|
|---|
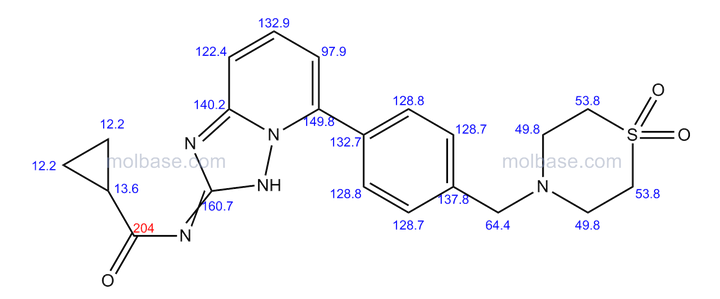
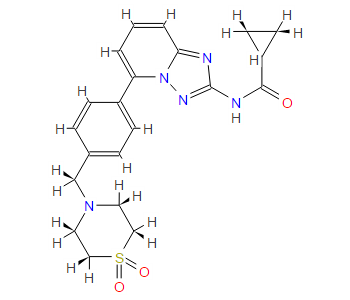
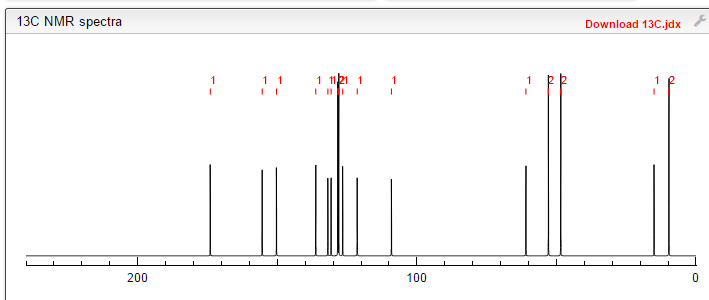
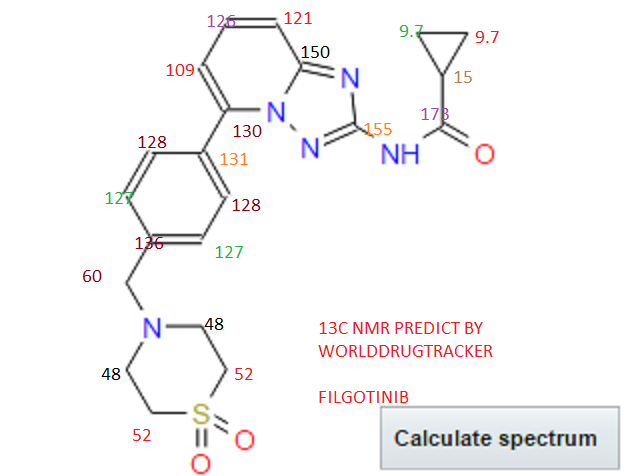
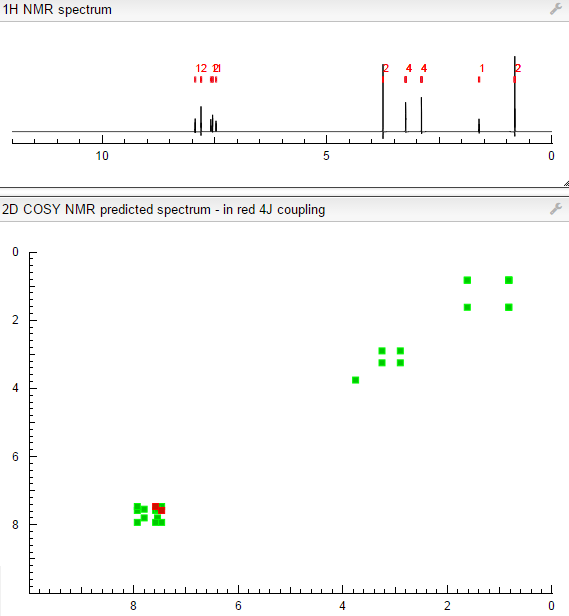
isopropyl (5Z)-7-{(1R,2R,3R,5S)-2-[(1E)-3,3-difluoro-4-phenoxybut-1-en-1-yl]-3,5-dihydroxycyclopentyl}hept-5-enoate,
Drug: Zioptan
Generic molecule: tafluprost
Company: Merck
Approval date: Feb. 10, 2012
The scoop: Merck says this is the first (get ready for a mouthful) preservative-free prostaglandin analog ophthalmic solution and is for treating elevated eye pressure in some patients with the most common form of glaucoma. Merck sells the ointment in the U.S. and most of Europe, while it licensed it to Japanese drugmaker Santen in Japan, Germany and northern Europe.
Tafluprost (trade names Taflotan, marketed by Santen Pharmaceutical Co. and Zioptan, by Merck (U.S.)) is a prostaglandin analogue used topically (as eye drops) to control the progression of glaucoma and in the management of ocular hypertension. It reduces http://en.wikipedia.org/wiki/Intraocular_pressure”; rel=”nofollow”>intraocular pressure by increasing the outflow of aqueous fluid from the eyes.[1][2]
Taflotan contains 15 µg/ml Tafluprost. Taflotan sine is a preservative-free, single-dose formulation containing 0.3 ml per dose.[3]

|
|
| Systematic (IUPAC) name | |
|---|---|
| isopropyl (5Z)-7-{(1R,2R,3R,5S)-2-[(1E)-3,3-difluoro-4-phenoxybut-1-en-1-yl]-3,5-dihydroxycyclopentyl}hept-5-enoate | |
| Clinical data | |
| Trade names | Saflutan, Taflotan, Tapros, Zioptan |
| AHFS/Drugs.com | International Drug Names |
| Pregnancy cat. | C (US) |
| Legal status | ℞-only (US) |
| Routes | Topical (eye drops) |
| Identifiers | |
| CAS number | 209860-87-7 |
| ATC code | S01EE05 |
| PubChem | CID 6433101 |
| ChemSpider | 8044182 |
| UNII | 1O6WQ6T7G3 |
| ChEBI | CHEBI:66899 |
| ChEMBL | CHEMBL1963683 |
| Chemical data | |
| Formula | C25H34F2O5 |
| Mol. mass | 452.531266 g/mol |


- Schubert-Zsilavecz, M, Wurglics, M, Neue Arzneimittel 2008/2009
- Santen Home Page
- Gelbe Liste (in German)

