Synthesis offers potential routes to analogues of vitamin-D-based drug
Paricalcitol, an A-ring-modified 19-nor analogue of 1α,25-dihydroxyvitamin D2, is currently used for the treatment and prevention of secondary hyperparathyroidism associated with chronic renal failure.
Read more
http://www.chemistryviews.org/details/ezine/6508291/New_Route_to_Paricalcitol.html

Zemplar; 131918-61-1; 19-Nor-1alpha,25-dihydroxyvitamin D2; Compound 49510; Paracalcin; Zemplar (TN); 19-Nor-1,25-(OH)2D2; CHEBI:7931
Molecular Formula: C27H44O3 Molecular Weight: 416.63646
Abbott (Originator), Tetrionics (Bulk Supplier)

launched 1998
(1R,3R)-5-[(2E)-2-[(1R,3aS,7aR)-1-[(E,2R,5S)-6-hydroxy-5,6-dimethylhept-3-en-2-yl]-7a-methyl-2,3,3a,5,6,7-hexahydro-1H-inden-4-ylidene]ethylidene]cyclohexane-1,3-diol
For treatment of secondary hyperparathyroidism associated with chronic kidney disease (CKD) Stage 3 and 4
Paricalcitol (chemically it is 19-nor-1,25-(OH)2-vitamin D2. Marketed by Abbott Laboratories under the trade name Zemplar) is a drugused for the prevention and treatment of secondary hyperparathyroidism (excessive secretion of parathyroid hormone) associated withchronic renal failure. It is an analog of 1,25-dihydroxyergocalciferol, the active form of vitamin D2 (Ergocalciferol).
Paricalcitol is a synthetic vitamin D analog. Paricalcitol has been used to reduce parathyroid hormone levels. Paricalcitol is indicated for the prevention and treatment of secondary hyperparathyroidism associated with chronic renal failure.

Medical uses
Its primary use in medicine is in the treatment of secondary hyperparathyroidism associated with chronic kidney disease.[2] In three placebo-controlled studies, chronic renal failure patients treated with paricalcitol achieved a mean parathyroid hormone (PTH) reduction of 30% in six weeks. Additionally there was no difference in incidence of hypercalcemia or hyperphosphatemia when compared to placebo.[3] A double-blind randomised study with 263 dialysis patients showed a significant advantage over calcitriol (also known as activated vitamin D3; a similar molecule to 1,25-dihydroxyergocalciferol, adding a methyl group on C24 and lacking a double-bond in the C22 position). After 18 weeks, all patients in the paricalcitol group had reached the target parathormone level of 100 to 300 pg/ml, versus none in the calcitriol group.[4] Combination therapy with paricalcitol and trandolapril has been found to reduce fibrosis inobstructive uropathy.[5] Forty-eight week therapy with paricalcitol did not alter left ventricular mass index or improve certain measures of diastolic dysfunction in 227 patients with chronic kidney disease.[6]
Patents
| Country |
Patent Number |
Approved |
Expires (estimated) |
| United States |
6136799 |
1998-10-08 |
2018-10-08 |
| United States |
5246925 |
1995-04-17 |
2012-04-17 |
Mechanism of action

