











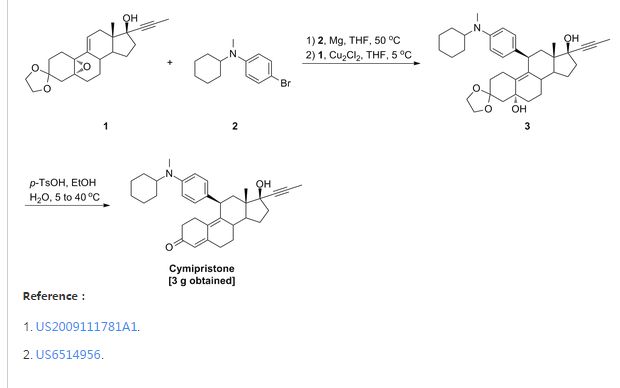
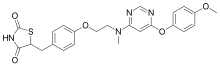
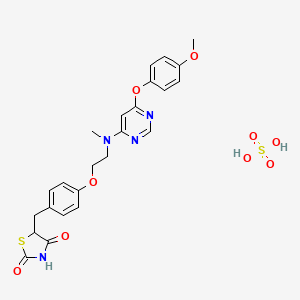
Cymipristone
- Estra-4,9-dien-3-one, 11-[4-(cyclohexylmethylamino)phenyl]-17-hydroxy-17-(1-propynyl)-, (11β,17β)- (9CI)
- (11β,17β)-11-[4-(Cyclohexylmethylamino)phenyl]-17-hydroxy-17-(1-propyn-1-yl)estra-4,9-dien-3-one
- Saimisitong
NDA Filed china
Shanghai Siniwest Pharmaceutical Chemical Technology Co., Ltd., Shanghai Zhongxi Pharmaceutical Co. Ltd., Xianju Pharmaceutical Co., Ltd,
A progesterone receptor antagonist potentially for termination of intrauterine pregnancy.
![]()
CAS No.329971-40-6
- Molecular FormulaC34H43NO2
- Average mass497.711 Da
- Steroid Compounds, a Method for Preparation thereof, Pharmaceutical Compositions Containing the Same and Use thereof
-
This invention relates to steroid compounds and pharmaceutical acceptable salts thereof, a method for preparation thereof, pharmaceutical compositions containing the same as active component, and their use in the preparation of medicines for treating diseases associated with progestogen dependence and for fertility control, abortion or contraception and for anticancer use.
-
Mifepristone (11β-[4-(N,N-dimethylamino)phenyl]-17α-(1-propinyl)-17β-hydroxy-4,9-estradiene-3-one) is a steroid compound which is disclosed in French Patent No. 2,497,807 to Rousell-Uclaf, published May 31, 1983. It is the first progesterone receptor antagonist put into clinical application and is a new type of anti-progestin. It binds to progesterone receptor and glucocorticoid receptor, having an affinity with progesterone receptor in rabbit endometrium five-fold higher than that of progesterone and thereby having strong anti-progesterone effect. It causes degeneration of pregnant villus tissue and decidual tissue, endogenous prostaglandin (PG) release, luteinizing hormone decrease, corpus luteum dissolution, and necrosis of embryo sac whose development depends on corpus luteum, leading to abortion. Therefore, it can be used as a non-surgical medicine for stopping early pregnancy. It can also be used, inter alia, in contraception and as an antineoplastic. (The Antiprogestin Steroid Ru486 and Human Fertility Control, 1985, New York: Plenum Press) .
-
Onapristone (11β-[4-(N,N-diemthylamino)phenyl]-17α-hydroxy-17β-(3-hydroxypropyl)-13α-4,9-estradiene-3-one), is a steroid compound which is disclosed in German Patent No. 3,321,826 to Schering AG, published Dec. 20, 1984. It has a strong antiprogestin activity and can be used in abortion (American Journal of Obstetrics and Gyencology, 1987, 157:1065-1074), anticancer (Breast Cancer Research and Treatment, 1989, 14:275-288), etc. It was reported that onapristone had toxicity to human liver (European Journal of Cancer, 1999, 35(2):214-218).
-
Lilopristone (11β-[4-(N,N-dimethylamino) phenyl]-17α-[3-hydroxy-1(Z)-propenyl]-17β-hydroxy-4,9-estradiene-3-one) is a steroid compound which is disclosed in German Patent No. 3,347,126 to Schering AG, published July 11, 1985. It has a strong antiprogestin activity and can be used in abortion, contraception (American Journal of Obstetrics and Gyencology, 1987, 157:1065-1074), etc. It was reported that the clinical effect of lilopristone in stopping early pregnancy was only equivalent to that of mifepristone (Human Reproduction, 1994, 9(1):57-63).
-
ZK112993 (11β-(4-acetylphenyl)-17α-(1-propinyl)-17β-hydroxy-4,9-estradiene-3-one) is as steroid compound which is disclosed in German Patent No. 3,504,421 to Schering AG, published Aug. 7, 1986. It has a potent antiprogestin activity and can be used in, inter alia, anticancer (Anticancer Res., 1990, 10:683-688).
-
In European Patent No. 321,010 to Akzo NV, The Netherland published June 21, 1989 are disclosed “11-arylsteroid compounds” having a strong antiprogestin activity.
PATENT
WO 2001018026
http://www.google.com/patents/EP1219632A1?cl=en