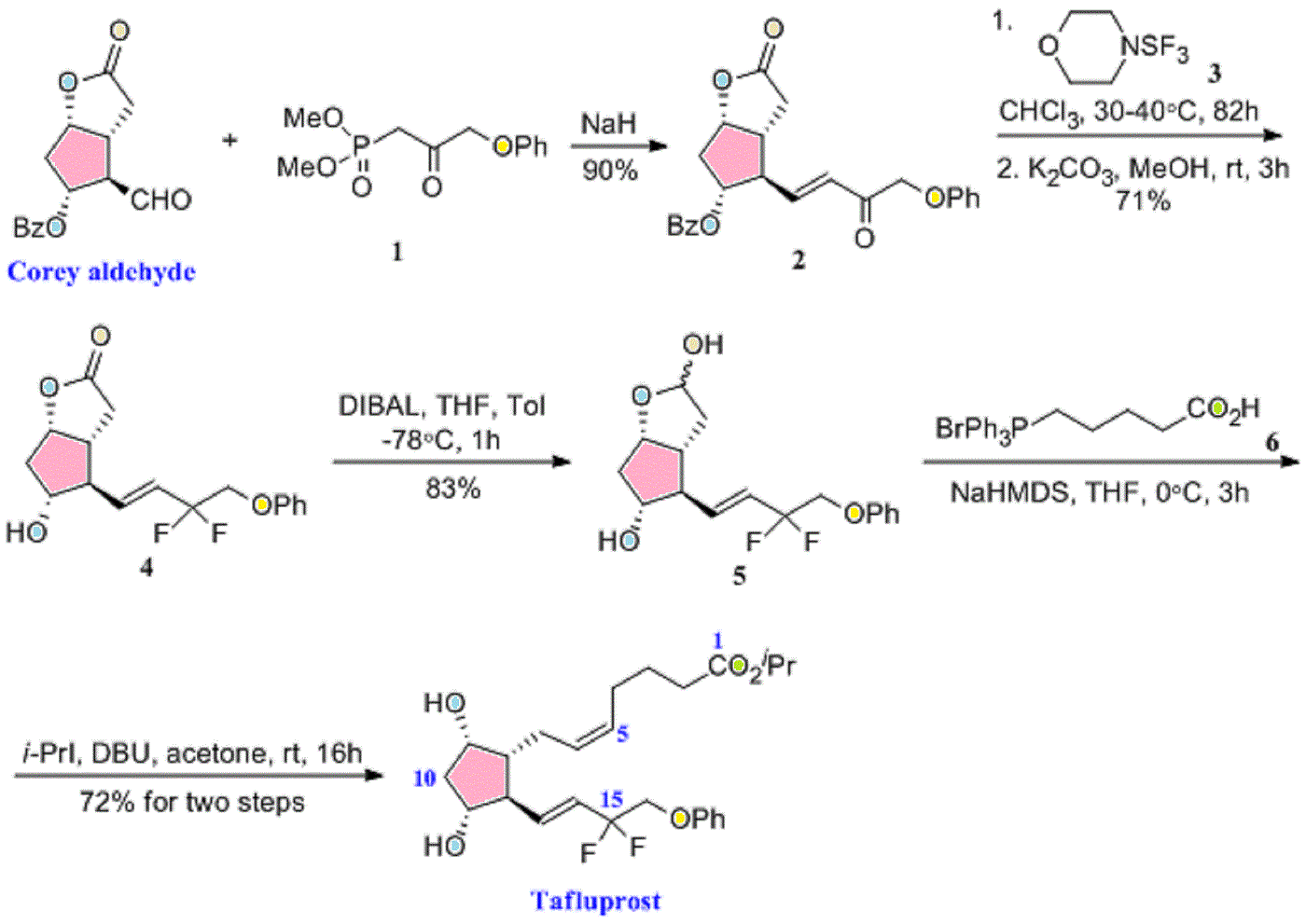
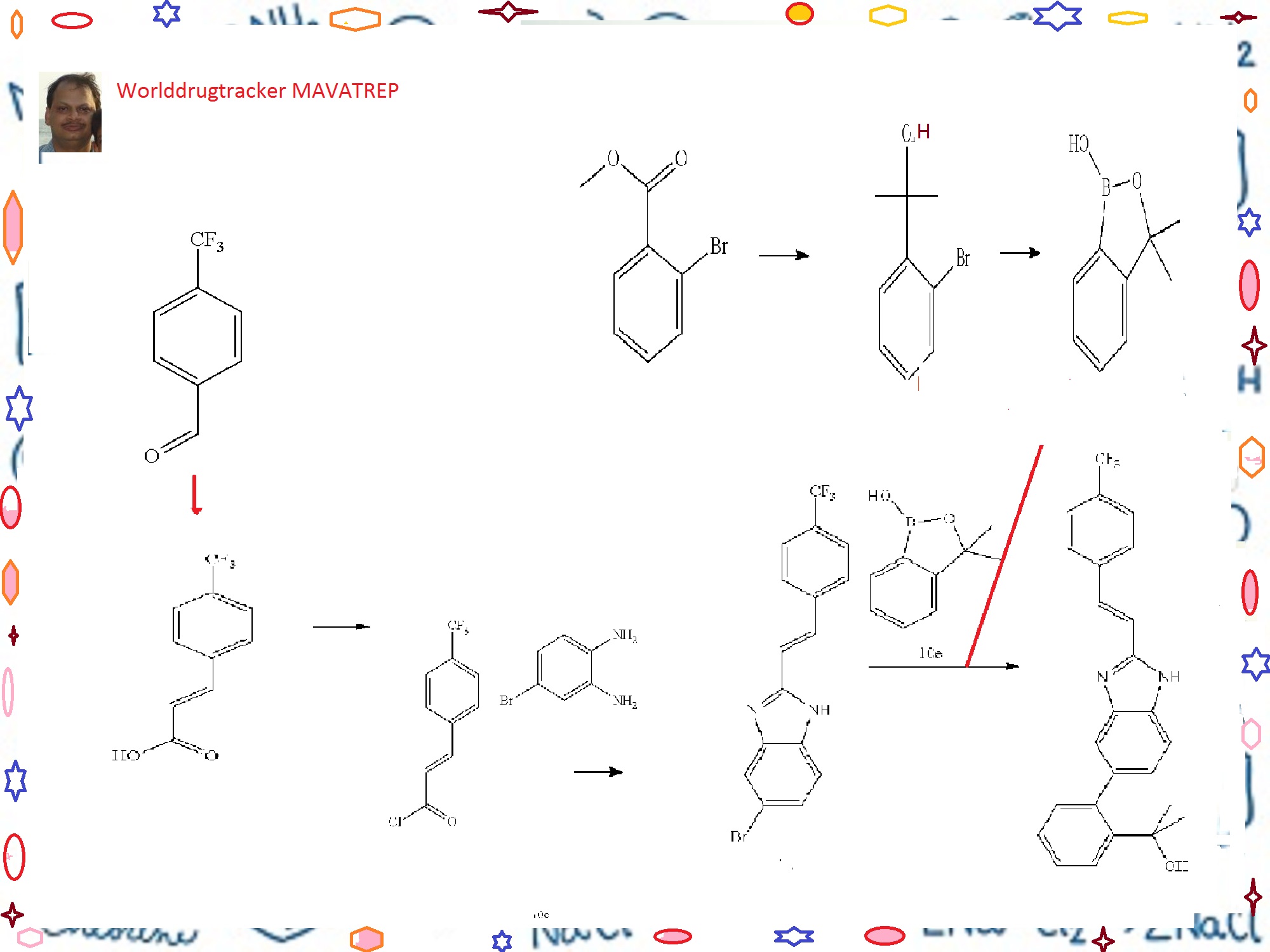
European Patent No. 8509621 discloses a process for the preparation of tafluprost. In the first step, (3afl,4fl,5fl,6aS)-4-formyl-2-oxohexahydro-2 — cyclopenta[b]furan-5-ylbenzoate (CTAF 1 (i)) is condensed with dimethyl (2-oxo-3- phenoxypropyl)-phosphonate in the presence of lithium chloride and triethylamine, to provide (3aft,4F?,5F?,6aS)-2-oxo-4-((£)-3-oxo-4-phenoxybut-1 -en-1 -yl)hexahydro-2H- cyclopenta[b]-furan-5-ylbenzoate (CTAF1 ). In the second step, CTAF 1 is reacted with morpholinosulfurtrifluoride to provide (3aH,4H,5H,6aS)-4-((£)-3,3-difluoro-4- phenoxybut-1 -en-1 -yl)-2-oxohexahydro-2 –cyclopenta-[b]furan-5-yl benzoate (CTAF2). CTAF 2 is debenzoylated by potassium carbonate in methanol, to provide (3aH,4H,5H,6aS)-4-((£)-3,3-difluoro-4-phenoxybut-1 -en-1 -yl)-5-hydroxyhexahydro-2H- cyclopenta[b]furan-2-one(CTAF 3), which is further reduced by diisobutyl aluminum hydride (DIBALH) to provide (3af?,4f?,5f?,6aS)-4-((£)-3,3-difluoro-4-phenoxybut-1 -en-1 – yl) hexahydro-2H-cyclopenta[b]furan-2,5-diol (CTAF 4). CTAF 4 is then treated with (4- carboxybutyl)triphenylphosphonium bromide, in the presence of potassium bis(trimethylsilyl)amide in THF, to provide (Z)-7-((1 f?,2f?,3f?,5S)-2-((£)-3,3-difluoro-4- phenoxybut-1 -en-1 -yl)-3,5-dihydroxycyclopentyl)hept-5-enoic acid (“tafluprost free acid,” CTAF5), which is reacted with isopropyl iodide in the presence of DBU to provide (Z)- isopropyl 7-((1 F?,2F?,3F?,5S)-2-((£)-3,3-difluoro-4-phenoxybut-1 -en-1 -yl)-3,5-dihydroxy- cyclopentyl)hept-5-enoate (“tafluprost,” CTAF 6). The reaction sequence is summarized in Scheme 1 .