3D structure of paricalcitol
Like 1,25-dihydroxyergocalciferol, paricalcitol acts as an agonist for the vitamin D receptor and thus lowers the bloodparathyroid hormone level.[1]
Pharmacokinetics
Within two hours after administering paricalcitol intravenous doses ranging from 0.04 to 0.24 µg/kg, concentrations of paricalcitol decreased rapidly; thereafter, concentrations of paricalcitol declined log-linearly. No accumulation of paricalcitol was observed with multiple dosing.[9]
vitamin D is a fat-soluble vitamin. It is found in food, but also can be formed in the body after exposure to ultraviolet rays. Vitamin D is known to exist in several chemical forms, each with a different activity. Some forms are relatively inactive in the body, and have limited ability to function as a vitamin. The liver and kidney help convert vitamin D to its active hormone form. The major biologic function of vitamin D is to maintain normal blood levels of calcium and phosphorus. Vitamin D aids in the absorption of calcium, helping to form and maintain healthy bones.
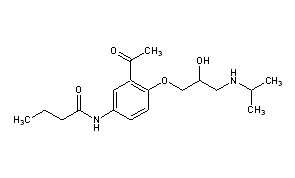
The 19-nor vitamin D analogue, Paricalcitol (I), is characterized by the following formula:
In the synthesis of vitamin D analogues, a few approaches to obtain a desired active compound have been outlined previously. One of the methods is the Wittig-Homer attachment of a 19-nor A-ring phosphine oxide to a key intermediate bicyclic-ketone of the Windaus-Grundmann type, to obtain the desired Paricalcitol, as is shown for example in U.S. Pat. Nos. 5,281,731 and 5,086,191 of DeLuca.
The synthesis of Paricalcitol requires many synthetic steps which produce undesired by-products. Therefore, the final product may be contaminated not only with a by-product derived from the last synthetic step of the process but also with compounds that were formed in previous steps. In the United States, the Food and Drug Administration guidelines recommend that the amounts of some impurities be limited to less than 0.1 percent.
U.S. Pat. Nos. 5,281,731 and 5,086,191 of DeLuca disclose a purification process of Paricalcitol by using a HPLC preparative method.
As the unwanted products have almost the same structure as the final product, it may difficult to get a sufficiently pure drug substance, vitamin D analogue, using this route to purify the drug substance. Moreover, the high polarity of Paricalcitol makes it very difficult to purify by HPLC and to recover the solid product. Furthermore, HPLC preparative methods are generally not applicable for use on industrial scale. There remains a need in the art to provide a method of preparing the vitamin D analogue Paricalcitol in a sufficiently pure form which is applicable for use on an industrial scale.
Paricalcitol (chemical name: 19-nor-1α,3β,25-trihydroxy-9,10-secoergosta-5(Z),7(Z),22(E)-triene; Synonyms: 19-nor-1,25-dihydroxyvitamin D2, Paracalcin) is a synthetic, biologically active vitamin D analog of calcitriol with modifications to the side chain (D2) and the A (19-nor) ring. Paricalcitol inhibits the secretion of parathyroids hormone (PTH) through binding to the vitamin D receptor (D. M. Robinson, L. J. Scott, Drugs, 2005, 65 (4), 559-576) and it is indicated for the prevention and treatment of secondary hyperparathyroidism (SHPT) in patients with chronic kidney disease (CKD).
Paricalcitol is marketed under the name Zemplar®, which is available as a sterile, clear, colorless, aqueous solution for intravenous injection (each mL contains 2 microgram (2 μg) or 5 μg paricalcitol as active ingredient) or as soft gelatin capsules for oral administration containing 1 μg, 2 μg or 4 μg paricalcitol.
The molecular formula of paricalcitol is C27H44O3 which corresponds to a molecular weight of 416.65. It is a white, crystalline powder and has the following structural formula:
Historically, nor-vitamin D compounds were described in 1990 as a new class of vitamin D analogs wherein the exocyclic methylene group C(19) in ring A has been removed and replaced by two hydrogen atoms (see e.g. WO 90/10620). So far, two different routes have been discovered for the synthesis of such 19-nor-vitamin analogs which specifically may be used for the preparation of paricalcitol.
The first synthesis of paricalcitol is disclosed in WO 90/10620 (additional patents from patent family: EP patent no. 0 387 077, U.S. Pat. No. 5,237,110, U.S. Pat. No. 5,342,975, U.S. Pat. No. 5,587,497, U.S. Pat. No. 5,710,294 and U.S. Pat. No. 5,880,113) and generally described in Drugs of the Future, 1998, 23, 602-606.
Example 3 of WO 90/10620 provides the preparation of 1α,25-dihydroxy-19-nor-vitamin D2 (Scheme 1) by using experimental conditions analogous to the preparation of 1α,25-dihydroxy-19-nor-vitamin D3. According to this description the starting material 25-hydroxyvitamin D2 is first converted to 1α,25-dihydroxy-3,5-cyclovitamin D2 (a2) using the procedures published by DeLuca et al. in U.S. Pat. No. 4,195,027 and Paaren et al. published in J. Org. Chem., 1980, 45, 3252. Acetylation of compound a2 followed by dihydroxylation of the exocyclic methylene group using osmium tetroxide in pyridine gives the 10,19-dihydroxy compound a4 which is converted with sodium metaperiodate (diol cleavage) to the 10-oxo-intermediate a5. Reduction of the 10-oxo group in a5 is carried out by treatment with sodium borohydride in a mixture of ethanol and water giving the corresponding 10-hydroxy derivative a6. Mesylation of the 10-hydroxy group in a6 (→a7) followed by reduction with lithium aluminium hydride in THF gives the 10-deoxy intermediate a8 wherein the 1-OAcyl group was simultaneously cleaved during the reduction step. Solvolysis (cycloreversion) of a8 by treatment with hot (55° C.) acetic acid results in the formation of two monoacetates (a9 and a10) which are separated and purified by using HPLC. Finally both monoacetates are saponified with aqueous potassium hydroxide in methanol yielding paricalcitol which is purified by HPLC.
The preparation of paricalcitol according to the method provided in WO 90/10620 has several drawbacks:
-
- (1) the starting material 25-hydroxyvitamin D2 is one of the major metabolites of vitamin D2 and not readily available in larger amounts. Additional efforts have to be made in order to synthesize the starting material in sufficient amounts resulting in a protractive and unattractive total synthesis of paricalcitol. Examples for the preparation of 25-hydroxyvitamin D2 are described e.g. in U.S. Pat. No. 4,448,721; WO 91/12240; Tetrahedron Letters, 1984, 25, 3347-3350; J. Org. Chem., 1984, 49, 2148-2151 and J. Org. Chem., 1986, 51, 1264-1269;
- (2) the use of highly toxic osmium tetroxide which requires special precaution for its handling;
- (3) use of HPLC for separation of isomers and purification of the final compound. As teached in WO 2007/011951 paricalcitol is difficult to purify by HPLC and as a preparative method HPLC is generally not applicable for use on industrial scale;
- (4) the yields for the preparation of paricalcitol are not described in WO 90/10620. Generally, the provided yields for the preparation of the analogue compound 1α,25-dihydroxy-19-nor-vitamin D3 are very low especially for the corresponding steps 7 to 11 (yield starting from 1α,25-dihydroxy-10-oxo-3,5-cyclo-19-nor-vitamin D3 1-acetate which is the vitamin D3 analogue to a5 in Scheme1: step 7: 63.4%, steps 8-10: 10.7%, step 11: 51.7%; overall yield starting with step 7: 3.5%).


