacetic acid (0.42 g) in methanol (10 ml) was added dicyclohexylamine (0.2 g) in methanol (5 ml). The resulting clear solution was stirred at room temperature for 15 minutes and then evaporated to dryness. Trituration of the residue with ether (30 ml) gave a white solid (0.38 g; m.p. 207° C.).EXAMPLE 20 3-(5-Trifluoromethylbenzothiazol-2ylmethyl)-4-oxo-3H-phthalazin-1-ylacetic acid, meglumine saltA solution of 3-(5-trifluoromethylbenzothiazol-2-ylmethyl)-4-oxo-phthalazin-1-ylacetic acid (419 mg) and meglumine (196 mg) in methanol (50 ml) was stirred at room temperature for an hour and then evaporated to dryness. The residue was triturated with ether (25 ml), filtered and the collected solid was air dried (610 mg; m.p. 157° C.)……………………………
Mylari, Banavara L.; Zembrowski, William J.; Beyer, Thomas A.; Aldinger, Charles E.; Siegel, Todd W.
Journal of Medicinal Chemistry, 1992 , vol. 35, 12 p. 2155 – 2162
………………………………..
Mylari; Beyer; Scott; Aldinger; Dee; Siegel; Zembrowski
Journal of Medicinal Chemistry, 1992 , vol. 35, 3 p. 457 – 465
…………………………….
Literature References:
Aldose reductase inhibitor. Prepn: B. L. Mylari et al.,EP222576; E. R. Larson, B. L. Mylari, US4939140(1987, 1990 both to Pfizer);
B. L. Mylari et al.J. Med. Chem.34, 108 (1991).
Pharmacology: B. Tesfamariam et al.,J. Cardiovasc.Pharmacol.21, 205 (1993); B. Tesfamariam et al.,Am. J. Physiol.265, H1189 (1993).
Clinical pharmacokinetics: P. B. Inskeep et al.,J. Clin. Pharmacol.34, 760 (1994).
COMBINATION OF AN ALDOSE REDUCTASE INHIBITOR AND A GLYCOGEN PHOSPHORYLASE INHIBITOR COMBINATION OF AN ALDOSE REDUCTASE INHIBITOR AND A GLYCOGEN PHOSPHORYLASE INHIBITOR
Nanotechnology is the use of tiny structures – less than 1,000 nanometres across – that are designed to have specific properties. Nanotechnology is an emerging field in science that is used in a wide range of applications, from consumer goods to health products.
In medicine, nanotechnology has only partially been exploited. It is being investigated as a way to improve the properties of medicines, such as their solubility or stability, and to develop medicines that may provide new ways to:
deliver medicines to the body;
target medicines in the body more accurately;
diagnose and treat diseases;
support the regeneration of cells and tissues.
Activities at the European Medicines Agency
The European Medicines Agency follows the latest developments in nanotechnology that are relevant to the development of medicines. Recommendations from the Agency’sCommittee for Medicinal Products for Human Use (CHMP) have already led to the approval of a number of medicines based on nanotechnology. These include medicines containing:
liposomes (microscopic fatty structures containing the active substance), such asCaelyx (doxorubicin), Mepact (mifamurtide) and Myocet (doxorubicin);
nano-scale particles of the active substance, such as Abraxane (paclitaxel), Emend(aprepitant) and Rapamune (sirolimus).
The development of medicines using newer, innovative nanotechnology techniques may raise new challenges for the Agency in the future. These include discussions on whether the current regulatory framework is appropriate for these medicines and whether existing guidelines and requirements on the way the medicines are assessed and monitored are adequate.
The Agency also needs to consider the acceptability of new testing methods and the availability of experts to guide the Agency’s opinion-making.
An overview of the initiatives taken by European Union (EU) regulators in relation to the development and evaluation of nanomedicines and nanosimilars was published in the scientific journal Nanomedicines. The article describes the regulatory challenges and perspectives in this field:
In 2009, the CHMP established an ad hoc expert group on nanomedicines.
This group includes selected experts from academia and the European regulatory network, who support the Agency’s activities by providing specialist input on new scientific knowledge and who help with the review of guidelines on nanomedicines. The group also helps the Agency’s discussions with international partners on issues concerning nanomedicines.
In 2011, the CHMP began to develop in 2011 a series of four reflection papers on nanomedicines to provide guidance to sponsors developing nanomedicines.
These documents cover the development both of new nanomedicines and of nanosimilars (nanomedicines that are claimed to be similar to a reference nanomedicine), since the first generation of nanomedicines, including liposomal formulations, iron-based preparations and nanocrystal-based medicines, have started to come off patent:
The fourth document, a draft reflection paper on the data requirements for intravenous iron-based nanocolloidal products developed with reference to an innovator medicine, will be released for a six-month public consultation in 2013.
International workshops on nanomedicines
The Agency organises workshops on nanomedicines to explore the scientific aspects of nanomedicines and enable the sharing of experience at an international level, in order to assist future developments in the field:
Molecular formula of calcitonin is C145H241N43O49S2
• Molecular weight is 3434.8 g/mol
Calcitonin-related polypeptide alpha
NMR solution structure of salmon calcitonin in SDS micelles.[1]
Calcitonin
CAS Registry Number: 9007-12-9
Additional Names: Thyrocalcitonin; TCA; TCT
Therap-Cat: Calcium regulator.
The structural formula
Calcitonin (also known as thyrocalcitonin) is a 32-amino acid linear polypeptide hormone that is produced in humansprimarily by the parafollicular cells (also known as C-cells) of the thyroid, and in many other animals in the ultimobranchial body.[2] It acts to reduce blood calcium (Ca2+), opposing the effects of parathyroid hormone (PTH).[3]
Calcitonin has been found in fish, reptiles, birds, and mammals. Its importance in humans has not been as well established as its importance in other animals, as its function is usually not significant in the regulation of normal calcium homeostasis.[4] It belongs to the calcitonin-like protein family.
UV – range
Conditions : Concentration – 53 mg / 100 ml
Solvent designation schedule
Methanol
Water
0.1М HCl
0.1M NaOH
The absorption maximum
278 nm
–
275 nm
–
4.9
–
4.4
–
with
1670
–
1500
–
IR – spectrum
Wavelength (μm)
Wavenumber (cm -1 )
Links
UV and IR Spectra. H.-W. Dibbern, R.M. Muller, E. Wirbitzki, 2002 ECV
NIST/EPA/NIH Mass Spectral Library 2008
Handbook of Organic Compounds. NIR, IR, Raman, and UV-Vis Spectra Featuring Polymers and Surfactants, Jr., Jerry Workman. Academic Press, 2000.
Handbook of ultraviolet and visible absorption spectra of organic compounds, K. Hirayama. Plenum Press Data Division, 1967.
Calcitonin-related polypeptide alpha
NMR solution structure of salmon calcitonin in SDS micelles.[1]
However, effects of calcitonin that mirror those of PTH include the following:
Inhibits phosphate reabsorption by the kidney tubules[11]
In its skeleton-preserving actions, calcitonin protects against calcium loss from skeleton during periods of calcium mobilization, such as pregnancy and, especially, lactation.
Other effects are in preventing postprandial hypercalcemia resulting from absorption of Ca2+. Also, calcitonin inhibits food intake in rats and monkeys, and may have CNS action involving the regulation of feeding and appetite.
Calcitonin was purified in 1962 by Copp and Cheney.[13] While it was initially considered a secretion of the parathyroid glands, it was later identified as the secretion of the C-cellsof the thyroid gland.[14]
It has been investigated as a possible non-operative treatment for spinal stenosis.[16]
The following information is from the UK Electronic Medicines Compendium[17]
General characteristics of the active substance
Salmon calcitonin is rapidly absorbed and eliminated. Peak plasma concentrations are attained within the first hour of administration.
Animal studies have shown that calcitonin is primarily metabolised via proteolysis in the kidney following parenteral administration. The metabolites lack the specific biological activity of calcitonin. Bioavailability following subcutaneous and intramuscular injection in humans is high and similar for the two routes of administration (71% and 66%, respectively).
Calcitonin has short absorption and elimination half-lives of 10–15 minutes and 50–80 minutes, respectively. Salmon calcitonin is primarily and almost exclusively degraded in the kidneys, forming pharmacologically inactive fragments of the molecule. Therefore, the metabolic clearance is much lower in patients with end-stage renal failure than in healthy subjects. However, the clinical relevance of this finding is not known. Plasma protein binding is 30% to 40%.
Characteristics in patients
There is a relationship between the subcutaneous dose of calcitonin and peak plasma concentrations. Following parenteral administration of 100 IU calcitonin, peak plasma concentration lies between about 200 and 400 pg/ml. Higher blood levels may be associated with increased incidence of nausea, vomiting, and secretory diarrhea.
Preclinical safety data
Conventional long-term toxicity, reproduction, mutagenicity, and carcinogenicity studies have been performed in laboratory animals. Salmon calcitonin is devoid of embryotoxic, teratogenic, and mutagenic potential.
An increased incidence of pituitary adenomas has been reported in rats given synthetic salmon calcitonin for 1 year. This is considered a species-specific effect and of no clinical relevance. Salmon calcitonin does not cross the placental barrier.
In lactating animals given calcitonin, suppression of milk production has been observed. Calcitonin is secreted into the milk.
Pharmaceutical manufacture
Calcitonin was extracted from the ultimobranchial glands (thyroid-like glands) of fish, particularly salmon. Salmon calcitonin resembles human calcitonin, but is more active. At present, it is produced either by recombinant DNA technology or by chemical peptide synthesis. The pharmacological properties of the synthetic and recombinant peptides have been demonstrated to be qualitatively and quantitatively equivalent.[17]
Oral calcitonin may have a chondroprotective role in osteoarthritis (OA), according to data in rats presented in December, 2005, at the 10th World Congress of the Osteoarthritis Research Society International (OARSI) in Boston, Massachusetts. Although calcitonin is a known antiresorptive agent, its disease-modifying effects on chondrocytes and cartilage metabolisms have not been well established until now.
This new study, however, may help to explain how calcitonin affects osteoarthritis. “Calcitonin acts both directly on osteoclasts, resulting in inhibition of bone resorption and following attenuation of subchondral bone turnover, and directly on chondrocytes, attenuating cartilage degradation and stimulating cartilage formation,” says researcher Morten Karsdal, MSC, PhD, of the department of pharmacology at Nordic Bioscience in Herlev, Denmark. “Therefore, calcitonin may be a future efficacious drug for OA.”[18]
Subcutaneous injections of calcitonin in patients suffering from mania resulted in significant decreases in irritability, euphoria and hyperactivity and hence calcitonin holds promise for treating bipolar disorder.[19] However no further work on this potential application of calcitonin has been reported.
Diagnostics
It may be used diagnostically as a tumor marker for medullary thyroid cancer, in which high calcitonin levels may be present and elevated levels after surgery may indicate recurrence. It may even be used on biopsy samples from suspicious lesions (e.g., lymph nodes that are swollen) to establish whether they are metastasis of the original cancer.
Cutoffs for calcitonin to distinguish cases with medullary thyroid cancer have been suggested to be as follows, with a higher value increasing the suspicion of medullary thyroid cancer:[20]
females: 5 ng/L or pg/mL
males: 12 ng/L or pg/mL
children under 6 months of age: 40 ng/L or pg/mL
children between 6 months and 3 years of age: 15 ng/L or pg/mL
When over 3 years of age, adult cutoffs may be used
Increased levels of calcitonin have also been reported for various other conditions. They include: C-cell hyperplasia, Nonthyroidal oat cell carcinoma, Nonthyroidal small cell carcinoma and other nonthyroidal malignancies, acute and chronic renal failure, hypercalcemia, hypergastrinemia and other gastrointestinal disorders, and pulmonary disease.[21]
Structure
Calcitonin is a polypeptide hormone of 32 amino acids, with a molecular weight of 3454.93 daltons. Its structure comprises a single alpha helix.[1] Alternative splicing of the gene coding for calcitonin produces a distantly related peptide of 37 amino acids, called calcitonin gene-related peptide (CGRP), beta type.[22]
The following are the amino acid sequences of salmon and human calcitonin:[23]
Compared to salmon calcitonin, human calcitonin differs at 16 residues.
Description: Cellular and molecular coordination of tissues which secrete chemical compounds to regulate growth, reproduction, metabolism, and ion homeostasis.
References
^ Jump up to:abPDB2glhAndreotti G, Méndez BL, Amodeo P, Morelli MA, Nakamuta H, Motta A (August 2006). “Structural determinants of salmon calcitonin bioactivity: the role of the Leu-based amphipathic alpha-helix”. J. Biol. Chem.281 (34): 24193–203.doi:10.1074/jbc.M603528200. PMID16766525.
Jump up^ Potts, John; Jüppner, Harald (2008). “Chapter 353. Disorders of the Parathyroid Gland and Calcium Homeostasis”. In Dan L. Longo, Dennis L. Kasper, J. Larry Jameson, Anthony S. Fauci, Stephen L. Hauser, and Joseph Loscalzo. Harrison’s Principles of Internal Medicine (18 ed.). McGraw-Hill.
Rhoades, Rodney (2009). Medical Physiology: Principles for Clinical Medicine. Philadelphia: Lippincott Williams & Wilkins. ISBN978-0-7817-6852-8.
Jump up^ Carney SL (1997). “Calcitonin and human renal calcium and electrolyte transport”.Miner Electrolyte Metab23 (1): 43–7. PMID9058369.
Jump up^ Copp DH, Cheney B (January 1962). “Calcitonin-a hormone from the parathyroid which lowers the calcium-level of the blood”. Nature193 (4813): 381–2.doi:10.1038/193381a0. PMID13881213.
Jump up^ Hirsch PF, Gauthier GF, Munson PL (August 1963). “Thyroid hypocalcemic principle and recurrent laryngeal nerve injury as factors affecting the response to parathyroidectomy in rats”. Endocrinology73 (2): 244–252. doi:10.1210/endo-73-2-244.PMID14076205.
Jump up^ Basuyau, J. -P.; Mallet, E.; Leroy, M.; Brunelle, P. (2004). “Reference Intervals for Serum Calcitonin in Men, Women, and Children”. Clinical Chemistry50 (10): 1828–1830.doi:10.1373/clinchem.2003.026963. PMID15388660. edit
Jump up^ Burtis CA, Ashwood ER, Bruns DE. Tietz Textbook of Clinical Chemistry and Molecular Diagnostics, 5th edition. Elsevier Saunders. p. 1774. ISBN978-1-4160-6164-9.