CTAF 1(i)
CTAF 1 CTAF 2

U.S. Patent Application Publication No. 2010/0105775A1 discloses amino acid salts of prostaglandins. The application also discloses a process for the preparation of prostaglandins, comprising forming an amino acid salt of a prostaglandin and converting the amino acid salt to the prostaglandin.
EXAMPLE 1 : Preparation of CTAF 1

CTAF1(i)
CTAF1
To a stirred suspension of sodium hydride (60% dispersion in mineral oil, 0.217 g, 5.429 mmol) in THF (5 ml_) was added a solution of dimethyl (2-oxo-3- phenoxypropyl)phosphonate(1 .21 g, 4.705 mmol) in THF (2 ml_), over 15 minutes at 0- 5°C under a nitrogen atmosphere. The mixture was warmed to 25-35 , 0.5 M zinc chloride solution in THF (9.4 ml_, 4.705 mmol) was added over 10 minutes, and then the mixture was stirred for 15 minutes at 25-35<€. CTAF1 (i) (3af?,4F?,5F?,6aS)-4-formyl-2- oxohexahydro-2 –cyclopenta[b]furan-5-yl benzoate (1 g) in dichloromethane (10 ml_) was added over 5 minutes at 25-35 °C. The temperature was raised to 35-40 °C and the mixture was stirred for 2hours under a nitrogen atmosphere. The mixture was cooled to 15°C and the reaction was quenched by adding acetic acid (0.2 mL), followed by adding saturated ammonium chloride solution (10 mL), and further stirring for 15 minutes. The organic layer was separated and the aqueous layer was extracted with ethyl acetate (5 mL). The combined organic layers were evaporated under reduced pressure below 50°C. The crude product was purified by column chromatography on silica gel (100-200 mesh) with 30% ethyl acetate in hexane, to afford the title compound (0.9 g, 61 %yield).
EXAMPLE 2: Preparation of CTAF 2

CTAF1 CTAF2
To a stirred solution of CTAF1 (5 g, 0.0123 mol) in dichloromethane (100 mL) was added diethylaminosulfurtrifluoride (13 mL, 0.09841 mol) at 0-5 °C under a nitrogen atmosphere. The temperature was raised to 25-35 °C and maintained for 24 hours under a nitrogen atmosphere at the same temperature. The mass was slowly added into a saturated sodium bicarbonate solution (75 mL) at 0-5 °C. Temperature was raised to 25- 35 °C, the layers were separated, and the aqueous layer was extracted with dichloromethane (2×25 mL). The combined organic layer was washed with water (25mL) and dried over sodium sulfate (5 g). The organic layer was evaporated to dryness under reduced pressure below 40 °C. The crude product was purified by column chromatography on silica gel (100-200 mesh) with 30% ethyl acetate in hexane, to afford the title compound (4.2 g, 79% yield). EXAMPLE 3: Preparation of CTAF 4

CTAF 2 CTAF 4
CTAF 2 (2.30 g, 5.37 mmol) was dissolved in toluene (25 mL) and the solution was cooled to -65 °C under nitrogen. Diisobutyl aluminum hydride (1 .5 M in toluene, 1 1 .8 mL, 17.7mmol) was added over 15 minutes at -61 to -65 . The mixture was stirred for 3hours and then the reaction was quenched by adding methanol (1 .5 mL). Sulfuric acid (1 M, 25 mL) was added and the temperature rose to -20°C during the addition. Methyl t-butyl ether (MTBE) (10 mL) was added and the mixture was allowed to warm to room temperature. The organic phase was separated and the aqueous phase was extracted with MTBE (2x 10 mL). The combined organic phase was washed with water (10 mL), saturated aqueous sodium bicarbonate (10 mL), and then brine (10 mL). The washes were back-extracted with MTBE (10 mL). The combined organic phases were dried with magnesium sulfate, filtered, and evaporated to give a colourless oil (2.20 g). The crude product was chromatographed on silica (60 g), eluting with a mixture of ethyl acetate and heptane (2:1 by volume), and then with ethyl acetate, to give CTAF 4 as a colourless oil (1 .71 g, 97% yield).
EXAMPLE 4: Preparation of CTAF 2