Another strategy for synthesizing 19-nor vitamin D compounds is disclosed in EP 0 516 410 (and corresponding U.S. Pat. No. 5,281,731, U.S. Pat. No. 5,391,755, U.S. Pat. No. 5,486,636, U.S. Pat. No. 5,581,006, U.S. Pat. No. 5,597,932 and U.S. Pat. No. 5,616,759). The concept is based on condensing of a ring-A unit, as represented by structure b1 (Scheme 2), with a bicyclic ketone of the Windaus-Grundmann type, structure b2, to obtain 19-nor-vitamin D compound (b3).

Specific methods for synthesizing compounds of formula b1 are shown in Schemes 3, 4 and 5. According to Scheme 3, the route starts with the commercially available (1R,3R,4R,5R)(−)quinic acid (b4). Esterification of b4 with methanol followed by protection of the l- and 3-hydroxygroup using tert.-butyldimethylsilyl chloride (TBDMSCl) gives compound b5. Reduction of the ethyl ester in b5 yields b6 which is subjected to a diol cleavage giving compound b7. The 4-hydroxy group is protected as trimethylsilylether resulting in the formation of b8 which is further converted in a Peterson reaction with ethyl (trimethylsilyl)acetate before being deprotected with dilute acetic acid in tetrahydrofurane (THF). The resulting compound b9 is treated with 1,1-thiocarbonyldiimidazole to obtain b10. Subsequent reaction with tributyltin hydride in the presence of a radical initiator (AIBN) gives b11. Compound b11 is then reduced with DIBAH to the allylalcohol b12 which is then reacted with NCS and dimethyl sulfide giving the allylchloride b13. Finally the ring A synthon b14 is prepared by treatment of the allychloride b13 with lithium diphenylphosphide followed by oxidation with hydrogen peroxide.
In an alternative method for synthesizing the ring A unit (Scheme 3), the intermediate b5 can be also subjected to radical deoxygenation using analogues conditions as previously described, resulting in the formation of b16. Reduction of the ester (→b17), followed by diol cleavage (→b18) and Peterson reaction gives intermediate b11 which can be further processed to b14 as outlined in Scheme 3.
Another modification for the preparation is shown in Scheme 5. As described, b7 can be also subjected to the radical deoxygenation yielding intermediate b18 which can be further processed to b14 as depicted in Schemes 3 and 4.
In EP 0 516 411 (and its counterpart, U.S. Pat. No. 5,086,191) is disclosed the preparation of intermediates useful for the synthesis of 19-nor vitamin D compounds (Scheme 6). The key step is the condensation of compounds c1 which can be prepared in an analogous manner as previously described for e.g. b14 (Scheme 3) with compounds c2, resulting in compounds of formula c3.
EP 0 516 411 discloses that Grignard coupling of hydroxy-protected 3-hydroxy-3-methylbutylmagnesium bromide with compound c5 (Scheme 7) can give hydroxy-protected 1α,25-dihydroxy-19-nor vitamin D3 or coupling of the corresponding 22-aldehyde c3 (X1=X2=TBDMS, R1=—CHO) with 2,3-dimethylbutyl phenylsulphone can give after desulfonylation, 1α-hydroxy-19-norvitamin d2 in hydroxy-protected form.
An additional method for preparation of 1α-hydroxy-19-nor-vitamin D compounds is provided in EP 0 582 481 (and corresponding U.S. Pat. No. 5,430,196, U.S. Pat. No. 5,488,183, U.S. Pat. No. 5,525,745, U.S. Pat. No. 5,599,958, U.S. Pat. No. 5,616,744 and U.S. Pat. No. 5,856,536) (Scheme 8). Similar to the strategy as described above and shown in schemes 3 to 7, the basis for preparing 1α-hydroxy-19-nor-vitamin D compounds is an independent synthesis of ring A synthon and ring C/D synthon which are finally coupled resulting in vitamin analogs.
Thus the synthesis of 1α-hydroxy-19-nor-vitamin D compounds comprises the coupling of either the ketone d1 with the acetylenic derivatives d2 or ketone d4 with acetylenic derivatives d3, yielding compounds of formula d5. Partial reduction of the triple bond giving d6 followed by reduction using low-valent titanium reducing agents results in the formation of 7,8-cis and 7,8-trans-double bond isomers (d7). Compounds of formula d7 can be also obtained directly from d5 by reaction of d5 with a metal hydride/titanium reducing agent. The isomeric mixture of compounds of formula d7 may be separated by chromatography to obtain separately the 7,8-trans-isomer. The 7,8-cis-isomer of structure d7 can be isomerized to yield the corresponding 7,8-trans-isomer. Finally any protecting groups, if present, can be then removed to obtain 1α-hydroxy-19-nor-vitamin D compounds.

The main disadvantage of the strategies as shown in Schemes 3 to 8 is the fact that ring A as well as ring C/D of the vitamin D derivative has to be separately synthesized before coupling them to compounds like 1α-hydroxy-nor-vitamin D or a protected precursor thereof. According to literature procedure, the ring fragment C/D can be prepared from vitamin D2 by ozonolysis (see e.g. J. C. Hanekamp et al., Tetrahedron, 1992, 48, 9283-9294) from which the ring A is cleaved (and disposed). This fragment has then to be separately synthesized e.g. by using other sources or starting materials like quinic acid in up to 10 steps or more. Therefore such strategies for the total synthesis of 1α-hydroxy-nor-vitamin D compounds become protractive and unattractive for large scale and according to the procedures provided in these patents, the final compounds are obtained only in amounts of <10 mg and in most cases even <1 mg.
Paricalcitol is an active Vitamin D Analog. Paricalcitol is used for the treatment and prevention of secondary hyperparathyroidism associated with chronic kidney disease.

It has been shown to reduce parathyroid hormone levels by inhibiting its synthesis and secretion.
…………………………….
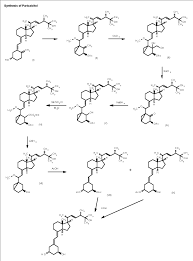
………………………………….