MacIntyre I, Alevizaki M, Bevis PJ, Zaidi M (1987). “Calcitonin and the peptides from the calcitonin gene”. Clin. Orthop. Relat. Res.&na; (217): 45–55. doi:10.1097/00003086-198704000-00007. PMID3549095.
Grani, G; Nesca, A; Del Sordo, M; Calvanese, A; Carbotta, G; Bianchini, M; Fumarola, A (Jun 2012). “Interpretation of serum calcitonin in patients with chronic autoimmune thyroiditis.”. Endocrine-related cancer19 (3): 345–9. doi:10.1530/ERC-12-0013.PMID22399011.
Calcium regulating hormone secreted from the mammalian thyroid gland and in non-mammalian species from the ultimobranchial gland. Postulation of a plasma-calcium lowering substance: Copp et al.,Endocrinology70, 638 (1962).
Recognition as a hormone: Hirsch et al.,ibid.73, 244 (1963); of thyroid origin: Foster et al.,Nature202, 1303 (1964).
Over-all action is to oppose the bone and renal effects of parathyroid hormone, q.v.; inhibits bone resorption of Ca2+, with accompanying hypocalcemia and hypophosphatemia and decreased urinary Ca2+ concentrations. Also abolishes the osteolytic effect of toxic doses of vitamins A and D. Calcitonin is highly active biologically, e.g. 50 mg/min infused into a 100 g rat leads to a significant (1 mg/100 ml) decrease in the concn of the plasma calcium within 60 min (together with a corresponding fall in plasma phosphate). Activity is destroyed by trypsin, chymotrypsin, pepsin, polyphenol oxidase; also by hydrogen peroxide oxidation, photooxidation, and treatment with N-bromosuccinimide. Calcitonin structures are single polypeptide chains containing 32 amino acid residues. Structure of porcine: Neher et al.,Helv. Chim. Acta51, 917 (1968); Potts et al.,Proc. Natl. Acad. Sci. USA59, 1321 (1968); Bellet al.,J. Am. Chem. Soc.90, 2704 (1968); eidem,Biochemistry9, 1665 (1970).
Synthesis of porcine: Rittel et al.,Helv. Chim. Acta51, 924 (1968); Guttmann et al.,ibid. 1155.
Isoln of human calcitonin from non-pathological thyroid glands: Haymovits, Rosen, Endocrinology81, 993 (1967); from medullary carcinoma of the thyroid: Neher et al.,Nature220, 984 (1968); Helv. Chim. Acta51, 1738 (1968); Neher, Riniker, DE1929957 (1970 to Ciba), C.A.73, 28902b (1970).
Structure of human: Neher et al.,Helv. Chim. Acta51, 1900 (1968). Synthesis of human: Sieber et al.,ibid. 2057; J. Hirt et al.,Rec. Trav. Chim.98, 143 (1979).
Biosynthetic studies: J. W. Jacobs et al.,J. Biol. Chem.254, 10600 (1979); S. G. Amara et al.,ibid.255, 2645 (1980).
Amino acid sequence differs among mammalian species, salmon calcitonin showing a marked difference from that of the higher vertebrae as well as a more potent biological activity. Mechanism of action: E. M. Brown, G. D. Aurbach, Vitam. Horm.38, 236 (1980). Anorectic activity in rats: W. J. Freed et al.,Science206, 850 (1979).
Growth inhibition of human breast cancer cells in vitro: Y. Iwasaki et al.,Biochem. Biophys. Res. Commun.110, 235 (1983).
Review of early literature: Munson, Hirsch, Clin. Orthop.49, 209 (1966).
Review of isoln, structure, synthesis: Behrens, Grinnan, Annu. Rev. Biochem.38, 83 (1969); Potts et al.,Vitam. Horm.29,41 (1971).
Comprehensive review: Calcitonin, Proc. Symp. on Thyrocalcitonin and the C Cells, S. Taylor, Ed. (Springer-Verlag, New York, 1968); Foster et al., “Calcitonin” in Clinics in Endocrinology and Metabolism, I. MacIntyre, Ed. (W. B. Saunders, Philadelphia, 1972) pp 93-124.
Review of pharmacology and therapeutic use: J. C. Stevenson, I. M. A. Evans, Drugs21, 257-272 (1981).
Literature References: Clinical trial in postmenopausal osteoporosis: C. H. Chesnut et al.,Am. J. Med.109, 267 (2000). LC determn in biological fluids: M. Aguiar et al., J. Chromatogr. B818, 301 (2005).
A long-acting decanoateester of haloperidol is used as an injection given every four weeks to people with schizophrenia or related illnesses who have poor adherence to medication regimens (most commonly due to them forgetting to take their medication, or due to poor insight into their illness) and suffer frequent relapses of illness, or to overcome the drawbacks inherent to its orally administered counterpart.[6] Such long acting injections are controversial because it can be seen as denying people their right to stop taking their medication.
Haloperidol was discovered by Paul Janssen.[70] It was developed in 1958 at the Belgian company Janssen Pharmaceutica and submitted to the first of clinical trials in Belgiumlater that year.[71]
Coincident with civil unrest in the United States in the 1960s and 1970s, schizophrenia was racialized to match the behavior of angry/violent black men. Haldol was promoted as a way to pacify them, and was marketed to appeal to feelings of racial unease. (cf. Metzl 2010. The Protest Psychosis)
Soviet dissidents, including medical staff, have reported several times on the use of haloperidol in the Soviet Union for punitive purposes or simply to break the prisoners’ will.[72][73][74] Notable dissidents who were administered haloperidol as part of their court-ordered treatment were Sergei Kovalev and Leonid Plyushch.[75] The accounts Plyushch gave in the West, after he was allowed to leave the Soviet Union in 1976, were instrumental in triggering Western condemnation of Soviet practices at the World Psychiatric Association‘s 1977 meeting.[76] The use of haloperidol in the Soviet Union’s psychiatric system was prevalent because it was one of the few psychotropic drugs produced in quantity in the USSR.[77]
Haloperidol has been used for its sedating effects during the deportations of immigrants by the United States Immigration and Customs Enforcement (ICE). During 2002-2008, federal immigration personnel used haloperidol to sedate 356 deportees. By 2008, following court challenges over the practice, it was given to only three detainees. Following lawsuits, U.S. officials changed the procedure so the drug is administered only by the recommendation of medical personnel and under court order.[78][79]
Brand names
Haloperidol is sold under the tradenames Aloperidin, Bioperidolo, Brotopon, Dozic, Duraperidol (Germany), Einalon S, Eukystol, Haldol (common tradename in the US and UK), Halosten, Keselan, Linton, Peluces, Serenace and Sigaperidol.
Veterinary use
Haloperidol is also used on many different kinds of animals. It appears to be particularly successful when given to birds, e.g., a parrot that will otherwise continuously pluck its feathers out.[80]
Jump up^ Joint Formulary Committee (2013). British National Formulary (BNF) (65 ed.). London, UK: Pharmaceutical Press. p. 229-230. ISBN978-0-85711-084-8. edit
^ Jump up to:ab Brayfield, A, ed. (13 December 2013). “Haloperidol”. Martindale: The Complete Drug Reference. London, UK: Pharmaceutical Press. Retrieved 29 May 2014.
Jump up^ Rossi, S, ed. (2013). Australian Medicines Handbook (2013 ed.). Adelaide: The Australian Medicines Handbook Unit Trust. ISBN978-0-9805790-9-3. edit
Jump up^ Giannini, A. James; Underwood, Ned A.; Condon, Maggie (2000). “Acute Ketamine Intoxication Treated by Haloperidol”. American Journal of Therapeutics7 (6): 389–91.doi:10.1097/00045391-200007060-00008. PMID11304647.
Jump up^ Giannini, A. James; Eighan, Michael S.; Loiselle, Robert H.; Giannini, Matthew C. (1984). “Comparison of Haloperidol and Chlorpromazine in the Treatment of Phencyclidine Psychosis”. The Journal of Clinical Pharmacology24 (4): 202–4.doi:10.1002/j.1552-4604.1984.tb01831.x. PMID6725621.
Jump up^ Cavanaugh, SV (1986). “Psychiatric emergencies”. The Medical clinics of North America70 (5): 1185–202. PMID3736271.
Jump up^ Irving, Claire B; Adams, Clive E; Lawrie, Stephen (2006). “Haloperidol versus placebo for schizophrenia”. In Irving, Claire B. Cochrane Database of Systematic Reviews (4): CD003082. doi:10.1002/14651858.CD003082.pub2. PMID17054159.
Jump up^ Allen, MH; Currier, GW; Hughes, DH; Reyes-Harde, M; Docherty, JP; Expert Consensus Panel for Behavioral Emergencies (2001). “The Expert Consensus Guideline Series. Treatment of behavioral emergencies”. Postgraduate Medicine (Spec No): 1–88; quiz 89–90. PMID11500996.
Jump up^ Allen, Michael H.; Currier, Glenn W.; Hughes, Douglas H.; Docherty, John P.; Carpenter, Daniel; Ross, Ruth (2003). “Treatment of Behavioral Emergencies: A Summary of the Expert Consensus Guidelines”. Journal of Psychiatric Practice9 (1): 16–38. doi:10.1097/00131746-200301000-00004. PMID15985913.
Jump up^ Allen, Michael H.; Currier, Glenn W.; Carpenter, Daniel; Ross, Ruth W.; Docherty, John P. (2005). “Introduction: Methods, Commentary, and Summary”. Journal of Psychiatric Practice11: 5. doi:10.1097/00131746-200511001-00002.
Jump up^ Oosthuizen, P.; Emsley, R. A.; Turner, J.; Keyter, N. (2001). “Determining the optimal dose of haloperidol in first-episode psychosis”. Journal of Psychopharmacology15 (4): 251–5. doi:10.1177/026988110101500403. PMID11769818.
Smith, Thomas J.; Ritter, Joseph K.; Poklis, Justin L.; Fletcher, Devon; Coyne, Patrick J.; Dodson, Patricia; Parker, Gwendolyn (2012). “ABH Gel is Not Absorbed from the Skin of Normal Volunteers”. Journal of Pain and Symptom Management43(5): 961–6. doi:10.1016/j.jpainsymman.2011.05.017. PMID22560361.
Weschules, Douglas J. (2005). “Tolerability of the Compound ABHR in Hospice Patients”. Journal of Palliative Medicine8 (6): 1135–43.doi:10.1089/jpm.2005.8.1135. PMID16351526.
Jump up^ Truven Health Analytics, Inc. DrugPoint® System (Internet) [cited 2013 Sep 29]. Greenwood Village, CO: Thomsen Healthcare; 2013.
Jump up^ Joint Formulary Committee. British National Formulary (BNF) 65. Pharmaceutical Pr; 2013.
^ Jump up to:ab Leucht, Stefan; Cipriani, Andrea; Spineli, Loukia; Mavridis, Dimitris; Örey, Deniz; Richter, Franziska; Samara, Myrto; Barbui, Corrado; Engel, Rolf R; Geddes, John R; Kissling, Werner; Stapf, Marko Paul; Lässig, Bettina; Salanti, Georgia; Davis, John M (2013). “Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: A multiple-treatments meta-analysis”. The Lancet382 (9896): 951–62.doi:10.1016/S0140-6736(13)60733-3. PMID23810019.
^ Jump up to:ab Silvestri, Simone; Seeman, Mary V.; Negrete, Juan-Carlos; Houle, Sylvain; Shammi, C.M.; Remington, Garry J.; Kapur, Shitij; Zipursky, Robert B.; Wilson, Alan A.; Christensen, Bruce K.; Seeman, Philip (2000). “Increased dopamine D 2 receptor binding after long-term treatment with antipsychotics in humans: A clinical PET study”.Psychopharmacology152 (2): 174–80. doi:10.1007/s002130000532.PMID11057521.
Jump up^ Dorph-Petersen, Karl-Anton; Pierri, Joseph N; Perel, James M; Sun, Zhuoxin; Sampson, Allan R; Lewis, David A (2005). “The Influence of Chronic Exposure to Antipsychotic Medications on Brain Size before and after Tissue Fixation: A Comparison of Haloperidol and Olanzapine in Macaque Monkeys”. Neuropsychopharmacology30 (9): 1649–61. doi:10.1038/sj.npp.1300710. PMID15756305.
Jump up^ Vernon, Anthony C.; Natesan, Sridhar; Modo, Mike; Kapur, Shitij (2011). “Effect of Chronic Antipsychotic Treatment on Brain Structure: A Serial Magnetic Resonance Imaging Study with Ex Vivo and Postmortem Confirmation”. Biological Psychiatry69 (10): 936–44. doi:10.1016/j.biopsych.2010.11.010. PMID21195390.
Jump up^ Dalton, Susanne Oksbjerg; Mellemkjaer, Lene; Thomassen, Lars; Mortensen, Preben B.; Johansen, Christoffer (2005). “Risk for cancer in a cohort of patients hospitalized for schizophrenia in Denmark, 1969–1993”. Schizophrenia Research75 (2–3): 315–24.doi:10.1016/j.schres.2004.11.009. PMID15885523.
Jump up^ Grinshpoon, Alexander; Barchana, Micha; Ponizovsky, Alexander; Lipshitz, Irena; Nahon, Daniella; Tal, Orna; Weizman, Abraham; Levav, Itzhak (2005). “Cancer in schizophrenia: Is the risk higher or lower?”. Schizophrenia Research73 (2–3): 333–41.doi:10.1016/j.schres.2004.06.016. PMID15653279.
Jump up^ Szarfman, Ana; Tonning, Joseph M; Levine, Jonathan G; Doraiswamy, P. Murali (2006). “Atypical Antipsychotics and Pituitary Tumors: A Pharmacovigilance Study”.Pharmacotherapy26 (6): 748–58. doi:10.1592/phco.26.6.748. PMID16716128.
Jump up^ Hippisley-Cox, Julia; Vinogradova, Y; Coupland, C; Parker, C (2007). “Risk of Malignancy in Patients with Schizophrenia or Bipolar Disorder”. Archives of General Psychiatry64 (12): 1368–76. doi:10.1001/archpsyc.64.12.1368. PMID18056544.
Jump up^ Levav, Itzhak; Kohn, Robert; Barchana, Micha; Lipshitz, Irena; Pugachova, Inna; Weizman, Abraham; Grinshpoon, Alexander (2009). “The risk for cancer among patients with schizoaffective disorders”. Journal of Affective Disorders114 (1–3): 316–20.doi:10.1016/j.jad.2008.06.010. PMID18675461.