CTAF1 CTAF2
To a stirred solution of CTAF1 (20 g, 0.0492 mol) in dichloromethane(400 mL) was added diethylaminosulfurtrifluoride (52 mL, 0.393 mol) at 0-10°C under a nitrogen atmosphere. The temperature was raised to 25-35 and maintained for 96hours under a nitrogen atmosphere at that temperature. The mass was slowly added to a saturated NaHCOs solution (600 mL) at 0-10°C. The mixture was heated to 25-35 <€ and filtered through aCelite bed. The layers were separated and the aqueous layer was extracted with DCM (2×100 mL). The combined organic layer was washed with 10% NaCI solution (100 mL) and evaporated to dryness under reduced pressure below 40°C. The residue was purified by column chromatography on silica gel (100-200 mesh) with 30% ethyl acetate in hexane.
Column purified material was dissolved in MTBE (80 mL) at 40°C and stirred for 30 minutes at that temperature. Diisopropyl ether (160 mL) was added at 35-40 and stirring continued for 30 minutes at 35-40 . Cooled the mass to 5-15°C and stirred for 30 minutes at that temperature. The solid was filtered, washed with a mixture of MTBE and diisopropyl ether (DIPE) (1 :2 by volume, 60 mL), and dried at 40°C under vacuum, to afford pure CTAF2 (12.0 g, 57% yield).
EXAMPLE 5: Preparation of CTAF 5

(4-Carboxybutyl)triphenylphosphonium bromide (10.32 g, 23.3 mmol, 4 eq) was suspended in THF (20 mL) under a nitrogen atmosphere and cooled to 5°C. NaHMDS solution (1 M in THF, 46.6 mL, 46.6 mmol, 8 eq) was added over 10 minutes. The red/orange mixture was stirred for 30 minutes. A solution of CTAF 4 (1 .90 g, 5.82 mmol) in THF (10 mL) was added over 30 minutes at 0-3 . The mixture was stirred for 1 .5hours and then the reaction was quenched by adding water (30 mL) and the masswas warmed to room temperature. The aqueous phase was separated and the organic phase was washed with water (20 mL). The combined aqueous phases were washed with MTBE (30 mL). The organic phases up to this point were discarded. The aqueous phase was acidified with 2M hydrochloric acid (14 mL, to pH 3-4) and extracted with ethyl acetate (2×30 mL). The combined ethyl acetate layers were washed with brine (20 mL), dried with magnesium sulfate, filtered, and evaporated under reduced pressure to give CTAF 5 asa yellow oil (8.60 g).
A 2.96 g sample was removed and the remainder (5.64 g) was chromatographed on silica (30 g) eluting with ethyl acetate to give purified CTAF 5 (1 .41 g) asa yellow oil. NMR analysis showed approximately 90% purity, remainder triphenyl phosphine oxide.
EXAMPLE 6: Preparation of CTAF 5 DCHA salt

CTAF 5 CTAF 5 DCHA sa t
CTAF5 (1 1 .72 g, 90% purity, 25.7 mmol, containing 1 .4% trans isomer) was dissolved in acetone (60 mL). Dicyclohexylamine (4.66 g, 25.7 mmol) was added and the mixture was stirred at room temperature overnight. The solid was filtered and washed with acetone (6 mL), then dried to give the DCHA salt (12.93 g, 85% yield, 0.29% trans-isomer).
A sample (7.03 g) was further purified by recrystallisation. It was dissolved in hot acetone (30 mL) and cooled to room temperature with stirring. The mixture was stirred for 3 hours, filtered and the solid was washed with acetone (3 mL) and dried to give a white solid (6.41 g, 91 % recovery, 0.1 1 % trans-isomer).
A PXRD pattern of the product is shown as Fig. 1 , obtained using copper Ka radiation. In the drawing, the y-axis is intensity units and the x-axis is the 2-theta angle, in degrees. EXAMPLE 7: Pre aration of CTAF 6

CTAF 5 DCH A sa l ^ I AI- O
CTAF 5 DCHA salt (5.80 g, 9.80 mmol) was suspended in ethyl acetate (20 mL). Sulfuric acid (1 M, 20 mL) was added and the mixture was stirred until a clear solution was obtained. The organic phase was separated and the aqueous phase was extracted with ethyl acetate (2×20 mL). The combined organic layers were washed with water (15 mL) and brine (15 mL), dried with magnesium sulfate, filtered, and evaporated. The residue was dissolved in acetone (40 mL) and charged into a jacketed vessel at 30°C. 1 ,8-Diazabicyclo[5.4.0]undec-7-ene (DBU) (8.95 g, 58.8 mmol) was added, then 2- iodopropane (10.0 g, 58.8 mmol) was added, and the mixture was stirred for 20hours. The mixture was concentrated under reduced pressure and the residue was partitioned between ethyl acetate (30 mL) and aqueous potassium dihydrogen orthophosphate (8 g) in water (50 mL). The organic phase was separated and the aqueous was extracted with ethyl acetate (30 mL). The combined organic phases were washed with brine (20 mL), dried with magnesium sulfate, filtered and evaporated to give a yellow oil (4.83 g). The crude product was chromatographed on silica (130 g), eluting with a mixture of ethyl acetate and heptane (2:1 by volume), to give CTAF 6 (3.98 g, 90% yield) as a colorless oil.


 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HEREJoin me on Facebook 

 Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com











.gif)