The 25-hydroxyvitamin D2 (I) is converted into the cyclovitamin D2 acetate (II) according to known methods. The dihydroxylation of the methylene group of (II) with OsO4 in pyridine gives vicinal diol (III), which is oxidized with NaIO4 yielding the ketonic cyclovitamin (IV). The reduction of the ketonic group of (IV) with NaBH4 in ethanol/water affords the corresponding hydroxy derivative (V), which is treated with mesyl chloride and triethylamine to give the mesylate (VI). The reduction of (VI) with LiAlH4 in THF yields the 19-nor-cyclovitamin D (VII), which is treated with hot acetic acid to afford both monoacetates (VIII) and (IX), separated by HPLC. Finally, both monoacetates (VIII) and (IX) are hydrolyzed with KOH in methanol.
…………………………
EXAMPLEShttp://www.google.com/patents/US20070149489
|
|
| HPLC method: |
|
|
| Column: |
Hypersyl Gold (250 × 4.6 5 μm) |
| Mobile phase: |
(A) water (95%) |
|
(B) acetonitrile (5%) |
| Gradient: |
From 0 to 10 min (A) isocraticaly |
|
From 10 to 30 min (B) increases from 0 to 55% |
|
From 30 to 40 min (A) isocraticaly |
|
From 30 to 40 min (B) increases from 55 to 100% |
| Detection: |
252 nm |
| Flow: |
2 mL/min |
| Detection limit: |
0.02% |
|
Example 1 Crystallization of Paricalcitol from Acetone
500 mg of Paricalcitol were dissolved in 75 ml of acetone in a sonicator at 28° C. over a period of 15 minutes. The clear solution was filtered through glass wool into another flask, and the solution was then concentrated by evaporation, until the volume was 57.5 ml acetone (control by weight). The solution was cooled to −18° C., and the temperature was maintained at −18° C. for 20 hours. The crystals were filtered and washed with 20 ml of cold (−18° C.) acetone, then dried at high vacuum in an oven at 28° C. for 22 hours to obtain a yield of 390 mg (purity of 98.54%).
………………………………………………………….
http://www.google.com/patents/US20110184199

