












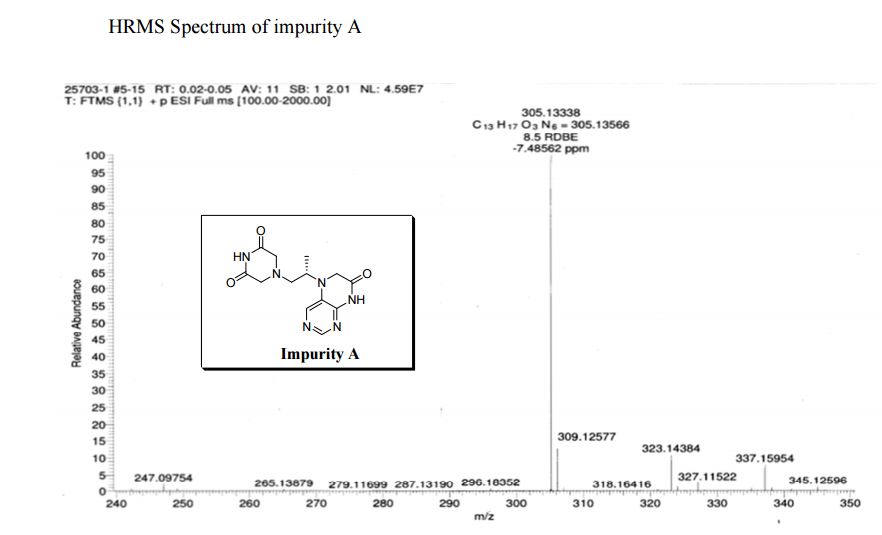
| CAS : | 156928-09-5 | ||
|---|---|---|---|
| Molecular Formula: | C6H10O3 | ||
| Molecular Weight: | 130.144 |
- Furo[2,3-b]furan-3-ol, hexahydro-, [3R-(3α,3aβ,6aβ)]-
- (3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-ol
- 3R,3AS,6aR-hexahydrofuro[2,3-b]furan-3-ol
- R,S,R-Bisfuran alcohol
WO2012075122 SP ROT= -13.2/1G/100ML, METHANOL
PATENT
http://www.google.com.ar/patents/WO2012070057A1?cl=en
The overall synthesis of the present invention is shown in the scheme 1:

Yet another aspect of present invention is to provide a process for the preparation of compound formula I as per below scheme 2.