Jump up^ Sandyk, R; Hurwitz, MD (1983). “Toxic irreversible encephalopathy induced by lithium carbonate and haloperidol. A report of 2 cases”. South African medical journal64 (22): 875–6. PMID6415823.
Jump up^ Bush, S. E.; Hatton, R. C.; Winterstein, A. G.; Thomson, M. R.; Woo, G. W. (2008). “Effects of concomitant amiodarone and haloperidol on Q-Tc interval prolongation”.American Journal of Health-System Pharmacy65 (23): 2232–6.doi:10.2146/ajhp080039. PMID19020191.
Jump up^ Igarashi, K.; Kasuya, F.; Fukui, M.; Usuki, E.; Castagnoli Jr, N. (1995). “Studies on the metabolism of haloperidol (HP): The role of CYP3A in the production of the neurotoxic pyridinium metabolite HPP+ found in rat brain following ip administration of HP”. Life Sciences57 (26): 2439–46. doi:10.1016/0024-3205(95)02240-5. PMID8847965.
Jump up^ Usuki, Etsuko; Pearce, Robin; Parkinson, Andrew; Castagnoli, Neal (1996). “Studies on the Conversion of Haloperidol and Its Tetrahydropyridine Dehydration Product to Potentially Neurotoxic Pyridinium Metabolites by Human Liver Microsomes”. Chemical Research in Toxicology9 (4): 800–6. doi:10.1021/tx960001y. PMID8831826.
Jump up^ Avent, Kathryn M.; Devoss, J. J.; Gillam, Elizabeth M. J. (2006). “Cytochrome P450-Mediated Metabolism of Haloperidol and Reduced Haloperidol to Pyridinium Metabolites”.Chemical Research in Toxicology19 (7): 914–20. doi:10.1021/tx0600090.PMID16841959.
Jump up^ Avent, Kathryn M.; Riker, Richard R.; Fraser, Gilles L.; Van Der Schyf, Cornelis J.; Usuki, Etsuko; Pond, Susan M. (1997). “Metabolism of haloperidol to pyridinium species in patients receiving high doses intravenously: Is HPTP an intermediate?”. Life Sciences61 (24): 2383–90. doi:10.1016/S0024-3205(97)00955-7. PMID9399630.
Jump up^ Kawashima, Hidekazu; Iida, Yasuhiko; Kitamura, Youji; Saji, Hideo (2004). “Binding of 4-(4-chlorophenyl)-1-[4-(4-fluorophenyl)-4-oxobutyl]pyridinium ion (HPP+), a metabolite of haloperidol, to synthetic melanin: Implications for the dopaminergic neurotoxicity of HPP+“. Neurotoxicity Research6 (7–8): 535–42. doi:10.1007/BF03033449.PMID15639785.
Jump up^ Bishnoi, Mahendra; Chopra, Kanwaljit; Kulkarni, Shrinivas K. (2008). “Activation of striatal inflammatory mediators and caspase-3 is central to haloperidol-induced orofacial dyskinesia”. European Journal of Pharmacology590 (1–3): 241–5.doi:10.1016/j.ejphar.2008.06.033. PMID18590723.
Jump up^ Eyles, Darryl W.; Avent, Kathryn M.; Stedman, Terry J.; Pond, Susan M. (1997). “Two pyridinium metabolites of haloperidol are present in the brain of patients at post-mortem”.Life Sciences60 (8): 529–34. doi:10.1016/S0024-3205(96)00656-X. PMID9042387.
Jump up^ Ulrich, Sven; Neuhof, Sabine; Braun, Verena; Danos, Peter; Pester, Uwe; Hoy, Ludwig (2000). “Disposition of Haloperidol Pyridinium and Reduced Haloperidol Pyridinium in Schizophrenic Patients: No Relationship with Clinical Variables During Short-Term Treatment”. Journal of Clinical Psychopharmacology20 (2): 210–9.doi:10.1097/00004714-200004000-00014. PMID10770460.
Jump up^ Ulrich, S.; Sandmann, U.; Genz, A. (2005). “Serum Concentrations of Haloperidol Pyridinium Metabolites and the Relationship with Tardive Dyskinesia and Parkinsonism: A Cross-Section Study in Psychiatric Patients”. Pharmacopsychiatry38 (4): 171–7.doi:10.1055/s-2005-871240. PMID16025420.
Jump up^ Seeman, P; Tallerico, T (1998). “Antipsychotic drugs which elicit little or no Parkinsonism bind more loosely than dopamine to brain D2 receptors, yet occupy high levels of these receptors”. Molecular Psychiatry3 (2): 123–34.doi:10.1038/sj.mp.4000336. PMID9577836.
Jump up^ Leysen, JE; Janssen, PM; Megens, AA; Schotte, A (1994). “Risperidone: A novel antipsychotic with balanced serotonin-dopamine antagonism, receptor occupancy profile, and pharmacologic activity”. The Journal of Clinical Psychiatry55 (Suppl): 5–12.PMID7520908.
Jump up^ Cobos, Enrique J.; Del Pozo, Esperanza; Baeyens, José M. (2007). “Irreversible blockade of sigma-1 receptors by haloperidol and its metabolites in guinea pig brain and SH-SY5Y human neuroblastoma cells”. Journal of Neurochemistry102 (3): 812–25.doi:10.1111/j.1471-4159.2007.04533.x. PMID17419803.
Jump up^ Colabufo, Nicolaantonio; Berardi, Francesco; Contino, Marialessandra; Niso, Mauro; Abate, Carmen; Perrone, Roberto; Tortorella, Vincenzo (2004). “Antiproliferative and cytotoxic effects of some σ2 agonists and σ1 antagonists in tumour cell lines”. Naunyn-Schmiedeberg’s Archives of Pharmacology370 (2): 106–13. doi:10.1007/s00210-004-0961-2. PMID15322732.
^ Jump up to:abcdefghijk Kroeze, Wesley K; Hufeisen, Sandra J; Popadak, Beth A; Renock, Sean M; Steinberg, Seanna; Ernsberger, Paul; Jayathilake, Karu; Meltzer, Herbert Y; Roth, Bryan L (2003). “H1-Histamine Receptor Affinity Predicts Short-Term Weight Gain for Typical and Atypical Antipsychotic Drugs”. Neuropsychopharmacology28 (3): 519–26.doi:10.1038/sj.npp.1300027. PMID12629531.
^ Jump up to:ab Kornhuber, Johannes; Schultz, Andreas; Wiltfang, Jens; Meineke, Ingolf; Gleiter, Christoph H.; Zöchling, Robert; Boissl, Karl-Werner; Leblhuber, Friedrich; Riederer, Peter (1999). “Persistence of Haloperidol in Human Brain Tissue”. The American Journal of Psychiatry156 (6): 885–90. PMID10360127.
Jump up^ Kornhuber, Johannes; Wiltfang, Jens; Riederer, Peter; Bleich, Stefan (2006). “Neuroleptic drugs in the human brain: Clinical impact of persistence and region-specific distribution”. European Archives of Psychiatry and Clinical Neuroscience256 (5): 274–80. doi:10.1007/s00406-006-0661-7. PMID16788768.
Jump up^ de Boer, S. P.; E. J. Driessen; H. L. Verhaar (1982). Biographical Dictionary of Dissidents in the Soviet Union, 1956-1975. The Hague: Martinus Nijhoff Publishers.ISBN90-247-2538-0.[page needed]
Pantoprazole is a proton pump inhibitor drug used for short-term treatment of erosion and ulceration of the esophagus caused by gastroesophageal reflux disease.
Use
Pantoprazole is used for short-term treatment of erosion and ulceration of the oesophagus caused by gastroesophageal reflux disease. Initial treatment is generally of eight weeks’ duration, after which another eight week course of treatment may be considered if necessary. It can be used as a maintenance therapy for long term use after initial response is obtained.
Adverse effects
Antacid preparations such as pantoprazole work by suppressing the acid-mediated breakdown of proteins. This leads to an elevated risk of developing food and drug allergies due to undigested proteins passing into the gastrointestinal tract where sensitisation occurs. It is unclear whether this risk occurs with short-term or only long-term use.[1]
Nutrition: May reduce the absorption of important nutrients, vitamins and minerals, as well as medications, leaving users at increased risk for pneumonia.[2]
Cardiovascular: Increase in a chemical that suppresses the production of nitric oxide by 25% in humans, which have proven to relax and protect arteries and veins. Causes blood vessels to constrict, a development that could lead to a number of cardiovascular problems if continued for a prolonged period of time.[2]
Pharmacology
Wyeth pantoprazole 20mg.
Pantoprazole is metabolized in the liver by the cytochrome P450 system.[3] Metabolism mainly consists of demethylation by CYP2C19followed by sulfation. Another metabolic pathway is oxidation by CYP3A4. Pantoprazole metabolites are not thought to have any pharmacological significance. Pantoprazole is relatively free of drug interactions;[4] however, it may alter the absorption of other medications that depend on the amount of acid in the stomach, such as ketoconazole or digoxin. Generally inactive at acidic pH of stomach, thus it is usually given with a pro kinetic drug. Pantoprazole binds irreversibly to H+K+ATPase (proton pumps) and suppresses the secretion of acid. As it binds irreversibly to the pumps, new pumps have to be made before acid production can be resumed. The drug’s plasma half-life is about 2 hours.[5]
Pharmacokinetics
Absorption
Bioavailability: (oral, delayed release tablets), approximately 77%
Effect of food: (oral, delayed-release tablets), AUC and Cmax no effect, Tmax variable, absorption delayed, no net effect
Effect of food: (oral, for-delayed-release suspension), administer 30 minutes before a meal
Tmax, Oral, delayed-release suspension: 2 to 2.5 h
Tmax, Oral, delayed-release tablets: 2.5 h
Tmax, Oral, delayed-release tablets: 1.5 to 2 hours (pediatrics)
Distribution
Protein binding: about 98% to primarily albumin
Vd, extensive metabolizers (IV): approximately 11 L to 23.6 L
Vd, pediatrics (oral): 0.21 to 0.43 L/kg.
Metabolism
Hepatic; cytochrome P450 CYP2C19; minor metabolism from CYP3A4, 2D6, and 2C9
Excretion
Fecal: (oral or IV, normal metabolizers), 18%
Renal: (oral or IV, normal metabolizers), approximately 71%, none as unchanged
Dialyzable: no (hemodialysis)
Total body clearance: (IV) 7.6 to 14 L/hour.
Total body clearance: (oral, pediatrics) 0.18 to 2.08 L/h/kg
Elimination Half Life
Oral or IV, 1 hour
Oral or IV, slow metabolizers, 3.5 to 10 hours
Pediatrics, 0.7 to 5.34 hours
Availability
Pantoprazole was developed by Altana (owned by Nycomed) and was licensed in the USA to Wyeth (which was taken over by Pfizer). It was initially marketed under the brand name Protonix by Wyeth-Ayerst Laboratories and now is available as a generic. It is available by prescription in delayed-release tablets. It is also available for intravenous use.
On 24 December 2007, Teva Pharmaceutical released an AB-rated generic alternative to Protonix.[6] This was followed by generic equivalents from Sun Pharma and Kudco Pharma. Wyeth sued all three for patent infringement and launched its own generic version of Protonix with Nycomed.[7][8]
Pantoprazole is the international non-proprietary name of the chemical product 5-(difluoromethoxy)-2-[[(3,4-dimethoxy-2- pyridinyl)methyl]sulfmyl]-lH-benzimidazole of formula
Pantoprazole This product is an active ingredient used in the treatment of gastric ulcers, usually in the form of its sodium salt.
The product was described for the first time in European patent application EP-A-0166287 that also describes several processes for the preparation of products assignable to a general formula among which pantoprazole is to be found. The reaction sequences of these processes, applied precisely to the preparation of pantoprazole, are given in Scheme 1.
Scheme 1
In Scheme 1, the variables Y, Z, Z’ and Z” are leaving groups, for example atoms of halogen, and the variables M and M’ are atoms of alkali metals.
Austrian patent AT-B-394368 discloses another process based on a different route of synthetis, the reaction sequence of which is given in Scheme 2.
Pantoprazole Scheme 2
Nevertheless, this process has obvious drawbacks, since the methylation can take place not only in OH in the 4-position of the pyridine ring, but also in the nitrogen linked to a hydrogen of the benzimidazole ring, which can give place to mixtures of the desired product with the two possible methylated isomers of the benzimidazole compounds obtained, 3- methyl or 1 -methyl, which means that additional chromatographic purification steps are needed and the yields obtained are low.
PCT application WO97/29103 discloses another process for the preparation of pantoprazole, the reaction sequence of which is given in Scheme 3.
Scheme 3 As may be seen, different synthesis strategies have been proposed for the preparation of pantoprazole, some of them recently, which is an indication that the preparation of the product is still not considered to be sufficiently well developed, whereby there is still a need for developing alternative processes that allow pantoprazole to be prepared by means of simpler techniques and more accessible intermediate compounds and with good chemical yields.
EXAMPLES
Example 1. – Preparation of compound (IX)
47.5 ml (0.502 mol) of acetic anhydride were mixed with 1.65 g (0.0135 mol) of 4-dimethylaminopyridine, giving a transparent yellow solution which was heated to 65° – 70°C. This temperature was held by cooling since the reaction is exothermic. 25 g (0.1441 mol) of 2-methyl-3- methoxy-4-chloropyridine N-oxide (X) were added over a period of about 70 minutes. Once the addition was completed, the reaction was held at 65° – 70°C for a further 2h 20 minutes and after this time it was allowed to cool down to below 65°C and 90 ml of methanol were added gradually, while holding the temperature below 65°C. The resulting reaction mass was distilled at reduced pressure in a rotavap to remove the volatile components and the residue containing compound (IX) was used as such for the following reaction. Thin layer chromatography on silica gel 60 F254, eluting with CHCl3/MeOH (15: 1), showed a single spot at Rf – 0.82, indicating that the reaction has been completed.