FIG. 3 is a flow chart showing a detailed example for the synthesis of paricalcitol according to route A1.
FIG. 4 is a flow chart showing the general synthesis of paricalcitol according to route A1.
FIG. 5 is a flow chart showing a detailed example for the synthesis of paricalcitol according to route B1.
FIG. 6 is a flow chart showing the general synthesis of paricalcitol according to route B1.
FIG. 7 is a flow chart showing the general synthesis of paricalcitol using Julia olefination for installation of the side chain according to route B2.
FIG. 8 is a flow chart showing a detailed example for the synthesis of paricalcitol according to route C1.
FIG. 9 is a flow chart showing the general synthesis of paricalcitol according to route C1.
FIG. 10 is a flow chart showing the general synthesis of paricalcitol using Julia olefination for installation of the side chain according to route C2.
Example B11Process Step 12Deprotection of IM-A10b(I) and IM-A10b(II) to Paricalcitol
A mixture consisting of IM-A10b(I) and IM-A10b(II) (41 mg, HPLC purity 54.8%) was dissolved in 1M TBAF in THF (1.5 mL) at temperature 20-25° C. and stirred for 2 h. Then, the reaction mixture was diluted with MeOH (1.5 mL) and 2M aqueous NaOH (0.3 mL) was added. The mixture was stirred for another 2 h and monitored by TLC. Then AcOEt (20 mL) and saturated aqueous NaHCO3 solution (20 mL) were added and the phases separated. The organic phase was washed with brine (20 mL), dried over MgSO4 and concentrated under reduced pressure. The product was purified by column chromatography on silica gel (15 g), with mobile phase cyclohexane/AcOEt (100:0 to 92:8).
Yield 11 mg (81%).
In an additional purification, the product (Paricalcitol, 11 mg) was dissolved in acetone (1 mL) at 35-40° C. The solution was filtered and then cooled to −18° C. to initiate crystallization. The obtained slurry was stirred for 15 min at room temperature (20-25° C.) and again cooled to −18° C. for 3.5 h. The solid material was filtered off, washed with cold (−18° C.) acetone (0.25 mL) and dried in vacuo (6 mbar, 40° C.).
Yield of paricalcitol: 4 mg (36%, HPLC purity 98.3%)
Example C7Process Step 12Hydrolysis of IM-A11a to Paricalcitol
To a solution of IM-A11a(I) and IM-A11a(II) (5.24 g, HPLC-purity 94.2%) in EtOH (80 mL) was added at room temperature (20-25° C.) 2M aqueous NaOH solution (8 mL). The reaction mixture was stirred for 1 h 20 min (TLC monitoring), then EtOAc (8 mL) was added and the mixture was concentrated under reduced pressure to a volume of 40 mL whereupon the crystallization started. Water (50 mL) was added to the suspension and after stirring for 75 min at room temperature the solid was isolated by filtration (pH of the mother liquor measured 8-9). The wet product was slurried in EtOH/H2O (24 g, 1:1) at room temperature, filtered, washed with EtOH/H2O (5 mL, 1:1) and dried (40° C., 10 mbar).
Yield of paricalcitol: 4.26 g (89.5%, HPLC-purity 97.7%).
…………………………………………………..
| US5854390 * |
Feb 6, 1996 |
Dec 29, 1998 |
Lek, Tovarna Farmacevtskih In Kemicnih Izdelkov, D.D. |
Chromatographic purification of vancomycin hydrochloride by use of preparative HPLC |
| US6448421 * |