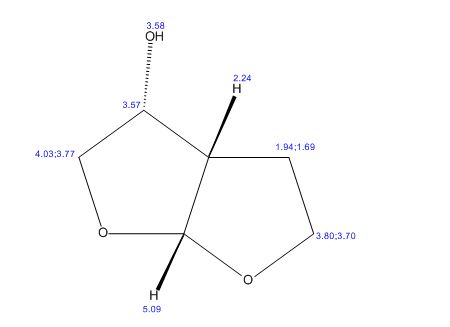
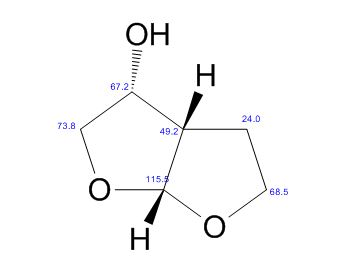
(3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-ol
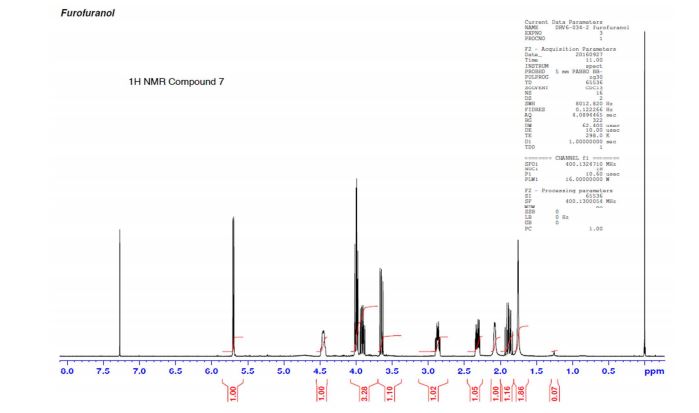
(3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-ol (7) as clear oil (7.8 g, 96.8 A% purity by GC-MS, 55.7 mmol, 74% yield). C6H10O3, GC-MS (EI): m/z 100 (M- H2CO).
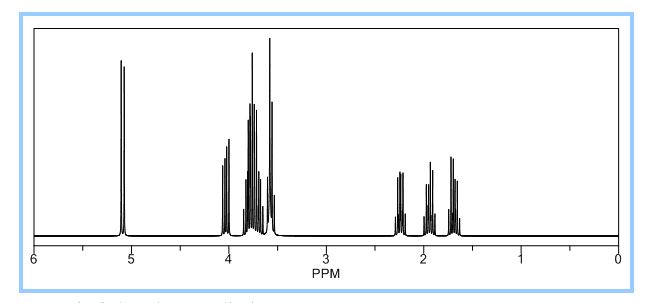
1H NMR (CDCl3): 1.88 (m, 1H), 2.08 (bd, 1H, −OH), 2.31 (m, 1H), 2.87 (m, 1H), 3.64 (dd, J = 9.2, 7.0 Hz, 1H), 3.87–4.02 (abx system, 3H), 4.45 (m, 1H), 5.70 (d, J = 5.2 Hz, 1H).
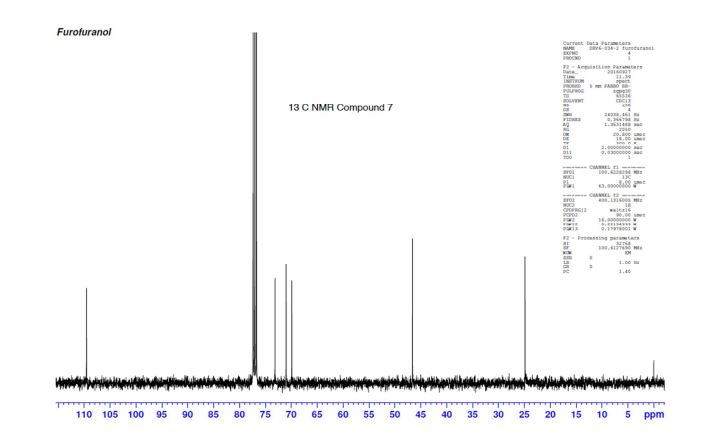
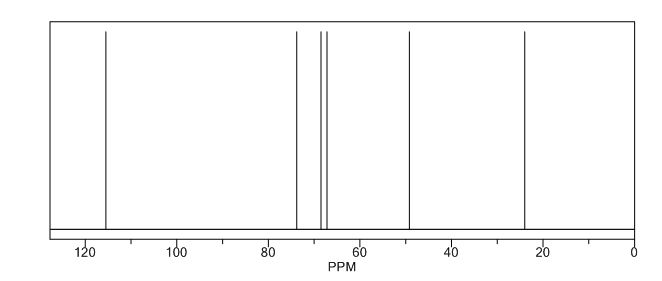
13C NMR (CDCl3): 109.54, 73.15, 71.00, 69.90, 46.58, 24.86.
Diastereomeric ratio of 7 to 12 = 98.2:1.8.
GC retention time of 7= 3.20 min; 12 = 3.09 min.

A practical synthesis of (3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-ol—a key intermediate in the synthesis of darunavir—from monopotassium isocitrate is described. The isocitric acid salt, obtained from a high-yielding fermentation fed by sunflower oil, was converted in several steps to a tertiary amide. This amide, along with the compound’s ester functionalities, was reduced with lithium aluminum hydride to give, on acidic workup, a transient aminal-triol. This was converted in situ to the title compound, the bicyclic acetal furofuranol side chain of darunavir, a protease inhibitor used in treatment of HIV/AIDS. Key to the success of this process was identifying an optimal amide that allowed for complete reaction and successful product isolation. N-Methyl aniline amide was identified as the most suitable substrate for the reduction and the subsequent cyclization to the desired product. Thus, the side chain is produced in 55% overall yield from monopotassium isocitrate.
Practical Synthesis of the Bicyclic Darunavir Side Chain: (3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-ol from Monopotassium Isocitrate

In particular, the following synthetic scheme (1) illustrates the present commercial method of synthesizing compound (I) . This synthesis is disclosed in detail in A.K. Ghosh et al . , Tetra- hedron Letters, 36 (4) , pp. 505-508 (1995), incorporated herein by reference. Also see, A.K. Ghosh et al., J. Med. Chem . , 39, pp. 3278-3290 (1996) for the synthesis of compound (I) and a related compound of structural formula (II) (i.e., (3S, 3aR, 7aS) -3- hydroxyhexahydrofuro [2, 3-b] pyran) .