Example 2. – Preparation of compound fVIII
(IX) (VIII)
11.5 ml methanol and 11.5 ml of water were added over the crude residue from Example 1 containing compound (IX), and thereafter, while holding the temperature to between 25° and 30°C with a water bath, the residual acetic acid contained in the crude residue was neutralized by the addition of 33% aqueous NaOH. Once the residual acid had been neutralized, 19 ml (0.2136 mol) of the 33% aqueous NaOH were added over 20 minutes, while holding the temperature to between 25° and 30°C, and, on completion of the addition, the hydrolysis reaction at pH 11.7 – 11.8 was held for 2h 30 minutes, to between 25° and 30°C. On completion of the reaction, the pH was adjusted to 7.0 – 7.5 by the addition of HC1 35%, while holding the temperature to 25°C. Thereafter, 50 ml of methylene chloride were added and, after stirring and allowing to rest, the phases were decanted. A further five extractions were carried out with 30 ml methylene chloride each and the pooled organic phases were dried with anhydrous sodium sulfate, were filtered and washed, and were evaporated at reduced pressure in a rotavap, providing a solid residue having a melting point around 73°C and containing compound (VIII). Thin layer chromatography on silica gel 60 F254, eluting with CHCl3/MeOH (15: 1), gave a main spot at Rf = 0.55, showing that the reaction was complete. The thus obtained crude residue was used as such in the following reaction.
Example 3. – Preparation of compound (VI)
24.5 g of the residue obtained in Example 2, containing approximately 0.142 mol of the compound 2-hydroxymethyl-3-methoxy-4-chloropyridine (VIII), were mixed with 0.5 ml of DMF and 300 ml of anhydrous methylene chloride, to give a brown solution which was cooled to 0° – 5°C in an ice water bath. Thereafter, a solution of 11.5 ml (0.1585 mol) of thionyl chloride in 50 ml of anhydrous methylene chloride was added over 20 minutes, while holding the above-mentioned temperature,. Once the addition was complete, the reaction was held at 0° – 5°C for a further 90 minutes and then 120 ml of water and NaOH 33% were added to pH 5 – 6, requiring approximately 29 ml of NaOH. The phases were then decanted and separated. The organic phase was extracted with a further 120 ml of water and the pooled aqueous phases were extracted with a further 4×25 ml of methylene chloride, in order to recover the greatest possible amount of product. The pooled organic phases were dried over anhydrous sodium sulfate, filtered and washed, and evaporated at reduced pressure in a rotavap, to give a residue containing the compound 2-chloromethyl-3- methoxy-4-chloropyridine (VI). Thin layer chromatography on silica gel 60 F254, eluting with CHCl3/MeOH (15:1), showed a main spot at Rf = 0.83, indicating that the reaction was complete. The thus obtained crude residue was used as such in the following reaction. Example 4. – Preparation of compound (III)
26.11 g of the residue obtained in the Example 3 containing approximately 0.136 mol of the compound 2-chloromethyl-3-methoxy-4- chloropyridine (VI) were mixed with 370 ml of methylene chloride, to give a brown solution over which were added, at 20° – 25°C, 29.3 g (0.136 mol) of 5-difluoromethoxy-2-mercaptobenzimidazole (VII) and 17.10 ml (0.136 mol) of tetramethylguanidine (TMGH). The mixture was stirred at this temperature for 2 hours, after which 450 ml of water were added, with the pH being held to between 9.5 and 10. Thereafter the phases were decanted and the organic phase was washed 5×50 ml of a IN NaOH aqueous solution and, thereafter, with 2×50 ml of water. The organic phase was treated with 50 ml of water and an amount of HC1 30% sufficient to adjust the pH to between 5 and 6. Thereafter, the phases were decanted, and the organic phase was dried over anhydrous sodium sulfate, was filtered and washed, and evaporated at reduced pressure in a rotavap, to give a solid residue of melting point 64° – 73 °C that contains the compound (III). Thin layer chromatography on silica gel 60 F254, eluting with CHCl3/MeOH (15: 1), presented a main spot at Rf = 0.52. Yield 82%. The thus obtained compound 5-(difluoromethoxy)-2-[[(3-methoxy-4-chlorine-2 pyridinyl)methyl]mercapto]- lH-benzimidazole (III) was used as such in the following reaction Example 5. – Preparation of compound (IV)
25.8 g (0.0694 mol) of the compound (III) obtained in the Example 4 were mixed with 88 ml of methanol, to give a brown solution to which 3.7 ml of water, 0.99 g of ammonium molybdate and 0.78 g of sodium carbonate were added. The system was cooled to 0°C – 5°C, 3.4 ml (0.0756 mol) of 60% hydrogen peroxide were added, and the reaction mixture was held at 0°C – 5°C for 1 – 2 days, the end point of the reaction being checked by thin layer chromatography on silica gel 60 F254, eluting with CHCl3/MeOH (15: l).
During the reaction the presence of hydrogen peroxide in the reaction medium was controlled by testing with potassium iodide, water and starch. When effected on a sample containing hydrogen peroxide, it provides a brown-black colour. If the assay is negative before the chromatographic control indicates completion of the reaction, more hydrogen peroxide is added.
On completion of the reaction, 260 ml of water were added, the system was cooled to 0°C – 5°C again and the mixture was stirred for 2 hours at this temperature. The solid precipitate was filtered, washed with abundant water, and dried at a temperature below 60°C, to give 5-(difluoromethoxy)-2-[[(3- methoxy-4-chlorine-2-pyridinyl)methyl]sulfinyl]-lH-benzimidazole (IV), melting point 130° – 136°C, with an 83.5% yield. Thin layer chromatography on silica gel 60 F254, eluting with CHCl3/MeOH (15: 1), gave a main spot at Rf = 0.5.
Compound (IV) can be purified, if desired, by the following crystallization method:
5 g of crude product was suspended in 16 ml of acetone and was heated to boiling until a dark brown solution was obtained. Thereafter the thus obtained solution was allowed to cool down to room temperature and then was then chilled again to -20°C, at which temperature the mixture was held for 23 hours without stirring. Thereafter the solid was filtered and washed with 6×4 ml of acetone chilled to -20°C. Once dry, the resulting white solid weighed 2.73 g, had a point of melting of 142°C and gave a single spot in thin layer chromatography. The IR spectrum of the compound on KBr is given in Figure 1.
The acetonic solution comprising the mother liquors of filtration and the washes was concentrated to a volume of 20 ml and a further 5 g of crude compound were added. The above described crystallization process was repeated to obtain a further 4.11 g of purified product of characteristics similar to the previous one.
The acetonic solution from the previous crystallization was concentrated to a volume of 17 ml and a further 4 g of crude compound were added. The above described crystallization process was repeated to obtain a further 2.91 g of purified product of similar characteristics to the previous ones.
The acetonic solution from the previous crystallization was concentrated to a volume of 15 ml and a further 4 g of crude compound were added. The above described crystallization process was repeated to obtain a further 3.3 g of purified product of similar characteristics to the previous ones.
The acetonic solution from the previous crystallization was concentrated to a volume of 16 ml and a further 4.36 g of crude compound were added. The above described crystallization process was repeated to obtain a further 3.62 g of purified product of similar characteristics to the previous ones.
Finally, the acetonic solution from the previous crystallization was concentrated to a volume of 10 – 12 ml and held at -20°C for two days without stirring. Thereafter, the solid was filtered and washed with 5×3 ml of acetone chilled to -20°C. Once dry, the solid weighed 1.26 g and had similar characteristics to the previous ones.
The total yield of all the crystallizations was 80%.
Example 6. – Preparation of pantoprazole
12.95 g (0.0334 mol) of compound (IV) purified by crystallization of Example 5 were mixed with 38 ml of N,N-dimethylacetamide and thereafter 7.03 g (0.1003 mol) of potassium methoxide were added, while holding the temperature to between 20°C and 30°C, whereby a dark brown mixture was obtained. The system was held at approximately 25°C for about 23 hours, after which, once the reaction was complete, the pH was adjusted to 7 with the addition of 3.82 ml of acetic acid. The N,N-dimethylacetamide was removed at reduced pressure at an internal temperature of not more than 75°C. 65 ml of water and 50 ml of methylene chloride were added over the thus obtained residue, followed by decantation of the phases. Once the phases were decanted, the aqueous phase was extracted a with further 3×25 ml of methylene chloride, the organic phases were pooled and the resulting solution dried over anhydrous sodium sulfate, was filtered and washed, and evaporated at reduced pressure in a rotavap, to give a crude residue over which 55 ml of water were added, to give a suspension (if the product does not solidify at this point the water is decanted and a further 55 ml of water are added to remove remains of N,N-dimethylacetamide that hinder the solidification of the product). The solid was filtered and, after drying, 11.61 g of crude pantoprazole of reddish brown colour were obtained (Yield 90%). The thus obtained crude product was decoloured by dissolving the crude product in 150 ml of methanol, whereby a dark brown solution was obtained. 7.5 g of active carbon were added, while maintaining stirring for 45 minutes at 25°C – 30°C, after which the carbon was filtered out and the filter was washed. The methanol was then removed in the rotavap at reduced pressure, a temperature below 40°C. 10.33 g of a solid residue were obtained and were mixed with 14.9 ml of methylethylketone, and the suspension was heated to 45°C for about 10 minutes, after which it was cooled, first to room temperature and then to -20°C. This temperature was held over night and thereafter the solid was filtered, washed with 6×5 ml of methylethylketone chilled to -20°C. Once dry, 7.75 g of a white solid, melting point 140°C – 141 °C, were obtained. Thin layer chromatography on silica gel F254, eluting with CHCl3/MeOH (15: 1), gave a single spot at Rf =
0.41 and a IR spectrum corresponding identically with that of pantoprazole.
The ketonic solution comprising the mother liquors of filtration and the washes, was concentrated to 9.7 ml, was heated to 40°C, was held at this temperature for about five minutes and was then cooled, first to room temperature and then to -20°C, this temperature being held for 4 hours. At the end of this time, the solid was filtered and was washed with 4×2 ml of methylethylketone chilled to -20°C. Once dry, 0.42 g of a white solid of similar characteristics to the previous one was obtained.
The ketone solution from the previous treatment was concentrated to 3.1 ml, was heated to 40°C, was held to this temperature for about five minutes and then was cooled, first to room temperature and then to -20°C, this temperature being held for 4 hours. At the end of this time, the solid was filtered and was washed with 5×3 ml of methylethylketone chilled to – 20°C. Once dry, 0.41 g of a white-beige solid of similar characteristics to the previous one was obtained. The total yield, including purifications, was 67%.
If a whiter solid is desired, one or several washes can be carried with isopropyl acetate as follows: 6.6 g of pantoprazole from the methylethylketone treatment were suspended in 50 ml of isopropyl acetate. The system (white suspension) was stirred for about 30 minutes at 25°C, was then cooled to 0°C – 5°C, was stirred for about 15 minutes at this temperature and the solid was then filtered, was washed with 3×15 ml of isopropyl acetate. Once dry, 6.26 g of a pure white solid were obtained.
Trade Names
Country
Trade name
Manufacturer
Germany
Pantozol
Nycomed
Rifun
– “-
France
Eupantol
Altana
Inipomp
Sanofi-Aventis
United Kingdom
Protium
ALTANA
Italy
Pantekta
Abbott
Pantopan
Pharmacia
Pantork
Altana
USA
Protonix
Wyeth
Ukraine
Kontrolok
Nycomed Oranienburg GmbH, Germany
Nolpaza
Krka
Pultset
Nobel Ilach Sanayi ve Ticaret AS, Turkey
Proksium
JSC “Lubnyfarm”, Ukraine
various generic drugs
Formulations
ampoule 40 mg;
Tablets 40 mg
UV – spectrum
Conditions : Concentration – 1 mg / 100 ml
Solvent designation schedule
Methanol
Water
0.1 M HCl
0.1M NaOH
The absorption maximum
289 nm
291nm
Observed
decay
295 nm
391
346
–
418
ε
16600
14700
–
17700
IR – spectrum
Wavelength (μm)
Wavenumber (cm -1 )
NMR Spectrum
will be added
Links
EP 134 400 (Byk Gulden Lomberg; appl. 1.5.1984; CH-prior. 3.5.1983).
US 4,555,518 (Byk Gulden Lomberg; 26.11.1985; appl. 1.5.1984; CH-prior. 3.5.1983).
US 4,758,579 (Byk Gulden Lomberg; 19.7.1988; appl. 28.4.1987; CH-prior. 16.6.1984).
UV and IR Spectra. H.-W. Dibbern, RM Muller, E. Wirbitzki, 2002 ECV
NIST / EPA / NIH Mass Spectral Library 2008
Handbook of Organic Compounds. NIR, IR, Raman, and UV-Vis Spectra Featuring Polymers and Surfactants, Jr., Jerry Workman.Academic Press, 2000.
Handbook of ultraviolet and visible absorption spectra of organic compounds, K. Hirayama. Plenum Press Data Division, 1967.
[Dr. John Cooke, chair of Methodist Hospital’s cardiovascular services] [Houston Chronicle Health Zone dated Thursday, July 11, 2013 chron.com/refluxmeds] (Journal: Circulation)
Jump up^ Meyer, U A (1996). “Metabolic interactions of the proton-pump inhibitors lansoprazole, omeprazole and pantoprazole with other drugs”. European journal of gastroenterology & hepatology8 (Suppl 1): S21–25. doi:10.1097/00042737-199610001-00005.
Steinijans, V. W.; Huber, R.; Hartmann, M.; Zech, K.; Bliesath, H.; Wurst, W.; Radtke, H. W. (1996). “Lack of pantoprazole drug interactions in man: An updated review”. International Journal of Clinical Pharmacology and Therapeutics34 (6): 243–262. PMID8793611.
Phase 3 drug, UncategorizedComments Off on Cortendo AB: First Patient Enrolled into NormoCort Phase 3 SONICS Trial Following a Successful EU Investigator Meeting
An antimicrobial agent that destroys fungi by suppressing their ability to grow or reproduce. Antifungal agents differ from industrial fungicides in that they defend against fungi present in human or animal tissues.
An antimicrobial agent that destroys fungi by suppressing their ability to grow or reproduce. Antifungal agents differ from industrial fungicides in that they defend against fungi present in human or animal tissues.