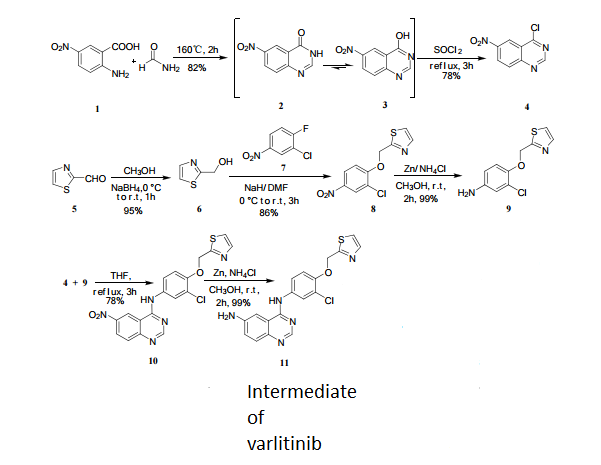
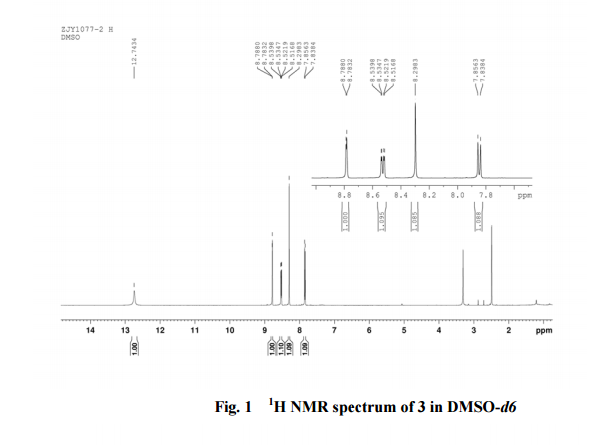
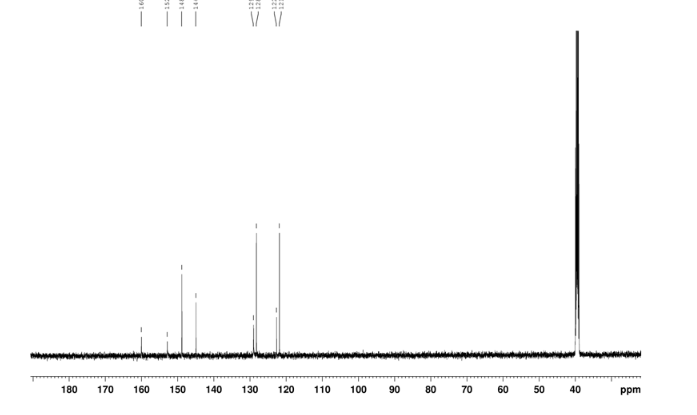
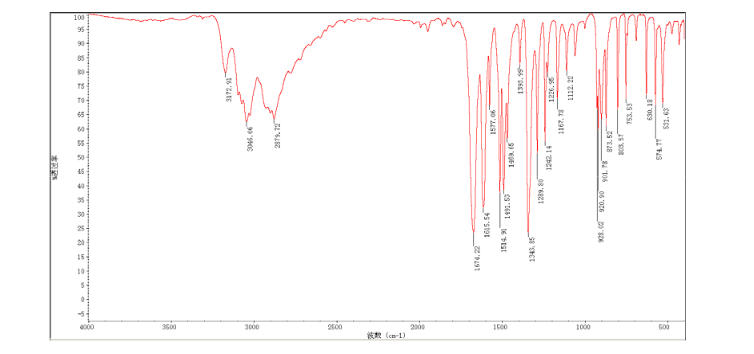
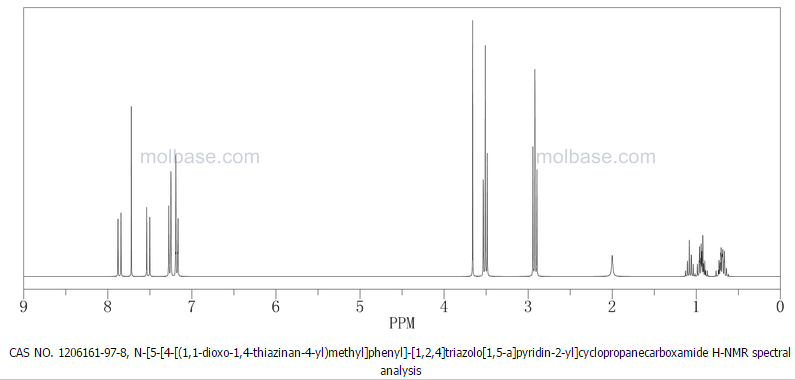
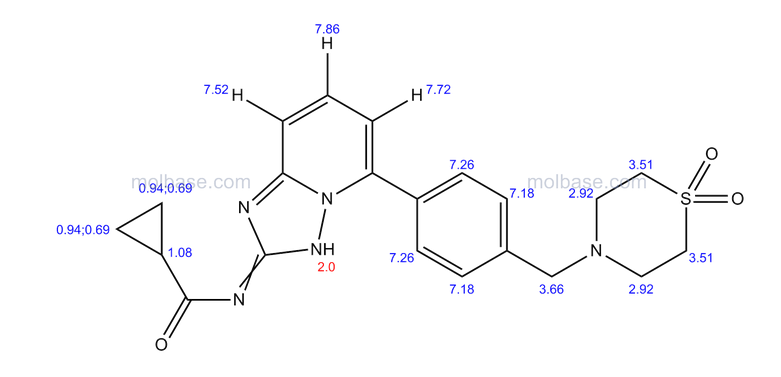
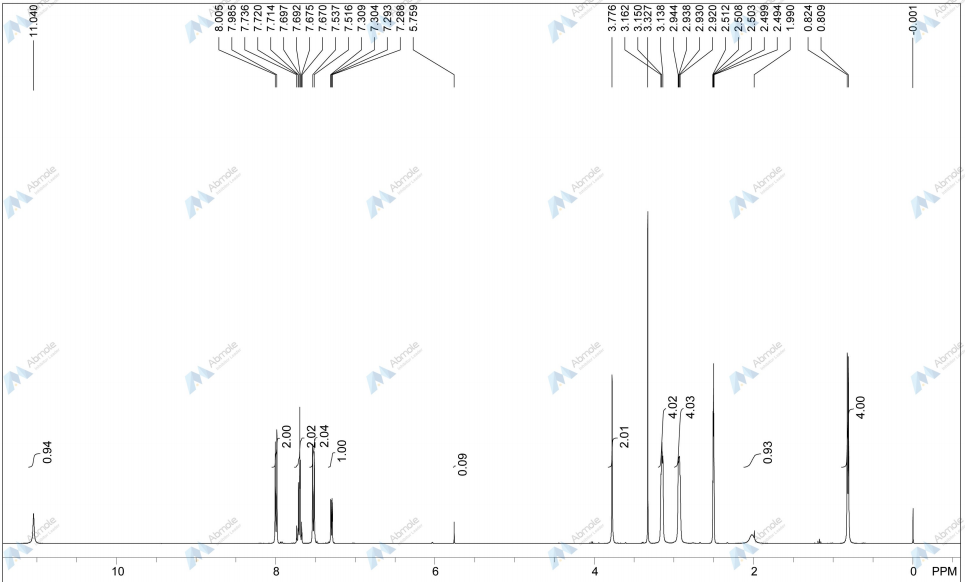
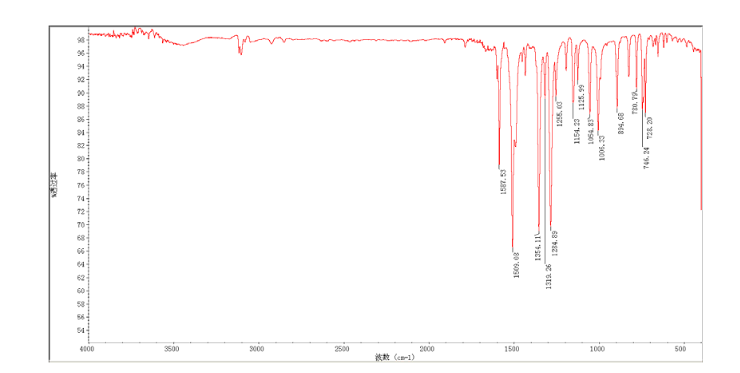