Scheme 1 (prior art)

(91%)
Cobaloxime (catalytic) , NaBH4, EtOH


Alternatively,
-OAc
0 Immobilized Lipase 30
0- pH 7 buffer 23°C, 24 h
(+)

R=Ac
MeLi, THF
^ R=H (Compound (I))
The present method of synthesizing bis-THF is summarized as follows:


<
(78-100%;

(70-90%)

(65-80%;

2. NaBH4, EtOH (65-75%) -15°C, 1-3 h Compound (I) (bis-THF) Another aspect of the present invention is to provide a method of preparing a compound having a structure

then utilizing the benzyl-protected 5-hydroxymethyl- 5H-furan-2-one in the synthesis of compound (I) .
Another aspect of the present invention is to provide a method of preparing compounds related to bis-THF by using a starting material having a following structure:


X
I
R R
The synthesis of bis-THF (compound (I) ) is summarized below:

(1) (2)

(3)

15) (6)

(I)
(3R, 3aS , 6aR) -3-Hydroxyhexahydrofuro [2 , 3-b] uran (I)

(3R, 3aS, 6aR) -3-Hydroxyhexahydrofuxo [2, 3- b] furan (I) : To a solution containing 250 mg (1.95 mmol) (3aS, 6aR) -3-oxyhexahydrofuro [2, 3-b] furan (6) in EtOH (25 mL) was added ’89 mg (2.35 mmol) NaBH4 at -18 °C. The reaction mixture was stirred at -18 °C for 2.5 hours, then the reaction was quenched with saturated NH4C1 solution (5 mL) and warmed to room temperature. The resulting mixture was concentrated under reduced pressure, and then 10 mL water was added. The aqueous layer was extracted with ethyl acetate (3 x 50 mL) and a solution of 70% CHC13, 20% MeOH, and 10% water (3 x 50 mL) . The combined organic extracts were dried over Na2S04. Column chromatography (silica gel 80 g, MeOH in CHC13 7%) gave compound (I) (178 mg. 70%) as a colorless solid, Rf=0.3, [α]25 D -12.4°, c 1.3, MeOH. IR (neat) 2951, 1641, 1211 cm“1; XH-NMR (400 MHz CDC13) δ: 1.85 (mc, IH) , 1.94 (bs, IH) , 2.27 (mc, IH) , 2.84 (mc, IH) , 3.63 (dd, IH, J=7.1 Hz, J=9.2 Hz), 3.89 (mc, IH) , 3.97 (mc, IH) , 4.43 (dd, IH, J=6.8 Hz, J=14.5 ” Hz), 5.68 (d, IH, J=5.2 Hz). 13C-NMR (125.8 MHz, CDC13, Dept) δ‘: 25.27 (-) , 46.97 (+) , 70.31 (-) , . 71.26 (-), 73.50 (+) , 109.93. (+) . C6H10O3; Exact Mass: 130.06; Mol. Wt . : 130.14; C, 55.37, H, 7.74, 0, 36.88.
Experimentals :
l-(Benzyloxy)-but-3-en-2-ol (±)-(8): To a solution of vinylmagnesium bromide (1 M in THF, 40 mL, 40 mmol) in THF (10 mL) at 0°C was added benz- yloxyacetaldehyde (7) (5 g, 33.3 mmol) dropwise. The mixture was stirred for 10 min at 0°C, and the reaction then was quenched with 20 L of saturated NaHC03 solution. The layers were separated, the aqueous layer was extracted with ethyl acetate (3 x 20 mL) , and the combined organic extracts were dried over sodium sulfate. Evaporation of solvent under reduced pressure, followed by column chromatography on silica gel (20% EtOAc in hexanes as the eluent) yielded alcohol (±)-8 (5.22 g, 88%) as a yellow oil, Rf=0.40 (30% EtOAc in hexanes); 1H-NMR (400 MHz, CDC13) δ: a 2.79 (bs, IH) , 3.39 (dd, IH, J=1.7, 7.85 Hz), 3.55 (dd, IH, J=3.35, 6.3 Hz), 4.35 (m, IH) , 4.58 (s, IH) , 5.21 (dt, IH, J=7.75, 1.4 Hz), 5.38 (dt, IH, J=14.18, 1.4 Hz), 5.84 (m, IH) , 7.30-7.38 (m, 5H) ; 13C-NMR (100.6 MHz, CDC13) δ: 71.52, 73.37, 74.02, 116.49, 127.85, 128.49, 136.58, 137.81. (S)-l-(Benzyloxy) -but-3-en-2-ol (9) and (R) -1- (benzyloxy) -but-3-en-2-oyl acetate (10):