Ketoconazole, 1-acetyl-4-[4-[[2-(2,4-dichlorophenyl)-2-[(1H-imidazol-1-yl)-methyl]-1,3– dioxolan-4-yl]methoxy]phenyl]piperazine, is a racemic mixture of the cis enantiomers (-)-(2S,4R) and (+)-(2R,4S) marketed as an anti-fungal agent. Ketoconazole inhibits fungal growth through the inhibition of ergosterol synthesis.(-)-Ketoconazole, the (2S,4R) enantiomer contained in the racemate of ketoconazole, is in phase III clinical trials at Cortendo for the treatment of endogenous Cushing’s syndrome. The company and licensee DiObex had also been developing the drug candidate for the treatment of type 2 diabetes; however, no recent development has been reported for this research.Preclinical studies have demonstrated the drug candidate’s ability to inhibit the synthesis of cortisol, resulting in substantial clinical benefits including lowering both blood pressure and cholesterol in addition to controlling glucose levels. It has also been shown that (-)-ketoconazole is responsible for virtually all of the cortisol synthesis inhibitory activity present in the racemate. Rights to the compound are shared with Cortendo.In 2012, orphan drug designation was assigned in the U.S. for the treatment of endogenous Cushing’s syndrome.
GÖTEBORG, Sweden.–(BUSINESS WIRE)–Cortendo AB (OSE:CORT) today announced that the first patient has been enrolled into the Phase 3 SONICS trial, i.e., “Study Of NormoCort In Cushing’s Syndrome.”
“The enrollment of the first patient into the SONICS trial represents a significant milestone for Cortendo”
The patient was enrolled by one of the trial’s lead principal investigators at a Pituitary Center from a prestigious institution in Baltimore, Maryland. “The enrollment of the first patient into the SONICS trial represents a significant milestone for Cortendo”, said Dr. Theodore R Koziol. ”The SONICS clinical trial team is acutely focused on the implementation of the trial following a successful EU Investigator’s meeting in Barcelona in July, which we believe further solidified the foundation for the trial.”
Cortendo successfully completed its European Investigator meeting supporting SONICS held in Barcelona, Spain on July 17-18. More than 35 investigators/study coordinators, including many of the world’s leading Cushing’s experts from 24 study sites, were in attendance and received training for the trial. Based on the positive feedback from the meeting, Cortendo has gained further confidence that NormoCort (COR-003) has the potential to be an important future treatment option for patients afflicted with Cushing’s Syndrome. A second US Investigator meeting is also being planned for later this year.
”It was gratifying to participate in the NormoCort SONICS trial investigator meeting in my home town of Barcelona with so many esteemed colleagues dedicated to treating patients with Cushing’s Syndrome”, said Susan Webb M.D. Ph.D. Professor of Medicine Universitat Autonoma de Barcelona. ”There remains a significant unmet medical need for patients, and I am delighted to be part of the development of this new therapy”.
Cortendo has also further strengthened its internal as well as external teams to support the study and to position the trial for an increased recruitment rate. In July, Cortendo added both an experienced physician and internal Clinical Operations Director to the NormoCort development team. Cortendo, working in concert with its CROs supporting the SONICS trial, now has a team of approximately 20 personnel on the NormoCort development program.
Cortendo has previously communicated its plan to meet the recruitment goal by increasing the number of study sites from 38 to 45 worldwide. The company is at various levels of activation with more than 30 study sites to date. Therein, Cortendo expects a large proportion of the sites to be activated by the end of the third quarter this year and remains confident that essentially all sites will be open by the end of 2014.
Risk and uncertainty
The development of pharmaceuticals carries significant risk. Failure may occur at any stage during development and commercialization due to safety or clinical efficacy issues. Delays may occur due to requirements from regulatory authorities not anticipated by the company.
About Cortendo
Cortendo AB is a biopharmaceutical company headquartered in Göteborg, Sweden. Its stock is publicly traded on the NOTC-A-list (OTC) in Norway. Cortendo is a pioneer in the field of cortisol inhibition and has completed early clinical trials in patients with Type 2 diabetes. The lead drug candidate NormoCort, the 2S, 4R-enantiomer of ketoconazole, has been re-focused to Cushing’s Syndrome, and has entered Phase 3 development. The company’s strategy is to primarily focus its resources within orphan drugs and metabolic diseases and to seek opportunities where the path to commercialization or partnership is clear and relatively near-term. Cortendo’s business model is to commercialize orphan and specialist product opportunities in key markets, and to partner non-specialist product opportunities such as diabetes at relevant development stages.
Alexander Lindström
Chief Financial Officer Office
+1 610 254 9200
Mobile : +1 917 349 7210
E-mail : alindstrom@cortendo.com
Ketoconazole, 1-acetyl-4- [4-[[2-(2,4-dichlorophenyl)-2-[(1H-imidazol-1-yl)-methyl]-1,3-dioxolan-4-yl] methoxy] phenyl] piperazine, is a racemic mixture of the cis enantiomers (-)-(2S, 4R) and (+)-(2R, 4S) marketed as an anti-fungal agent. Ketoconazole inhibits fungal growth through the inhibition of ergosterol synthesis. Ergosterol is a key component of fungal cell walls.
More recently, ketoconazole was found to decrease plasma cortisol and to be useful, alone and in combination with other agents, in the treatment of a variety of diseases and conditions, including type 2 diabetes, Metabolic Syndrome (also known as the Insulin Resistance Syndrome, Dysmetabolic Syndrome or Syndrome X), and other medical conditions that are associated with elevated cortisol levels. SeeU.S. Patent Nos. 5,584,790 ; 6,166,017 ; and 6,642,236 , each of which is incorporated herein by reference. Cortisol is a stress-related hormone secreted from the cortex of the adrenal glands. ACTH (adenocorticotropic hormone) increases cortisol secretion. ACTH is secreted by the pituitary gland, a process activated by secretion of corticotropin releasing hormone (CRH) from the hypothalamus.
Cortisol circulates in the bloodstream and activates specific intracellular receptors, such as the glucocorticoid receptor (GR). Disturbances in cortisol levels, synthetic rates or activity have been shown to be associated with numerous metabolic complications, including insulin resistance, obesity, diabetes and Metabolic Syndrome. Additionally, these metabolic abnormalities are associated with substantially increased risk of cardiovascular disease, a major cause of death in industrialized countries. See Mårin P et al., “Cortisol secretion in relation to body fat distribution in obese premenopausal women.” Metabolism 1992; 41:882-886, Bjorntorp, “Neuroendocrine perturbations as a cause of insulin resistance.” Diabetes Metab Res Rev 1999; 15(6): 427-41, and Rosmond, “Role of stress in the pathogenesis of the metabolic syndrome.” Psychoneuroendocrinology 2005; 30(1): 1-10, each of which is incorporated herein by reference.
While ketoconazole is known to inhibit some of the enzymatic steps in cortisol synthesis, such as, for example, 17α hydroxylase (Wachall et al., “Imidazole substituted biphenyls: a new class of highly potent and in vivo active inhibitors of P450 17 as potential therapeutics for treatment of prostate cancer.” Bioorg Med Chem 1999; 7(9): 1913-24, incorporated herein by reference) and 11b-hydroxylase (Rotstein et al., “Stereoisomers of ketoconazole: preparation and biological activity.” J Med Chem 1992; 35(15): 2818-25) and 11β-hydroxy steroid dehydrogenase (11β-HSD) (Diederich et al., “In the search for specific inhibitors of human 11β-hydroxysteroid-dehydrogenases (11β-HSDs): chenodeoxycholic acid selectively inhibits 11β-HSD-L” Eur J Endocrinol 2000; 142(2): 200-7, incorporated herein by reference) the mechanisms by which ketoconazole decreases cortisol levels in the plasma have not been reported. For example, there is uncertainty regarding the effect of ketoconazole on the 11β-hydroxy steroid dehydrogenase (11β-HSD) enzymes. There are two 11β-HSD enzymes. One of these, 11β-HSD-I, is primarily a reductase that is highly expressed in the liver and can convert the inactive 11-keto glucocorticoid to the active glucocorticoid (cortisol in humans and corticosterone in rats). In contrast, the other, 11β-HSD-II, is primarily expressed in the kidney and acts primarily as an oxidase that converts active glucocorticoid (cortisol in humans and corticosterone in rats) to inactive 11-keto glucocorticoids. Thus, the plasma concentration of active glucocorticoid is influenced by the rate of synthesis, controlled in part by the activity of adrenal 11β-hydroxylase and by the rate of interconversion, controlled in part by the relative activities of the two 11β-HSD enzymes. Ketoconazole is known to inhibit these three enzymes (Diederich et al., supra) and the 2S,4R enantiomer is more active against the adrenal 11β-hydroxylase enzyme than is the 2R,4S enantiomer (Rotstein et al., supra). However, there are no reports describing the effect of the two ketoconazole enantiomers on either of 11β-HSD-I or 11β-HSD-II, so it is not possible to predict what effects, if any, the two different ketoconazole enantiomers will each have on plasma levels of the active glucocorticoid levels in a mammal.
Ketoconazole has also been reported to lower cholesterol levels in humans (Sonino et al. (1991). “Ketoconazole treatment in Cushing’s syndrome: experience in 34 patients.” Clin Endocrinol (Oxf). 35(4): 347-52; Gylling et al. (1993). “Effects of ketoconazole on cholesterol precursors and low density lipoprotein kinetics in hypercholesterolemia.” J Lipid Res. 34(1): 59-67) each of which is incorporated herein by reference). The 2S,4R enantiomer is more active against the cholesterol synthetic enzyme 14 αlanosterol demethylase than is the other (2R,4S) enantiomer (Rotstein et al infra). However, because cholesterol level in a human patient is controlled by the rate of metabolism and excretion as well as by the rate of synthesis it is not possible to predict from this whether the 2S,4R enantiomer of ketoconazole will be more effective at lowering cholesterol levels.
The use of ketoconazole as a therapeutic is complicated by the effect of ketoconazole on the P450 enzymes responsible for drug metabolism. Several of these P450 enzymes are inhibited by ketoconazole (Rotsteinet al., supra). This inhibition leads to an alteration in the clearance of ketoconazole itself (Brass et al., “Disposition of ketoconazole, an oral antifungal, in humans.” Antimicrob Agents Chemother 1982; 21(1): 151-8, incorporated herein by reference) and several other important drugs such as Glivec (Dutreix et al., “Pharmacokinetic interaction between ketoconazole and imatinib mesylate (Glivec) in healthy subjects.” Cancer Chemother Pharmacol 2004; 54(4): 290-4) and methylprednisolone (Glynn et al., “Effects of ketoconazole on methylprednisolone pharmacokinetics and cortisol secretion.” Clin Pharmacol Ther 1986; 39(6): 654-9). As a result, the exposure of a patient to ketoconazole increases with repeated dosing, despite no increase in the amount of drug administered to the patient. This exposure and increase in exposure can be measured and demonstrated using the “Area under the Curve” (AUC) or the product of the concentration of the drug found in the plasma and the time period over which the measurements are made. The AUC for ketoconazole following the first exposure is significantly less than the AUC for ketoconazole after repeated exposures. This increase in drug exposure means that it is difficult to provide an accurate and consistent dose of the drug to a patient. Further, the increase in drug exposure increases the likelihood of adverse side effects associated with ketoconazole use.
[0008]
Rotstein et al. (Rotstein et al., supra) have examined the effects of the two ketoconazole cis enantiomers on the principal P450 enzymes responsible for drug metabolism and reported “…almost no selectivity was observed for the ketoconazole isomers” and, referring to drug metabolizing P450 enzymes: “[t]he IC50 values for the cis enantiomers were similar to those previously reported for racemic ketoconazole”. This report indicated that both of the cis enantiomers could contribute significantly to the AUC problem observed with the ketoconazole racemate.
One of the adverse side effects of ketoconazole administration exacerbated by this AUC problem is liver reactions. Asymptomatic liver reactions can be measured by an increase in the level of liver specific enzymes found in the serum and an increase in these enzymes has been noted in ketoconazole treated patients (Sohn, “Evaluation of ketoconazole.” Clin Pharm 1982; 1(3): 217-24, and Janssen and Symoens, “Hepatic reactions during ketoconazole treatment.” Am J Med 1983; 74(1B): 80-5, each of which is incorporated herein by reference). In addition 1:12,000 patients will have more severe liver failure (Smith and Henry, “Ketoconazole: an orally effective antifungal agent. Mechanism of action, pharmacology, clinical efficacy and adverse effects.” Pharmacotherapy 1984; 4(4): 199-204, incorporated herein by reference). As noted above, the amount of ketoconazole that a patient is exposed to increases with repeated dosing even though the amount of drug taken per day does not increase (the “AUC problem”). The AUC correlates with liver damage in rabbits (Ma et al., “Hepatotoxicity and toxicokinetics of ketoconazole in rabbits.” Acta Pharmacol Sin 2003; 24(8): 778-782 incorporated herein by reference) and increased exposure to the drug is believed to increase the frequency of liver damage reported in ketoconazole treated patients.
Additionally, U.S. Patent No. 6,040,307 , incorporated herein by reference, reports that the 2S,4R enantiomer is efficacious in treating fungal infections. This same patent application also reports studies on isolated guinea pig hearts that show that the administration of racemic ketoconazole may be associated with an increased risk of cardiac arrhythmia, but provides no data in support of that assertion. However, as disclosed in that patent, arrhythmia had not been previously reported as a side effect of systemic racemic ketoconazole, although a particular subtype of arrhythmia, torsades de pointes, has been reported when racemic ketoconazole was administered concurrently with terfenadine. Furthermore several published reports (for example, Morganroth et al. (1997). “Lack of effect of azelastine and ketoconazole coadministration on electrocardiographic parameters in healthy volunteers.” J Clin Pharmacol. 37(11): 1065-72) have demonstrated that ketoconazole does not increase the QTc interval. This interval is used as a surrogate marker to determine whether drugs have the potential for inducing arrhythmia. US Patent Number 6,040,307 also makes reference to diminished hepatoxicity associated with the 2S,4R enantiomer but provides no data in support of that assertion. The method provided in US Patent Number 6,040,307 does not allow for the assessment of hepatoxicity as the method uses microsomes isolated from frozen tissue.
DIO-902 is the single enantiomer 2S,4R ketoconazole and is derived from racemic ketoconazole. It is formulated using cellulose, lactose, cornstarch, colloidal silicon dioxide and magnesium stearate as an immediate release 200 mg strength tablet. The chemical name is 2S,4R cis-1-acetyl-4-[4-[[2-(2,4-dichlorophenyl)-2-(1H-imidazol-1-ylmethyl)-1,3-dioxolan-4-yl] methoxyl]phenyl] piperazine, the formula is C26H28Cl2N4O4, and the molecular weight is 531.44. The CAS number is 65277-42-1, and the structural formula is provided below. The chiral centers are at the carbon atoms 2 and 4 as marked.
[0132]
Ketoconazole is an imidazole-containing fungistatic compound. DIO-902 is an immediate release tablet to be taken orally and formulated as shown in the table below.