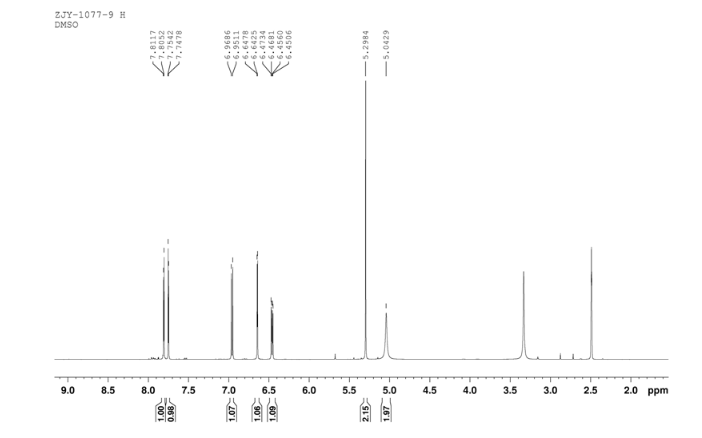
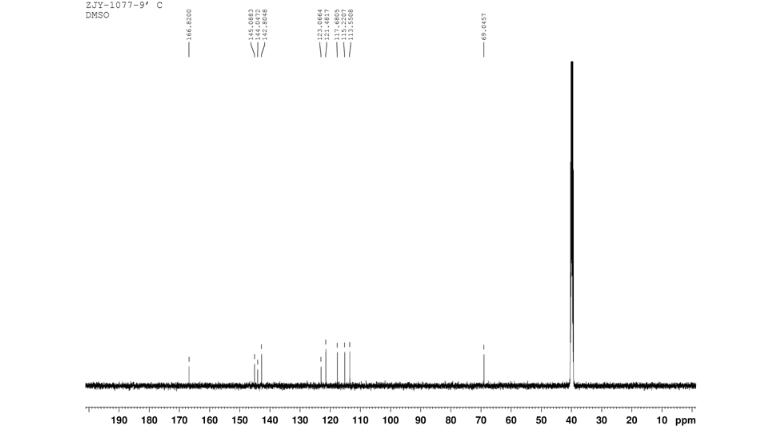
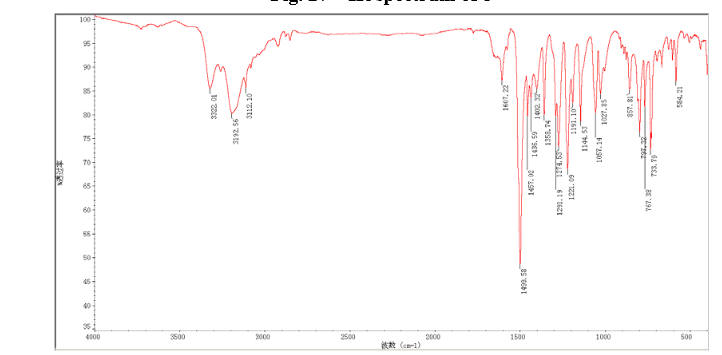
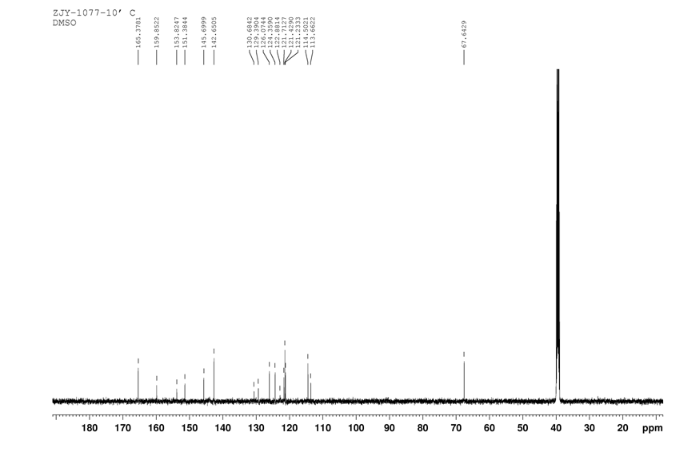
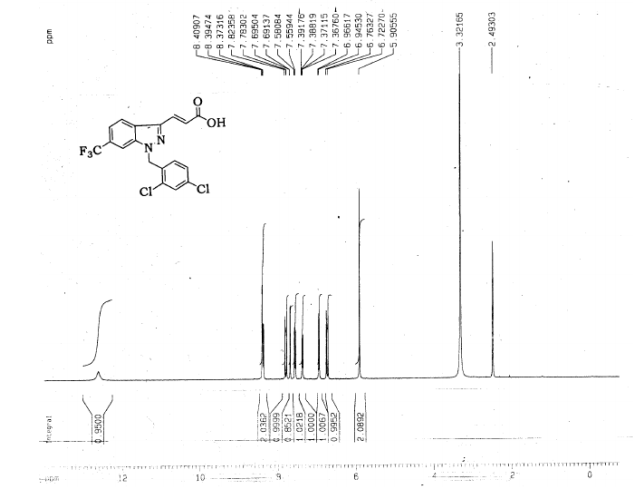
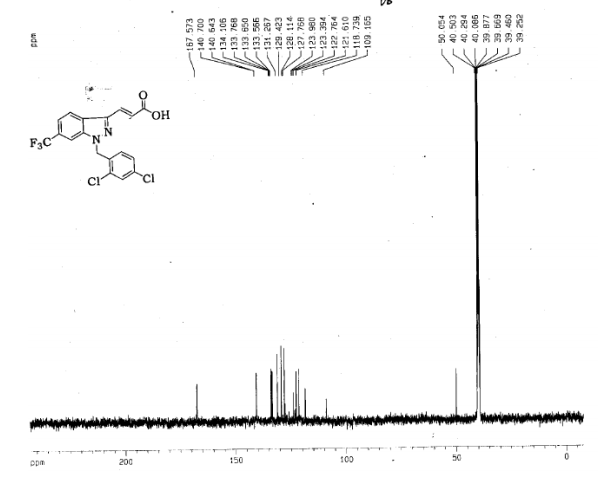
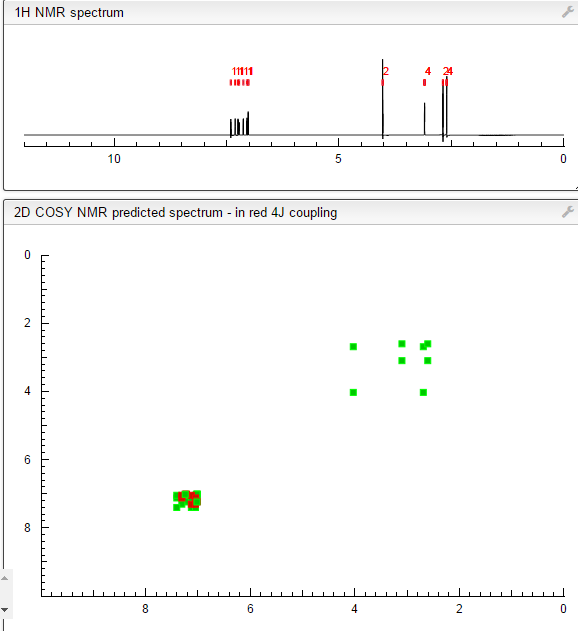
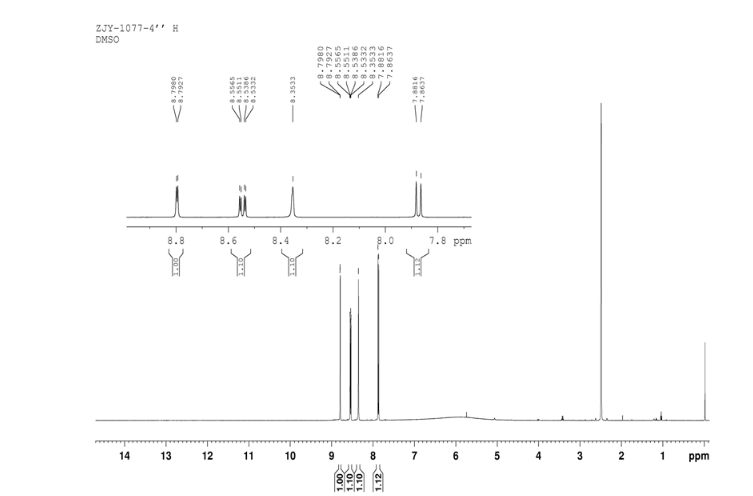 4 nmr dmsod6