The preparation method of the present invention includes the following single- or multi-step procedures:
1. Method for the preparation of 11β-[4-(N-methyl-N-cyclohexylamino)phenyl]-17α-(1-propinyl)-17β-hydroxy-4,9-estradiene-3-one (IV) which includes the following steps:
(1) Preparation of Grignard reagent (III)

4-bromo-N-methyl-N-cyclohexylaniline (II) is reacted with magnesium in tetrahydrofuran (THF) to obtain Grignard reagent of formula (III).
(2) C11 additive reaction

Compound of formula (IV) and the Grignard reagent of formula (III) prepared in step (1) are brought to an additive reaction to obtain compound of formula (V).
(3) Hydrolytic reaction

The compound of formula (V) prepared in step (2) is subjected to a hydrolytic reaction to obtain compound of form (VI).
2. Method for preparation of 11β-[4-(N-cyclohexylamino)phenyl]-17α-(1-propinyl)-17β-hydroxy-4,9-estradiene-3-one (XI) which includes the following steps:
(1) Preparation of Grignard reagent of formula (IX)

4-bromo-N-cyclohexylaniline (VII) is first protected by trimethylchlorosilane, then reacted with magnesium in THF to obtain Grignard reagent of formula (IX).
(2) C11 additive reaction

Compound of formula (IV) and the Grignard reagent of formula (IX) prepared in step (1) are brought to an additive reaction to obtain compound of formula (X).
(3) Hydrolytic reaction

The compound of formula (X) prepared in step (2) is subjects to a hydrolytic reaction to obtain compound of formula (XI).
Example 2:
-

-
9g 4-bromo-N-cyclohexylaniline (VII) (CA registration number [113388-04-8], see Synthetic Communications, 1986, 16(13): 1641-1645 for its preparation) was placed into a four-necked flask and 15 ml (1.5 mol/L) n-BuLi solution in n-hexane. The mixture was stirred for 30 min at room temperature. Then 8 g trimethylsilyl chloride (Me3SiCl) was added and the mixture was stirred for 1 hour. Solvent and excessive Me3SiCl was evaporated under reduced pressure to yield 4-bromo-(N-cyclohexyl-N-trimethylsilylaniline) (VIII) which was formulated into a solution with 7.5 ml anhydrous tetrahydrofuran for further use.
-
1.3 g magnesium was placed into a four-necked flask and a small amount of the above solution was added dropwise and slowly at 40°C. After completion of addition, the temperature was kept for 1 hour to yield a solution of 4-(N-cyclohexyl-N-trimethylsilylamino)phenylmagnesium bromide (IX) in tetrahydrofuran for further use.
- Preparation of 11β-[4-(N-cyclohexylamino)phenyl]-17α-(1-propinyl)-17β-hydroxy-4,9-estradiene-3-one (XI)(1) Preparation of 4-(N-cyclohexyl-N-trimethylsilylamino)phenyl magnesium bromide (IX)
(2) Preparation of 3,3-ethylenedioxy-5α,17β-dihydroxy-11β-[4-(N-cylohexylamino)phenyl]-17α-(1-propinyl)-9(10)-estrene(X).