Component
Percentage
2S,4R ketoconazole;
DIO-902
50%
Silicified Microcrystalline Cellulose, NF
(Prosolv HD 90)
16.5
Lactose Monohydrate, NF (316 Fast-Flo)
22.4
Corn Starch, NF (STA-Rx)
10
Colloidal Silicon Dioxide, NF (Cab-O-Sil M5P)
0.5
Magnesium Stearate, NF
0.6
The drug product may be stored at room temperature and is anticipated to be stable for at least 2 years at 25° C and 50% RH. The drug is packaged in blister packs.
A new process has been developed to separate ketoconazole (KTZ) enantiomers by membrane extraction, with the oppositely preferential recognition of hydrophobic and hydrophilic chiral selectors in organic and aqueous phases, respectively. This system is established by adding hydrophobic l-isopentyl tartrate (l-IPT) in organic strip phase (shell side) and hydrophilic sulfobutylether-β-cyclodextrin (SBE-β-CD) in aqueous feed phase (lumen side), which preferentially recognizes (+)-2R,4S-ketoconazole and (−)-2S,4R-ketoconazole, respectively. The studies performed involve two enantioselective extractions in a biphasic system, where KTZ enantiomers form four complexes with SBE-β-CD in aqueous phase and l-IPT in organic phase, respectively. The membrane is permeable to the KTZ enantiomers but non-permeable to the chiral selector molecules. Fractional chiral extraction theory, mass transfer performance of hollow fiber membrane, enantioselectivity and some experimental conditions are investigated to optimize the separation system. Mathematical model of I/II = 0.893e0.039NTU for racemic KTZ separation by hollow fiber extraction, is established. The optical purity for KTZ enantiomers is up to 90% when 9 hollow fiber membrane modules of 30 cm in length in series are used.
I, (−)-2S,4R-ketoconazole;
II, (+)-2R,4S-ketoconazole;
CDs, cyclodextrin derivatives;
l-IPT, l-isopentyl tartrate;
d-IPT, d-isopentyl tartrate;
HP-β-CD, hydroxypropyl-β-cyclodextrin;
Me-β-CD, methyl-β-cyclodextrin;
β-CD, β-cyclodextrin;
NTU, number of transfer units;
HTU, height of a transfer unit;
PVDF,polyvinylidene fluoride
…………………….
Stereoselective synthesis of both enantiomers of ketoconazole from (R)- and (S)-
Pelayo Camps, Xavier Farrés, Ma Luisa García, Joan Ginesta, Jaume Pascual, David Mauleón, Germano Carganico
Bromobenzoates (2R,4R)- and (2S,4S)-18, prepared stereoselectively from (R)- and (S)-epichlorohydrin, were transformed into (2R,4S)-(+)- and (2S,4R)-(−)-Ketoconazole, respectively, following the known synthetic protocols for the racemic mixture.
Tetrahedron Asymmetry 1995, 6(6): 1283
Stereoselective syntheses of both enantiomers of ketoconazole (1) from commercially available (R)- or (S)-epichlorohydrin has been developed. The key-step of these syntheses involves the selective substitution of the methylene chlorine atom by benzoate on a mixture of and or of their enantiomers, followed by crystallization of the corresponding cis-benzoates, (2S,4R)-18 or(2S,4S)-18, from which (+)- or (−)-1 were obtained as described for (±)-1. The ee’s of (+)- and (−)-ketoconazole were determined by HPLC on the CSP Chiralcel OD-H.
The incidence of fungal infections has considerably increased over the last decades. Notwithstanding the utility of the antifungal compounds commercialized in the last 15 years, the investigation in this field is however very extensive. During this time, compounds belonging to the azole class have beer, commercialized for both the topical and oral administrations, such a class including imidazoles as well as 1,2,4-triazoles. Some of these compounds car. show m some degree a low gastrointestinal tolerance as well as hepatotoxycity.
A large number of pharmaceutically active compounds are commercialized as stereoisomeric mixtures. On the other hand, the case in which only one of said stereoisomers is pharmaceutically active is frequent.
The undesired enantiomer has a lower activity and it sometimes may cause undesired side-effects.
Ketoconazole (1-acetyl-4-[4-[[2-(2,4-dichlorophenyl)-2-[(1H-imidazol-1-yl)methyl]-1,3-dioxolane-4-yl]methoxy]phenyl]piperazine), terconazole (1-[4-[[2(2,4-dichlorophenyl)-2-[(1H-1 , 2 ,4-triazol-1-yl)methyl]-1,3-dioxolane-4-yl]methoxy]phenyl]-4-(1-methylethyl)piperazine) and other related azole antifungal drugs contain in their structure a substituted 1,3-dioxolane ring, in which carbon atoms C2 and C4 are stereogenic centres, therefore four possible stereoisomers are possible. These compounds are commercialized in the form or cis racemates which show a higher antifungal activity than the corresponding trans racemates.
The cis homochiral compounds of the present invention, which are intermediates for the preparation of enantiomerically pure antifungal drugs, have been prepared previously in the racemic form and transformed into the different azole antifungal drugs in the racemic form [J. Heeres et al., J . Med . Chem . , 22 , 1003 (1979). J . Med . Chem . , 26, 611 (1983), J . Med . Chem . , 27 , 894 (1984) and US 4,144,346, 4,223,036, 4,358,449 and 4,335,125].
Scheme 1 shows the synthesis described for racemic ketoconazole [J. Heeres et al., J . Med . Chem . , 22 , 1003 (1979)]. Scheme 1
)
The synthesis of racemic terconazole [J. Heeres et al., J. Med . Chem . , 26 , 611 11983)] is similar. differing in the introduction of a 1 H- 1 , 2,4-triazol-1-yl substituent in place of 1H-imidazol-1-yl and in the nature of the phenol used in the last step of the synthetic sequence, which phenol is 1-methylethyl-4-(4- hydroxyphenyl)piperazme instead of 1-acetyl-4-(4-nydroxyphenyl)piperazine.
The preparation of racemic itraconazole [J. Heeres et al., J. Med . Chem. , 27 , 894 (1984)] is similar to that of terconazole, differing only in the nature of the phenol used in the last step of the synthetic sequence.
In the class of azoles containing a 1,3-dioxolane ring and a piperazine ring and moreover they are pure enantiomers, only the preparation of (+)- and (-)-ketoconazole has been described [D. M. Rotstein et al., J. Med . Chem . , 35, 2818 (1992)] (Scheme 2) starting from the tosylate of (+)- and (-) 2,2-dimethyl-1,3-dioxolane-4-methanol.
Scheme 2
This synthesis suffers from a series of drawbacks, namely: a) the use of expensive, high molecular weight starting products which are available only on a laboratory scale, and b) the need for several chromatographies during the process in order to obtain products of suitable purity, which maKes said synthesis economically unattractive and difficult to apply industrially.
Recently (N. M. Gray, WO 94/14447 and WO 94/14446) the use of (-)-ketoconazole and (+)-ketoconazole as antifungal drugs causing less side-effects than (±)-ketoconazole has been claimed.
The industrial preparation of enantiomerically pure antifungal drugs with a high antifungal activity and less side-effects is however a problem in therapy. The present invention provides novel homochiral compounds which are intermediates for the industrial preparation of already known, enantiomerically pure antifungal drugs such as ketoconazole enantiomers, or of others which have not yet been reported in literature, which are described first in the present invention, such as (+)-terconazole and (-)-terconazoie, which show the cited antifungal action, allowing to attain the same therapeutical effectiveness using lower dosages than those required for racemic terconazole
Example 14 : (2S,4R)-(-)-1-acetyl-4-[4-[ [2-(2,4-dichlorophenyl)-2-[(1H-imidazol-1-yl)-methyl]-1,3-dioxolane-4-yl]methoxy]phenyl]piperazine, (2S,4R) -(- )-ketoconazole.
This compound is prepared following the process described above for (2R,4S)-(+)-ketoconazole. Starting from HNa (60-65% dispersion in paraffin, 32 mg, 0.80 mmol), 1-acetyl-4-(4-hydroxyphenyl)piperazine (153 mg, 0.69 mol) and (2S,4S)-(-)-IV (Ar = 2,4-dichlorophenyl, Y = CH, R = CH3) (250 mg, 0.61 mmol), upon crystallization from an acetone:ethyl acetate mixture, (2S,4R) -(-)-ketoconazole is obtained [(2S,4R)-V Ar = 2,4-dichlorophenyl, Y = CH, Z = COCH3] (196 mg, 61% yield) as a solid, m.p. 153-155ºC (lit. 155-157ºC); [α]D20 = -10.50 (c = 0.4, CHCl3) (lit. [α]D25 = -10.58. c = 0.4, CHCl3) with e.e. > 99% (determined by HPLC using the chiral stationary phase CHIRALCEL OD-H and ethanol:hexane 1:1 mixtures containing 0.1 % diethylamine as the eluent).
+ KETOCONAZOLE…. UNDESIRED
Example 7: (2 R ,4S)-(+)-1-acetyl-4-[4-[[2-(2,4-dichlorophenyl)-2-[(1H-imidazol-1-yl)methyl]-1,3-dioxolane-4-yl]methoxy]phenyl]piperazine (22, 4 S)-(+)-ketoconazole.
To a suspension of NaH (dispersed in 60-65% paraffin, 19.2 mg, 0.48 mmol) in anhydrous DMSO (3 ml),
1-acetyl-4-(hydroxyphenyl)piperazine (102 mg, 0.46 mmol) is added and the mixture is stirred for 1 hour at room temperature. Then, a solution of (2R,4R) – (+)-IV (Ar = 2,4-dichlorophenyl, Y = CH, R = CH3) (160 mg, 0.39 mmol) in anhydrous DMSO (5 ml) is added, and the mixture is heated at 80ºC for 4 hours. The reaction mixture is allowed to cool to room temperature, diluted with water
(20 ml) and extracted with CH2Cl2 (3 × 25 ml). The combined organic phases are washed with water (3 × 25), dried with Na2SO4 and the solvent is evaporated off under vacuum. The oily residue thus obtained is crystallized from an acetone:ethyl acetate mixture to give (2R,4S)-(+)-ketoconazole ( (2R, 4 S) -V , Ar 2,4-dichlorophenyl, Y = CH , Z = COCH3 ) ( 110 mg , 5 3 % yie ld ) as a white solid, m.p. 155-156°C (lit. 154-156ºC), [α]D20 = + 8.99 (c = 0.4, CHCl3) (lit. [α]D25 = + 8.22, c = 0.4, CHCl3), with e.e. > 99% (determined by HPLC using the chirai stationary phase CHIRALCEL OD-H and ethanol:hexane 1:1 mixtures containing 0.1% of diethylamine, as the eluent; (+)-Ketoconazole retention time 73,28 min. (-)-Ketoconazole, retention time 79.06 min).
Experimental and theoretical analysis of the interaction of (+/-)-cis-ketoconazole with beta-cyclodextrin in the presence of (+)-L-tartaric acid
J Pharm Sci 1999, 88(6): 599
1H NMR spectroscopy was used for determining the optical purity of cis-ketoconazole enantiomers obtained by fractional crystallization. The chiral analysis was carried out using β-cyclodextrin in the presence of (+)-l-tartaric acid. The mechanism of the chiral discrimination process, the stability of the complexes formed, and their structure in aqueous solution were also investigated by 1H and 13C chemical shift analysis, two-dimensional NOE experiments, relaxation time measurements, and mass spectrometry experiments. Theoretical models of the three-component interaction were built up on the basis of the available NMR data, by performing a conformational analysis on the relevant fragments on ketoconazole and docking studies on the components of the complex. The model derived from a folded conformation of ketoconazole turned out to be fully consistent with the molecular assembly found in aqueous solution, as inferred from NOE experiments. An explanation of the different association constants for the complexes of the two enantiomers is also provided on the basis of the interaction energies.
The U.S. Food and Drug Administration today approved Cologuard, the first stool-based colorectal screening test that detects the presence of red blood cells and DNA mutations that may indicate the presence of certain kinds of abnormal growths that may be cancers such as colon cancer or precursors to cancer.
Colorectal cancer primarily affects people age 50 and older, and among cancers that affect both men and women, it is the third most common cancer and the second leading cause of cancer-related death in the United States, according to the Centers for Disease Control and Prevention (CDC). Colorectal cancer screening is effective at reducing illness and death related to colon cancer. The CDC estimates that if everyone age 50 or older had regular screening tests as recommended, at least 60 percent of colorectal cancer deaths could be avoided.
Colorectal cancer occurs in the colon (large intestine) or rectum (the passageway that connects the colon to the anus). Most colorectal cancers start as abnormal raised or flat tissue growths on the wall of the large intestine or rectum (polyps). Some very large polyps are called advanced adenomas and are more likely than smaller polyps to progress to cancer.
Using a stool sample, Cologuard detects hemoglobin, a protein molecule that is a component of blood. Cologuard also detects certain mutations associated with colorectal cancer in the DNA of cells shed by advanced adenomas as stool moves through the large intestine and rectum. Patients with positive test results are advised to undergo a diagnostic colonoscopy.
“This approval offers patients and physicians another option to screen for colorectal cancer,” said Alberto Gutierrez, Ph.D., director of the Office of In Vitro Diagnostics and Radiological Health at the FDA’s Center for Devices and Radiological Health. “Fecal blood testing is a well-established screening tool and the clinical data showed that the test detected more cancers than a commonly used fecal occult test.”
Today’s approval of the Cologuard does not change current practice guidelines for colorectal cancer screening. Stool DNA testing (also called “fecal DNA testing”) is not currently recommended as a method to screen for colorectal cancer by the United States Preventive Services Task Force (USPSTF). Among other guidelines, the USPSTF recommends adults age 50 to 75, at average risk for colon cancer, be screened using fecal occult blood testing, sigmoidoscopy, or colonoscopy.
The safety and effectiveness of Cologuard was established in a clinical trial that screened 10,023 subjects. The trial compared the performance of Cologuard to the fecal immunochemical test (FIT), a commonly used non-invasive screening test that detects blood in the stool. Cologuard accurately detected cancers and advanced adenomas more often than the FIT test. Cologuard detected 92 percent of colorectal cancers and 42 percent of advanced adenomas in the study population, while the FIT screening test detected 74 percent of cancers and 24 percent of advanced adenomas. Cologuard was less accurate than FIT at correctly identifying subjects negative for colorectal cancer or advanced adenomas. Cologuard correctly gave a negative screening result for 87 percent of the study subjects, while FIT provided accurate negative screening results for 95 percent of the study population.