4 nmr dmsod6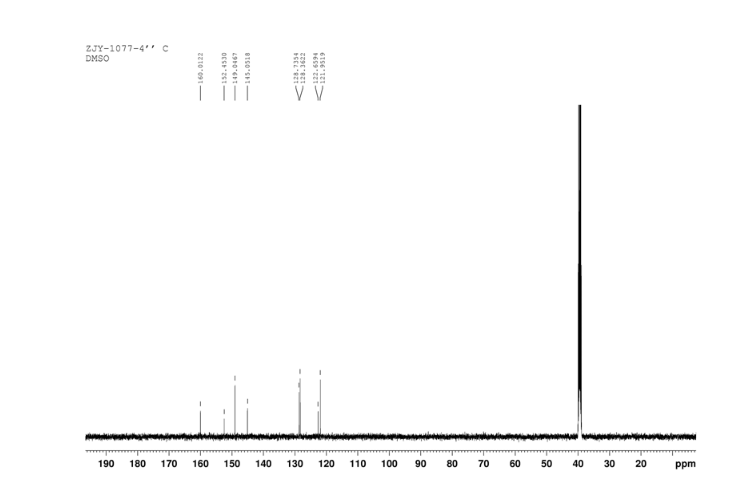
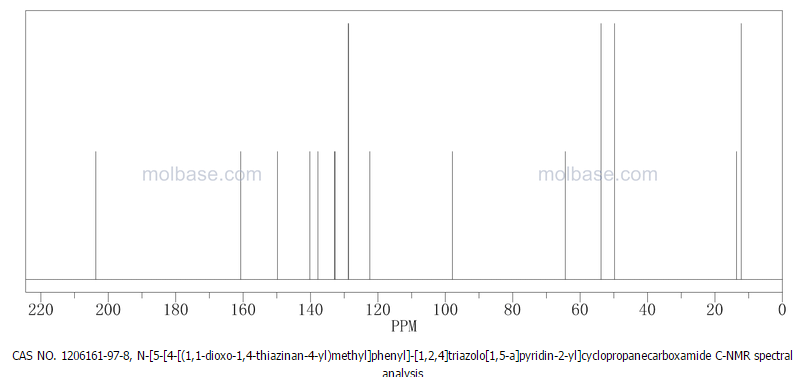
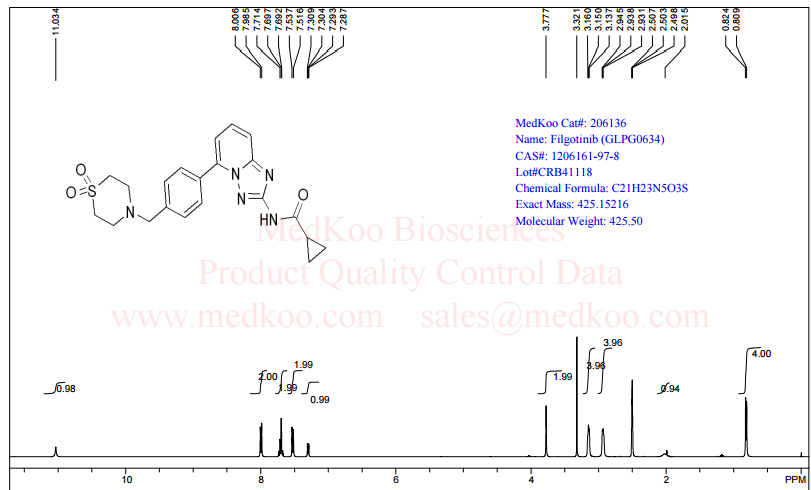
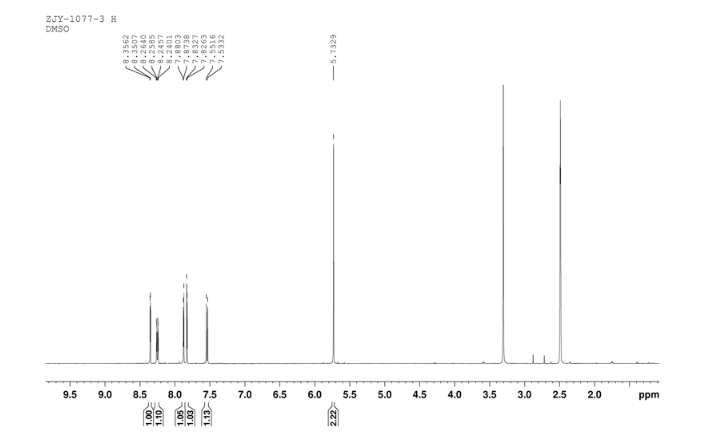
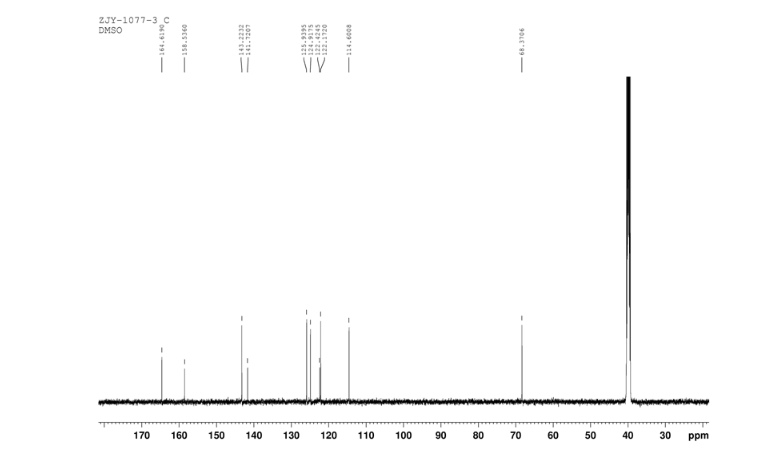
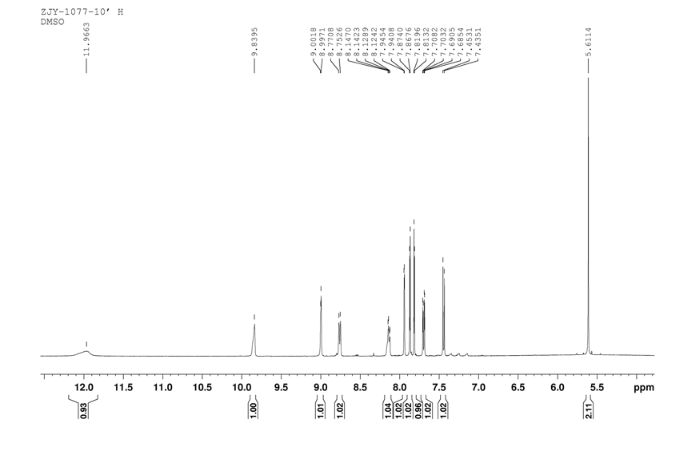
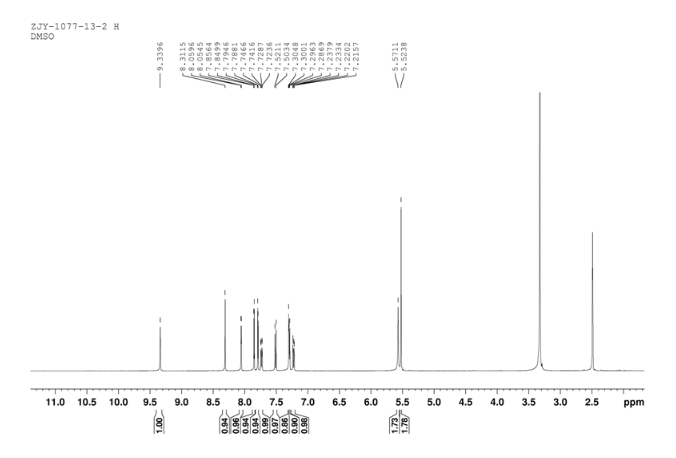
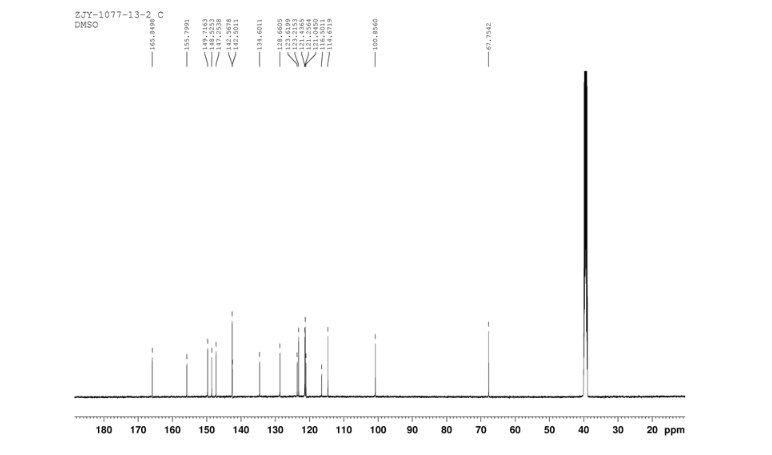