Jun 16, 1997 |
Sep 10, 2002 |
Chugai Seiyaku Kabushiki Kaisha |
Purifying a crude product derivative through a reverse phase chromatography and then crystallizing from an organic solvent; oxy gonane and indene, cyclohexyl derivatives |
| US20070149489 * |
Jul 18, 2006 |
Jun 28, 2007 |
Anchel Schwartz |
Preparation of paricalcitol |
| US7795459 * |
Apr 28, 2009 |
Sep 14, 2010 |
Alphora Research Inc. |
Paricalcitol purification |
| US20110137058 * |
Feb 15, 2011 |
Jun 9, 2011 |
Formosa Laboratories, Inc. |
Preparation of paricalcitol |
| DE102009013609A1 |
Mar 17, 2009 |
Nov 5, 2009 |
Formosa Laboratories, Inc. |
Herstellung von Paricalcitol |
References
- “Zemplar (paricalcitol) dosing, indications, interactions, adverse effects, and more”. Medscape Reference. WebMD. Retrieved 26 January 2014.
- Rossi, S, ed. (2013). Australian Medicines Handbook (2013 ed.). Adelaide: The Australian Medicines Handbook Unit Trust. ISBN 978-0-9805790-9-3. edit
- “Zemplar: Drug Information”
- Schubert-Zsilavecz, M, Wurglics, M, Neue Arzneimittel 2005/2006 (in German).
- Tan, X; He, W; Liu, Y (2009). “Combination therapy with paricalcitol and trandolapril reduces renal fibrosis in obstructive nephropathy”. Kidney international 76 (12): 1248–57.doi:10.1038/ki.2009.346. PMID 19759524.
- Thadhani, R; Appelbaum, E; Pritchett, Y; Chang, Y; Wenger, J; Tamez, H; Bhan, I; Agarwal, R et al. (2012). “Vitamin D Therapy and Cardiac Structure and Function in Patients With Chronic Kidney Disease – The PRIMO Randomized Controlled Trial”. JAMA 307 (7): 674–684. doi:10.1001/jama.2012.120. PMID 22337679.
- “PARICALCITOL capsule, liquid filled [Teva Pharmaceuticals USA Inc]” (PDF). DailyMed. Teva Pharmaceuticals USA Inc. September 2013. Retrieved 26 January 2014.
- “Zemplar Soft Capsules 1 mcg – Summary of Product Characteristics”. electronic Medicines Compendium. AbbVie Limited. 15 April 2013. Retrieved 26 January 2014.
- Rxlist: Zemplar
-
- Anchel Schwartz, Alexei Ploutno, Koby Wolfman, “Preparation of paricalcitol.” U.S. Patent US20070149489, issued June 28, 2007.US20070149489
| Systematic (IUPAC) name |
| (1R,3R,7E,17β)-17-[(1R,2E,4S)-5-hydroxy-1,4,5-trimethylhex-2-en-1-yl]-9,10-secoestra-5,7-diene-1,3-diol |
| Clinical data |
| Trade names |
Zemplar |
| AHFS/Drugs.com |
monograph |
| MedlinePlus |
a682335 |
| Pregnancy cat. |
|
| Legal status |
|
| Routes |
Oral, Intravenous |
| Pharmacokinetic data |
| Bioavailability |
72%[1] |
| Protein binding |
99.8%[1] |
| Metabolism |
Hepatic[1] |
| Half-life |
14-20 hours[1] |
| Excretion |
Faeces (74%), urine (16%)[1] |
| Identifiers |
| CAS number |
131918-61-1  |
| ATC code |
H05BX02 |
| PubChem |
CID 5281104 |
| IUPHAR ligand |
2791 |
| DrugBank |
DB00910 |
| ChemSpider |
4444552 |
| UNII |
6702D36OG5 |
| |
|
| ChEMBL |
CHEMBL1200622 |
| Synonyms |
(1R,3S)-5-[2-[(1R,3aR,7aS)-1-[(2R,5S)-6-hydroxy-5,6-dimethyl-3E-hepten-2-yl]-7a-methyl-2,3,3a,5,6,7-hexahydro-1H-inden-4-ylidene]ethylidene]-cyclohexane-1,3-diol |
| Chemical data |
| Formula |
C27H44O3 |
| Mol. mass |
416.636 g/mol |
more………….






























































































 just an animation
just an animation