Today the Centers for Medicare & Medicaid Services (CMS) issued a proposed national coverage determination for Cologuard. Cologuard is the first product reviewed through a joint FDA-CMS pilot program known as parallel review where the agencies concurrently review medical devices to help reduce the time between the FDA’s approval of a device and Medicare coverage. This voluntary pilot program is open to certain premarket approval applications for devices with new technologies and to medical devices that fall within the scope of a Part A or Part B Medicare benefit category and have not been subject to a national coverage determination.
“Parallel review allows the last part of the FDA process to run at the same time as the CMS process, cutting as many as six months from the time from study initiation to coverage,” said Nancy Stade, CDRH’s deputy director for policy. “The pilot program is ongoing, but we will apply what we have learned to improve the efficiency of the medical device approval pathway for devices that address an important public health need.”
“This is the first time in history that FDA has approved a technology and CMS has proposed national coverage on the same day,” said Patrick Conway, chief medical officer and deputy administrator for innovation and quality for CMS. “This parallel review represents unprecedented collaboration between the two agencies and industry and most importantly will provide timely access for Medicare beneficiaries to an innovative screening test to help in the early detection of colorectal cancer.”
CMS proposes to cover the Cologuard test once every three years for Medicare beneficiaries who meet all of the following criteria:
age 50 to 85 years,
asymptomatic (no signs or symptoms of colorectal disease including but not limited to lower gastrointestinal pain, blood in stool, positive guaiac fecal occult blood test or fecal immunochemical test), and
average risk of developing colorectal cancer (no personal history of adenomatous polyps, of colorectal cancer, or inflammatory bowel disease, including Crohn’s Disease and ulcerative colitis; no family history of colorectal cancers or an adenomatous polyp, familial adenomatous polyposis, or hereditary nonpolyposis colorectal cancer).
Cologuard is manufactured by Exact Sciences in Madison, Wisconsin.
Why India is becoming a preferable place for foreign companies to run a pharma franchise?
Various pharma companies in abroad are looking for partners in India for their pharma franchise business. There are many reasons that influence them to show interest in India based pharma franchise industry. We all are aware of the fact that India is a place where one can easily get cheap labor and innovation. The great minds working for technology development come up with latest equipments and machineries based of better technology for production of pharma products.
Cefuroxime Axetil (1-(acetyloxy) ethyl ester of cefuroxime, is (RS)-1-hydroxyethyl (6R,7R)-7-[2-(2-furyl)glyoxyl-amido]-3-(hydroxymethyl)-8-oxo-5-thia-1-azabicyclo[4.2.0]-oct-2-ene-2-carboxylate, 7 2 -(Z)-(O-methyl-oxime), 1-acetate 3-carbamate.
Its molecular formula is C 20 H 22 N 4 O 10S, and it has a molecular weight of 510.48.
Cefuroxime Axetil is used orally for the treatment of patients with mild-to-moderate infections, caused by susceptible strains of the designated microorganisms.
Cefuroxime axetil is a second generation oral cephalosporinantibiotic. It was discovered by Glaxo now GlaxoSmithKline and introduced in 1987 as Zinnat.[1] It was approved by FDA on Dec 28, 1987.[2] It is available by GSK as Ceftin in US[3] and Ceftum in India.[4]
It is an acetoxyethyl ester prodrug of cefuroxime which is effective orally.[5] The activity depends on in vivohydrolysis and release of cefuroxime.
Cefuroxime is chemically (6R, 7R)-3-carbamoyloxymethyl-7-[(Z)-2-(fur-2-yl)-2-methoxy-iminoacetamido] ceph-3-em-4-carboxylic acid and has the structural Formula II:
Cefuroxime axetil having the structural Formula I:
is the 1-acetoxyethyl ester of cefuroxime, a cephalosporin antibiotic with a broad spectrum of activity against gram-positive and gram negative micro-organisms.
This compound as well as many other esters of cefuroxime, are disclosed and claimed in U.S. Pat. No. 4,267,320. According to this patent, the presence of an appropriate esterifying group, such as the 1-acetoxyethyl group of cefuroxime axetil, enhances absorption of cefuroxime from the gastrointestinal tract, whereupon the esterifying group is hydrolyzed by enzymes present in the human body.
Because of the presence of an asymmetric carbon atom at the 1-position of the 1-acetoxyethyl group, cefuroxime axetil can be produced as R and S diastereoisomers or as a racemic mixture of the R and S diastereoisomers. U.S. Pat. No. 4,267,320 discloses conventional methods for preparing a mixture of the R and S isomers in the crystalline form, as well as for separating the individual R and S diastereoisomers.
The difference in the activity of different polymorphic forms of a given drug has drawn the attention of many workers in recent years to undertake the study on polymorphism. Cefuroxime axetil is the classical example of amorphous form exhibiting higher bioavailability than the crystalline form.
U.S. Pat. No. 4,562,181 and the related U.S. Pat. Nos. 4,820,833; 4,994,567 and 5,013,833, disclose that cefuroxime axetil in amorphous form, essentially free from crystalline material and having a purity of at least 95% aside from residual solvents, has a higher bioavailability than the crystalline form while also having adequate chemical stability.
These patents disclose that highly pure cefuroxime axetil can be recovered in substantially amorphous form from a solution containing cefuroxime axetil by spray drying, roller drying, or solvent precipitation. In each case, crystalline cefuroxime axetil is dissolved in an organic solvent and the cefuroxime axetil is recovered from the solution in a highly pure, substantially amorphous form.
Another U.S. Pat. No. 5,063,224 discloses that crystalline R-cefuroxime axetil which is substantially free of S-isomer is readily absorbed from the stomach and gastrointestinal tract of animals and is therefore ideally suited to oral therapy of bacterial infections.
According to this patent, such selective administration of R-cefuroxime axetil results in surprisingly greater bioavailability ability of cefuroxime, and thus dramatically reduces the amount of unabsorbable cefuroxime remaining in the gut lumen, thereby diminishing adverse side effects attributable to cefuroxime.
British Patent Specification No. 2,145,409 discloses a process for obtaining pure crystalline cefuroxime axetil and is said to be an improvement over British Patent Specification No. 1,571,683. Sodium cefuroxime is used as the starting material in the disclosed specification, which in turn, is prepared from either 3-hydroxy cefuroxime or cefuroxime.
Said process involves an additional step of preparing sodium cefuroxime, and therefore is not economical from commercial point of view.
CEFTIN (cefuroxime axetil) Tablets and CEFTIN (cefuroxime axetil) for Oral Suspension contain cefuroxime as cefuroxime axetil. CEFTIN (cefuroxime axetil) is a semisynthetic, broad-spectrum cephalosporin antibiotic for oral administration.
Chemically, cefuroxime axetil, the 1-(acetyloxy) ethyl ester of cefuroxime, is (RS)-1-hydroxyethyl (6R,7R)-7-[2-(2-furyl)glyoxyl-amido]-3-(hydroxymethyl)-8-oxo-5-thia-1-azabicyclo[4.2.0]-oct-2-ene-2-carboxylate, 72-(Z)-(O-methyl-oxime), 1-acetate 3-carbamate. Its molecular formula is C20H22N4O10S, and it has a molecular weight of 510.48.
Cefuroxime axetil is in the amorphous form and has the following structural formula:
CEFTIN (cefuroxime axetil) Tablets are film-coated and contain the equivalent of 250 or 500 mg of cefuroxime as cefuroxime axetil. CEFTIN (cefuroxime axetil) Tablets contain the inactive ingredients colloidal silicon dioxide, croscarmellose sodium, hydrogenated vegetable oil, hypromellose, methylparaben, microcrystalline cellulose, propylene glycol, propylparaben, sodium benzoate, sodium lauryl sulfate, and titanium dioxide.
CEFTIN (cefuroxime axetil) for Oral Suspension, when reconstituted with water, provides the equivalent of 125 mg or 250 mg of cefuroxime (as cefuroxime axetil) per 5 mL of suspension. CEFTIN (cefuroxime axetil) for Oral Suspension contains the inactive ingredients acesulfame potassium, aspartame, povidone K30, stearic acid, sucrose, tutti-frutti flavoring, and xanthan gum.
Dicyclohexylamine (17.2 g) in N,N-dimethylacetamide (50 ml) was added to a solution of cefuroxime acid (42.4 g) in N,N-dimethylacetamide (300 ml) at about −10° C. (R,S)1-Acetoxethylbromide (33.4 g) in N,N-dimethylacetamide (50 ml) was added to the above solution and the reaction mixture was stirred for 45 minutes at about −3 to 0° C. Potassium carbonate (1.1 g) was added to the reaction mixture and it was further stirred at that temperature for about 4 hours. The reaction mixture was worked up by pouring into it ethyl acetate (1.0 It), water (1.2 It) and dilute hydrochloric acid (3.5% w/w, 200 ml). The organic layer was separated and the aqueous layer was again extracted with ethyl acetate. The combined organic extracts were washed with water, dilute sodium bicarbonate solution (1%), sodium chloride solution and evaporated in vacuo to give a residue. Methanol was added to the residue and the crude product was precipitated by adding water.
The resulting precipitate was filtered off and recrystallized from the mixture of ethylacetate, methanol and hexane. The precipitated product was filtered, washed and dried to give pure crystalline cefuroxime axetil (42.5 g).
Assay (by HPLC on anhydrous basis)-98.2% w/w; Diastereoisomer ratio-0.53; Total related substances-0.48% w/w.
The present invention relates to an improved method for synthesis of cefuroxime axetil of formula (I) in high purity substantially free of the corresponding 2-cephem(Δ2)-ester of formule (II) and other impurities. The compound produced is valuable as a prodrug ester of the corresponding cephalosporin- 4-carboxylic acid derivative i. e. cefuroxime, particularly suitable for oral administration in various animal species and in man for treatment of infections caused by gram-positive and gram-negative bacteria.
BACKGROUND OF THE INVENTION
[0002]
One of the ways to improve the absorption of cephalosporin antibiotics which are poorly absorbed through the digestive tract is to prepare and administer the corresponding ester derivatives at the 4-carboxylic acid position. The esters are then readily and completely hydrolysed in vivoby enzymes present in the body to regenerate the active cephalosporin derivative having the free carboxylic acid at the 4-position.
[0003]
Among the various ester groups that can be prepared and administered only a selected few are biologically acceptable, in addition to possessing high antibacterial activity and broad antibacterial spectrum. Clinical studies on many such potential “prodrug esters” such as cefcanel daloxate (Kyoto), cefdaloxime pentexil tosilate (Hoechst Marion Roussel) and ceftrazonal bopentil (Roche), to name a few have been discontinued, while ceftizoxime alapivoxil ((Kyoto) in under Phase III clinical studies. The cephalosporin prodrug esters which have been successfully commercialised and marketed include cefcapene pivoxil (Flomox® , Shionogi), cefditoren pivoxil (Spectracef®, Meiji Seika), cefetamet pivoxil (Globocef®, Roche), cefotiam hexetil (Taketiam®, Takeda), cefpodoxime proxetil (Vantin®, Sankyo), cefteram pivoxil (Tomiron®, Toyama) and cefuroxime axetil (Ceftin® and Zinnat®, Glaxo Wellcome).
[0004]
Typically, such (3,7)-substituted-3-cephem-4-carboxylic acid esters represented by formula (I A) are synthesised by reacting the corresponding (3,7)-substituted-3- cephem-4-carboxylic acid derivative of formula (III A), with the desired haloester compound of formula (IV A) in a suitable organic solvent. The synthesis is summarised in Scheme-I, wherein in compounds of formula (I A), (II A), (III A) and (IV A) the groups R1 and R2 at the 3- and 7-positions of the β-lactam ring are substituents useful in cephalosporin chemistry ; R3 is the addendum which forms the ester function and X is halogen.
[0005]
However, the esterification reaction which essentially involves conversion of a polar acid or salt derivative to a neutral ester product invariably produces the corresponding (3,7)-substituted-2-cephem (Δ2)-4-carboxylic acid ester derivative of formula (II A) in varying amounts, arising out of isomerisation of the double bond from the 3-4 position to the 2-3 position as well as other unidentified impurities.
[0006]
It has been suggested [D. H. Bentley, et. al., Tetrahedron Lett., 1976, 41, 3739] that the isomerisation results from the ability of the 4-carboxylate anion of the starting carboxylic acid to abstract a proton from the 2-position of the 3-cephem-4-carboxylic acid ester formed, followed by reprotonation at 4-position to give the said Δ2-ester. It has also been suggested [R. B. Morin, et. al., J. Am. Chem. Soc., 1969, 91, 1401 ; R. B. Woodward, et. al., J. Am. Chem. Soc., 1966, 88, 852] that the equilibrium position for isomerisation is largely determined by the size of the ester addendum attached at the 4-carboxylic acid position.
[0007]
The 2-cephem-4-carboxylic acid esters of formula (II A) are not only unreactive as antibacterial agents but are undesired by-products. Pharmacopoeias of many countries are very stringent about the presence of the 2-cephem analogues in the finished sample of (3,7)-substituted-3-cephem-4-carboxylic acid esters and set limits for the permissible amounts of these isomers. Due to the structural similarity of the 2-cephem and 3-cephem analogues it is very difficult to separate the two isomers by conventional methods, such as chromatography as well as by fractional crystallisation. In addition to this removal of other unidentified impurities formed in the reaction, entails utilisation of tedious purification methods, thus overall resulting in,
a) considerable loss in yield, increasing the cost of manufacture and
b) a product of quality not conforming to and not easily amenable for upgradation to pharmacopoeial standards.
[0008]
Several methods are reported in the prior art for synthesis of cefuroxime axetil of formula (I) and various (3,7)-substituted-3-cephem-4-carboxylic acid esters of formula (I A), with attempts to minimise the unwanted Δ2-isomers formed in such reactions as well as conversion of the Δ2-isomer thus formed back to the desired Δ3– isomer. The prior art methods can be summarised as follows:
i) US Patent No, 4 267 320 (Gregson et. al.) describes a method for synthesis of cefuroxime axetil comprising reaction of cefuroxime acid or its alkali metal salts or onium salts with (R,S)-1-acetoxyethyl bromide in an inert organic solvent selected from N,N-dimethylacetamide, N,N-dimethylformamide, dimethyl sulfoxide, acetone, acetonitrile and hexamethylphosphoric triamide at a temperature in the range of -50 to +1150° C. The patent mentions that when alkali metal salts, specially potassium salt of cefuroxime acid are employed the reaction can be carried out in a nitrile solvent in the presence of a crown ether. When cefuroxime acid is employed the reaction is carried out in the presence of a weak inorganic base such as sodium carbonate or potassium carbonate, which is added prior to the addition of the haloester. The patent further mentions that the use of potassium carbonate in conjunction with the haloester, specially the bromo or iodo ester is preferred since it helps to minimise the formation of the Δ2-isomer. Ideally, substantially equivalent amounts of cefuroxime acid and the base is employed.