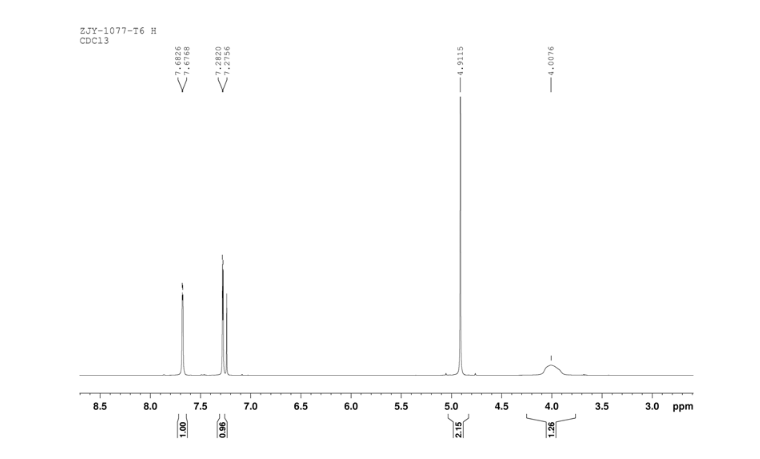 6 in dmsod6 1H NMR
6 in dmsod6 1H NMR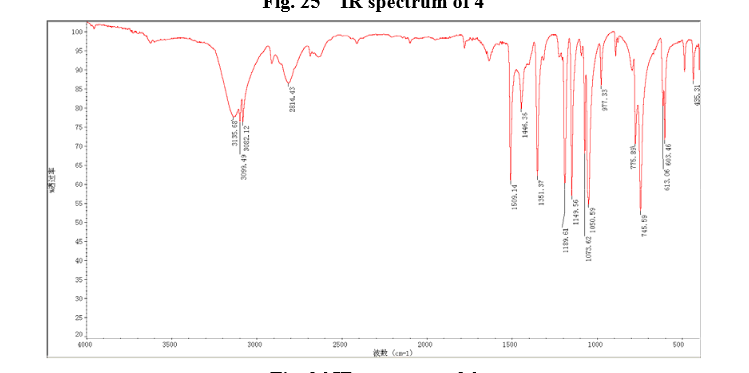
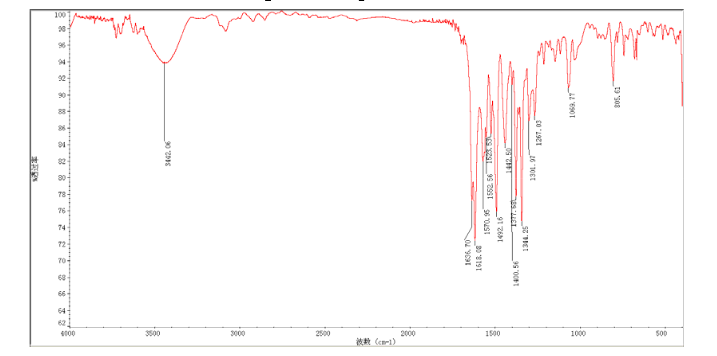



 carl fith
carl fith












 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..

 LIONEL MY SON
LIONEL MY SON





















 3D
3D