- 5g 3,3-ethylenedioxy-5,10-epoxy-17α-(1-propinyl)-17β-hydroxy-9(11)-estrene (IV) was placed into a four-necked flask and 10 ml anhydrous tetrahydrofuran and a catalytic amount of cuprous chloride (Cu2Cl2) added. Then solution of 4-(N-cyclohexyl-N-trimethylsilylamino)phenyl magnesium bromide (IX) in tetrahydrofuran was added dropwise and slowly while controlling the temperature below 5°C. After completion of addition, the mixture was allowed to react for 2 hours at room temperature and to stand overnight. Saturated ammonium chloride aqueous solution was added and the tetrahydrofuran layer separated which was washed with saturated ammonium chloride solution. The solution in tetrahydrofuran was washed with saturated saline and dried over anhydrous sodium sulfate. Evaporation of tetrahydrofuran under reduced pressure yielded a residual which was chromatographed on silica gel column using cyclohexane: acetone (5:1) as developing agent to yield 3 g 3,3-ethylenedioxy-5α,17β-dihydroxy-11β-[4-(N-cyclohexylamino)phenyl]-17α-(1-propinyl)-9(10)-estrene(X).
-
IR (KBr) cm-1: 3420 (C5, C17-OH), 1610, 1510 (benzene backbone), 840, 808 (ArH).
1H NMR (CDCl3) δ ppm: 0.52(3H, S, C13-CH3), 2.72(3H, S, N-CH3), 3.92(4H, m, -O-CH2CH2-O-), 4.24(1H, m, C11-H), 6.65-7.00 (4H, ArH).
(3) Preparation of 11β- [4- (N-cyclohexylamino)phenyl] -17α- (1-propinyl) -17β-hydroxy-4,9-estradiene-3-one (XI).

- 1.5g 3,3-ethylenedioxy-5,17β-dihydroxy-11β-[4-(N-cyclohexylamino)phenyl]-17α-(1-propinyl)-9(10)-estrene (X) and 0.75 g para-toluenesulfonic acid (PTS) were dissolved in 15 ml 90 % ethanol (v/v). The mixture was stirred for 2 hours while controlling the temperature at 40°C-50°C. After completion of the reaction, the reactant was poured into diluted sodium hydroxide aqueous solution, extracted with dichloroethane, washed with water to neutrality, and dried over anhydrous sodium sulfate. Evaporation of the solvent and chromatography on silica gel column using cyclohexane: ethyl acetate (5:1) as developing agent yielded 0.9 g 11β-[4-(N-cyclohexylamino)phenyl]-17α-(1-propinyl)-17β-hydroxy-4,9-estradiene-3-one (XI).
-
IR (KBr) cm-1: 3400 (C17-OH), 1658 (unsaturated ketone), 1613, 1514 (benzene backbone), 865, 810 (ArH).
1H NMR (CDCl3) δ ppm: 0.50 (3H, S, C13-CH3), 1.76 (3H, S, C≡C-CH3), 4.32(1H, S, C11-H), 5.75(1H, S, C4-H), 6.9-7.10 (4H, ArH).
PATENT
PATENT
Example 1
Race meters mifepristone synthetic routes:

Epoxy adduct match rice mifepristone
(N- hexylamino methylcyclohexyl) phenyl magnesium bromide (1) 4-
In the four-necked flask, 1.4 g of magnesium into pieces (Mg) and 10 ml of anhydrous tetrahydrofuran (THF), no iodine or add a little change, at about 50 ° C, a solution of 10.86 g of 4-bromo-methyl -N- cyclohexyl aniline (dissolved in 24 ml of anhydrous tetrahydrofuran) dropwise Bi, incubation was continued for 1 hour with stirring to give 4- (N- methyl-cyclohexylamino) phenyl magnesium bromide tetrahydrofuran solution (to be used in the next step an addition reaction ).
(2) 3,3-ethylenedioxy -5 α, 17 β – dihydroxy -11 β – [4- (Ν- methyl -Ν- cyclohexylamino) phenyl] -17 α – (1- propyl block-yl) -9 (10) – Preparation of estra-ene (adduct) of
In the four-necked flask, into 5 g of 3,3-ethylenedioxy-5,10-epoxy -17 α – (1- propynyl) – 17 (3 – hydroxy – 9 (11) – estra-ene (epoxy), 29.1 ml anhydrous tetrahydrofuran (THF) and 0.1 g cuprous chloride (of Cu 2 of Cl 2 ), a solution of 4- (N- methyl -N-cyclohexylamino) phenyl magnesium bromide tetrahydrofuran
Nan solution, temperature control 5. C, the drop was completed, the incubation was continued for 5 hours, the reaction was completed, the reaction solution was poured into saturated aqueous ammonium chloride solution, points to the water layer, the organic layer was washed with saturated ammonium chloride solution, the aqueous layer extracted with ethyl acetate number times, the organic layers combined, washed with saturated aqueous sodium chloride, dried over anhydrous sodium sulfate, and concentrated under reduced pressure, a silica gel column, eluent cyclohexane: acetone = (5: 1) to give 3,3-ethylene dioxo -5 α, 17 β – dihydroxy -11 β – [4- (- methyl -Ν- cyclohexylamino) phenyl] -17 α – (1- propynyl) -9 (10) – female steroidal women (adduct) solid 6 grams.
IR. ‘KBi cm- ^ SlS OI ^ ^ -OH lS jSlS benzene backbone), 819 (aromatic hydrogen). NMR Ή: (CDC1 3 ) ppm by [delta]: 0.47 (3H, the S, the C IR CH 3 ), 1.88 (3H, the S, the C ≡ the C-CH 3 ), 2.72 (3H, the S, the N-CH 3 ), 6.65- 7.03 (4H, ArH) O
(3) 11 β – [4- (N- methyl -N- cyclohexylamino) phenyl] -17 α – (1- propynyl) -17 β – hydroxy-estra-4,9-diene – Preparation of 3-one (match rice mifepristone) of
‘2.5 g of p-toluenesulfonic acid (PTS) and 5 grams of 3,3-ethylenedioxythiophene -5 α, 17 β – dihydroxy -11 β – [4- (Ν- methyl cyclohexylamino) phenyl] -17 α – (1- propynyl) -9 (10) – estra-ene (adduct) was dissolved in 50 ml of ethanol 90% (V / V), and at 5 ° C – 40 ° C the reaction was stirred 3 hours, the reaction solution was poured into dilute aqueous sodium hydroxide solution, the precipitated solid was suction filtered, washed with water until neutral, the filter cake was dissolved in 50 ml of ethyl acetate, then with saturated aqueous sodium chloride solution to the water layer was evaporated part of the solvent, the precipitated solid was suction filtered, and dried to give a pale yellow solid 11 β – [4- (Ν- -N- methyl-cyclohexylamino)] -17 α – (1- propynyl) -17 β – hydroxy estra-4,9-dien-3-one (match rice mifepristone) 3 grams.
^ Cm & lt IRCKB 1 : 3447 (the C . 17 -OH), among 1655 (unsaturated ketone), 1607,1513 (benzene backbone), 865,819 (aromatic hydrogen).
NMR ¾: (CDC1 3 ) ppm by [delta]: 0.56 (3H, the S 5 the C 13 -CH 3 ), 1.89 (3H, the S 5 -C ≡ the C-the CH3), 2.74 (3H, the S, the N-the CH3), 4.34 ( lH, the S, the C N -H), 5.75 (lH, the S, the C 4 -H), 6.68-6.99 (4H, ArH).
PATENT
PATENT
PAPER
Volume 878, Issues 7–8, 1 March 2010, Pages 719–723
Determination of cymipristone in human plasma by liquid chromatography–electrospray ionization-tandem mass spectrometry
doi:10.1016/j.jchromb.2010.01.027
Abstract
A rapid, specific and sensitive liquid chromatography–electrospray ionization-tandem mass spectrometry method was developed and validated for determination of cymipristone in human plasma. Mifepristone was used as the internal standard (IS). Plasma samples were deproteinized using methanol. The compounds were separated on a ZORBAX SB C18 column (50 mm × 2.1 mm i.d., dp 1.8 μm) with gradient elution at a flow-rate of 0.3 ml/min. The mobile phase consisted of 10 mM ammonium acetate and acetonitrile. The detection was performed on a triple-quadruple tandem mass spectrometer by selective reaction monitoring (SRM) mode via electrospray ionization. Target ions were monitored at [M+H]+m/z 498 → 416 and 430 → 372 in positive electrospray ionization (ESI) mode for cymipristone and IS, respectively. Linearity was established for the range of concentrations 0.5–100 ng/ml with a coefficient correlation (r) of 0.9996. The lower limit of quantification (LLOQ) was identifiable and reproducible at 0.5 ng/ml. The validated method was successfully applied to study the pharmacokinetics of cymipristone in healthy Chinese female subjects.
CHEMICAL ABSTRACTS, vol. 115, no. 25, 23 December 1991 (1991-12-23) Columbus, Ohio, US; abstract no. 270851g, X. ZHAO ET AL.: “Synthesis and terminating early pregnancy effect of mifepristone derivatives” page 117; XP002219009 & ZHONGGUO YAOKE DAXUE XUEBAO, vol. 22, no. 3, 1991, pages 133-136,
//////////Cymipristone, Saimisitong, NDA Filed , china, Shanghai Siniwest Pharmaceutical Chemical Technology Co., Ltd., Shanghai Zhongxi Pharmaceutical Co. Ltd., Xianju Pharmaceutical Co., Ltd,



























































{kind=link}