The US Patent No. 4 267 320 also describes methods, wherein the said esterification is carried out in the presence of an acid binding agent, which serve to bind hydrogen halide liberated in the reaction, thereby controlling the formation of the Δ2-isomer. The acid binding agents that are utilised include a tertiary amine base such as triethylamine or N, N-dimethylamine ; an inorganic base such as calcium carbonate or sodium bicarbonate and an oxirane compound such as ethylene oxide or propylene oxide.
However, from the examples provided in the above patent the yield of cefuroxime axetil and other (3,7)-substituted-3-cephem-4-carboxylic acid esters obtained is found to be only of about 50%, implying formation of substantial amounts of impurities in the reaction. Indeed, when cefuroxime acid is reacted with (R,S)-1-acetoxyethyl bromide in the presence of 0.55 molar equivalents of sodium carbonate or potassium carbonate in N,N-dimethylacetamide as solvent, as per the process disclosed in this patent, it is found that substantial amounts of the Δ2-isomer in a proportion ranging from 10-22% is formed, in addition to other unknown impurities. Also, substantial amounts of the starting cefuroxime acid remains unreacted even after 5 hrs of reaction. Isolation of the product generally affords a gummy material, which resists purification even after repeated crystallisations.
Moreover, the use of the acid binding agents mentioned in the above patent, specially tertiary amines and inorganic bases lead to cleavage of the β-lactam ring and also promote the undesired Δ2-isomerisation, thereby enhancing the level of impurities formed in the reaction.
ii) GB Patent No. 2 218 094 describes a method by which the Δ2-isomers formed during esterification can be converted back to the desired Δ3-isomers. The method comprises of oxidation of the dihydrothiazine ring in the mixture of Δ2– and Δ3– cephalosporin acid esters to the corresponding sulfoxide derivatives with suitable oxidising agents, whereby the Δ2-isomer gets isomerised to the corresponding Δ3-isomer during oxidation and the Δ3– cephalosporin acid ester sulfoxide is isolated. The sulfide group is regenerated back by reduction of the sulfoxide function with suitable reducing agents.
Typically, the oxidation is carried out using m-chloroperbenzoic acid and the reduction achieved by use of an alkali metal halide in presence of acetyl chloride in presence of an inert organic solvent or by use of a phosphorous trihalide.
Although, this method provides the desired Δ3-isomers in good purity, it cannot be considered as an industrially feasible method since it involves a two step process of oxidation and reduction, isolation of the intermediate products at each stage and necessary purifications, all resulting in considerable loss of the desired product and increase in the cost of manufacture. Moreover, the use of acetyl halide and phosphorous trihalide in the reduction step cannot be applied to cephalosporin derivatives that are sensitive to these reagents.
A similar method has been reported by Kaiser et. al. in J. Org. Chem., 1970, 35, 2430.
(iii)Mobasherry et. al. in J. Org. Chem., 1986, 51, 4723 describe preparation of certain Δ3-cephalosporin-4-carboxylic acid esters by reaction of the corresponding 3-cephem-4-carboxylic acids (in turn prepared form the corresponding carboxylic acid alkali metal salts) with an haloester in presence of 1.1 eq of sodium carbonate in the presence 1.2-1.5 eq of an alkyl halide and in presence of a solvent comprising of a mixture of N,N-dimethylformamide and dioxane. The authors claim that the method provides of Δ3– cephalosporin-4-carboxylic acid esters unaccompanied by the corresponding Δ2-isomer.
However, the method involves an additional step in that the starting 3-cephem-4-carboxylic acid ester derivatives are obtained from the corresponding alkali metal salts prior to reaction. In addition, longer reaction times of about 24 hrs coupled with the fact that it utilises dioxane, a potent carcinogen, not recommended by International Conference on Harmonisation (ICH) on industrial scale renders the method unattractive commercially.
Moreover, on duplication of the method exactly as described in the article it is found that about 3-4% of the corresponding Δ2-isomer is indeed formed in the reaction in addition to other unidentified impurities. Also, substantial amounts of the starting cephalosporin carboxylic acid is recovered unreacted.
(iv)Shigeto et. al. in Chem. Pharm. Bull., 1995, 43(11), 1998 have carried out the esterification of certain 7-substituted-3-cephem-4-carboxylic acid derivatives with 1-iodoethyl isopropyl carbonate in a solvent system containing a mixture of N, N-dimethylformamide and dioxane in a 3:5 ratio. A conversion to the corresponding 3-cephem- 4-carboxylate ester was achieved in only 34%, out of which the Δ2-isomer amounted to about 8%.
Esterification of 7-formamido-3-(N,N-dimethylcarbamoyloxy)methyl-3-cephem-4-carboxylic acid sodium salt with a suitable haloester in presence of solvents such as N, N-dimethylacetamide and N, N-dimethylformamide, with formation of about 0.8 to 3.0% of the Δ2-isomer is also reported in the above article by Shigeto et. al. The 7-formamido group was cleaved under acidic conditions to give the corresponding 7-amino derivative contaminated with only about 0.4% of the corresponding Δ2-isomer. The minimisation of the percentage of Δ2-isomer is attributed to the relative unstability of 7-amino-2-cephem-4-carboxylic acid esters in acidic conditions, facilitating isomerisation of the 2-cephem intermediate to the 3-cephem derivative.
However, the method does not have a general application, especially for synthesis of commercially valuable cephalosporin derivatives containing hydroxyimino or alkoxyimino substituents in the 7-amino side chain addendum, since these oxyimino functions exhibit a tendency to isomerise from the stable (Z)-configuration to the relatively undesirable(E)-configuration under acidic conditions. This would render separation of the two isomers cumbersome. Moreover, longer reaction times of about 18-20 hrs to effect the isomerisation of the double bond from the 2- position to the 3-position and use of toxic dioxane as solvent impose further limitations on the method.
(v) Demuth et. al. in J. Antibiotics, 1991, 44, 200 have utilised the N, N-dimethylformamide-dioxane system in the coupling of 1-iodocephem-4-nitrobenzyl ester with naldixic acid sodium salt and recommend use of dioxane since it reduces the basicity of the quinolone carboxylate and lowers the polarity of the reaction medium.
However, low yields of about 35% and use of toxic dioxane makes the method of little industrial application.
(vi) Wang et. al. in US Patent No. 5 498 787 claim a method for preparation of certain (3,7)-substituted-3-cephem-4-carboxylic acid prodrug esters, unaccompanied by the analogous 2-cephem esters comprising reaction of the corresponding (3,7)-substituted-3-cephem-4-carboxylic acid alkali metal salts with suitable haloesters in the presence of catalytic amounts of a quaternary ammonium or quarternary phosphonium salt. Among the prodrug esters covered in this patent is cefuroxime axetil. US Patent No. 5 498 787 claims that among the quarternary ammonium salts, such salts with acid counter ion, specially tetrabutyl ammonium sulfate (TBA+HSO4–) is the most preferred. When the molar ratio of TBA+HSO4–/cefuroxime sodium was above 0.40 no Δ2-isomer was detected, when the said molar ratio was below 0.40 and near about 0.20 the molar ratio of Δ2/Δ3 isomers formed was about 2.0%. When no TBA+HSO4– was added the molar ratio of Δ2/Δ3 isomers formed was about 10.0%. Examples 1 and 2 of this patent illustrate the esterification of cefuroxime sodium in presence of TBA+HSO4– and indicate that the Δ2-isomer was not detected after 3-12 hours of reaction. The same patent also establishes the superiority of TBA+HSO4– over other salts, specially tetrabutyl ammonium iodide (TBA+I–) since use of the latter salt resulted in considerable isomerisation of the double bond giving the undesired Δ2-isomer in predominant amounts.
The present inventors have, however, found that when cefuroxime sodium is reacted with (R,S)- 1-acetoxyethyl bromide in the presence of tetrabutylammonium sulfate (TBA+HSO4–) as per the method covered in US Patent No. 5,498 787 the same did not necessarily result in the production of the desired Δ3isomer free of the undesired Δ2 isomer and other impurities. Also, such process had limitations in that the reaction could not be completed at times even at the end of 5.0hrs. Moreover, the separation of the impurities; from the product proved cumbersome and could not be removed from the product even after successive crystallisations.
(vii) H. W. Lee et. al., Syntheic Communications, 1998, 28(23), 4345-4354 have demonstrated a method essentially similar to that claimed in US Patent No. 5 498 787 . The method of preparation of various esters of cefotaxime consists of reacting cefotaxime sodium with the requisite haloester compound in a suitable solvent and in presence of quarternary ammonium salts as phase transfer catalysts. It is claimed that when no quarternary ammonium salts are added the molar ratio (%) of Δ2/Δ3 isomers formed is about 10%. The formation of Δ2– isomer is minimised when quarternary ammonium salts are added and particularly when the molar ratio of TBA+HSO4–/cefotaxime sodium employed is 0.80 the formation of the Δ2– isomer is completely inhibited.
However, this method requires long hours (~18-24 hrs) and is carried out at higher temperatures (40-45° C) and as such may not be suitable for cephalosporin derivatives that are sensitive to heat.
(viii)H. W. Lee et. al. in Synthetic Communications, 1999, 29(11), 1873-1887 demonstrate a method for preparation of number of (3,7)-substituted-3-cephem-4-carboxylic acid esters comprising reacting the corresponding (3,7)-substituted-3-cephem-4-carboxylic acid derivatives with a base selected form cesium carbonate or cesium bicarbonate either used alone or in combination with potassium carbonate, sodium carbonate, potassium bicarbonate and sodium bicarbonate. The authors established that the formation of Δ2– isomers could be minimised by utilisation of a solvent combination ofN, N-dimethyl formamide and dioxane. The use of the latter mentioned solvent i. e. dioxane was expected to lower polarity of the reaction medium and thereby reduce the basicity of the transient 3-cephem-4-carboxylate anion formed in the reaction and thus preventing the isomerisation of the double bond from the 3-4 position to the 2-3 position.
The formation of the Δ2– isomer was found to be dependent on the amount of dioxane in the solvent mixture, the more the proportion of dioxane lesser the degree of isomerisation.
However, yields of representative esters obtained by the method are in the range of 45-85 %, implying that the reaction is accompanied by formation of substantial amounts of impurities and that the isomerisation is dependent on the nature of the substituent at 3α-position of the cephalosporin nucleus as well as on the nature of the haloester employed. Moreover, the method utilises dioxane, not desirable for reasons mentioned herein earlier and expensive cesium salts. This method, therefore, also has limited application.
(ix) Y.S. Cho et. al., in Korean J. Med. Chem., 1995, 5(1), 60-63 describe synthesis of several cephalosporin prodrug esters and their efficacy on oral administration. The esters were synthesised by reacting the corresponding cephalosporin-4-carboxylic acid derivative with the respective haloester derivative in presence of cesium carbonate and N, N-dimethylacetamide. The yields of the ester derivatives obtained are in the range of only 25-56%, indicating formation of substantial amounts of impurities in the reaction.
Example – 1
Preparation of (R, S -1-Acetoxyethyl-3-carbamoyloxymethyl-7-[(Z)-2-(fur-2-yl)-2-methoxyiminoacetamido]ceph-3-em-4-carboxylate (Cefuroxime axetil, I) :
Without use of GrouplI
/
II metal phosphate and C1-4 alcohol
[0045]
(R, S)-1-Acetoxyethyl bromide (1.6gms; 0.0094moles) was added to a mixture of cefuroxime acid (2gms; 0.0047moles) and potassium carbonate (0.326gms; 0.00235moles) in N,N-dimethylacetamide (10 ml) at 5°C and stirred at 0 to 20° C for 180 minutes Ethyl acetate was added to the reaction mixture, followed by 3% aqueous sodium bicarbonate solution (15ml). The organic layer containing the title product, Δ2 isomer (8.51%) and unidentified impurities (X1-1.86% and X2 – 3.54%) was separated and washed with 10% aqueous NaCl solution. The organic solvent was evaporated off under vacuum to give 1.08gms (44.90%) of the title compound as a gummy solid.
The reaction of 3-hydroxymethyl-7- [2- (2-furyl) -2-methoxyiminoacetamido] -3-cephem-4-carboxylic acid (I) with chlorosulfonyl isocyanate (II) and then with sodium 2-ethylhexanoate gives sodium cefuroxime (III), which is then treated with 1-bromoethyl acetate (IV) in DMA.
















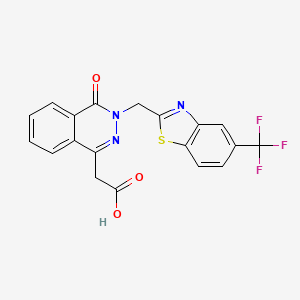
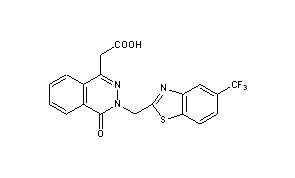









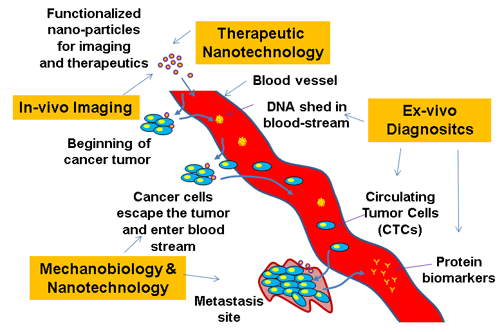



















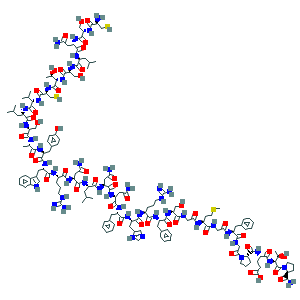
 cas 57014-02-5
cas 57014-02-5