A: To a solution of alcohol (±)-(8) (5.21 g, 29.3 mmol) in acetic anhydride (14 mL, 147 mmol) and tert-butyl methyl ether (70 mL, 586 mmol) was added immobilized lipase PS-30 (5.3 g ) on Celite 521 (Aldrich) . The mixture was stirred at room temperature for 20 h, and then filtered through Celite. Removal of solvent under reduced pressure followed, by column chromatography on silica gel (10 and 15% EtOAc in hexanes as the eluents) yielded acetate (10) (3.81 g, 54%) Rf=0.57 (30% EtOAc in hexanes) as a clear oil, [of]25 D -2° (c 1, CHC13) ; NMR (500 MHz, CDC13) δ: 2.10 (s, 3H) , 3.55-3.59 (m, 2H) , 4.56 (q, 2H, J=12.2, 14.0 Hz), 5.24 (d, IH, J-10.6 Hz), 5.32 (d, IH, J=17.3 Hz), 5.50 (m, IH) , 5.84 (m, IH) , 7.25-7.36 (m, 5H) ; 13C-NMR (125.8 MHz, CDC13) δ: 21.62, 71.67, 73.57, 73.59, 118.39, 128.14, 128.84, 133.77, 138.32, 170.63; alcohol 9 (2.34 g, 45%) as a yellow oil, Rf=0.40 (30% EtOAc in hexanes), [α]25 D– 8.3° (c 1.06, MeOH) .
B: To a solution of alcohol (±)-(8) (3.92 g, 22.0 mmol) in vinyl acetate (46 mL, 499 mmol) and ethylene glycol dimethyl ether (46 mL, 440 mmol) was added immobilized lipase PS-30 (4 g ) on Celite-545 (Aldrich) . The mixture was stirred at room temperature for 28 h, and then filtered through celite. Removal of solvent under reduced pressure, followed by column chromatography on silica gel (10 and 15% EtOAc in hexanes as the eluents) yielded acetate
(10) (2.20 g, 45%) Rf=0.57 (30% EtOAc in hexanes) as a clear oil, [ ]25 D -2.7° (c 1.35, MeOH); alcohol (9) (2.00 g, 51%) as a yellow oil, Rf=0.40 (30% EtOAc in hexanes), [α]25 D -11.4° (c 1.6, MeOH).
C: To a solution of alcohol (+)-(8) (30 mg, 0.168 mmol) in isopropenyl acetate (375 μL, 3.36 mmol) and ethylene glycol dimethyl ether (375 μL, 3.61mmol) was added immobilized lipase PS-30 (35 mg) on Celite-545 (Aldrich) . The mixture was stirred at room temperature for 23 h, and then filtered through celite. Removal of solvent under reduced pressure, followed by column chromatography on silica gel (10) and 15% EtOAc in hexanes as the eluents) yielded acetate 10 (20.3 mg, 54%) as an oil, Rf=0.57 (30% EtOAc in hexanes), [α]25 D -1.4° (c 1.02, MeOH); alcohol (9) (13 mg, 43%) as a yellow oil, Rf=0.40
(30% EtOAc in hexanes), [ ]25 D -13.5° (c 1.3, MeOH). (R) -1- (Benzyloxy) -but-3-en-2-ol (11): To a solution of acetate (10) (3.7 g, 16.9 mmol) in methanol (20 mL) was added K2C03 (7 g, 50.6 mmol). The mixture was stirred at room temperature for 35 min. Methanol then was removed under reduced pressure. The resulting solid residue was dissolved in ethyl acetate, washed with saturated NH4C1 solution and brine, and dried over sodium sulfate. Removal of ethyl acetate under reduced pressure yielded the crude alcohol (11) (3 g, 100%) as a yellow oil, Rf=0.40 (30% EtOAc in hexanes), [α]25 D 8.3° (c 1.06, MeOH) .
(S)-l- (Benzyloxy) -but-3-en-2-ol (9) from (11): To a solution of crude alcohol (5) (2 g, 11.2 mmol), triphenylphosphine (5.88 g , 22.4 mmol), and 4-nitrobenzoic acid (2.81 g, 16.8 mmol) in benzene (35 mL) was added at room temperature diisopropyl azodicarboxylate (4.35 mL, 22.4 mmol) dropwise. The mixture was stirred for 40 min, followed by the re- moval of solvent under reduced pressure. All of the crude ester then was dissolved in a mixture of MeOH:Et3N:H20 (20ml) in the ratio of 4:3:1 and reacted with LiOH (1.64 g, 39.3 mmol) at room temperature. The mixture was stirred for 2 h, followed by the removal of solvent. Column chromatography on silica gel (15% EtOAc in hexanes as the eluent) yielded alcohol (3) (1.64 g, 82%) as a yellow oil, Rf=0.40 (30% EtOAc in hexanes), [o;]25 D -7.3° (c 0.82, MeOH) . (S) -1- (Benzyloxy) -but-3-en-2-yl acrylate
(12): To a solution of alcohol (3) (1 g, 5.61 mmol) in CH2C12 (20 L) was added acryloyl chloride (685 μL, 8.41 mmol) dropwise, followed by the addition of Et3N (1.56 mL, 11.2 mmol). The resulting mixture was stirred for 10 min, and the solvent then was removed under reduced pressure. Filtration of the concentrated crude acrylate through a pad of silica gel using 15% EtOAc in hexanes, followed by the removal of solvent, yielded acrylate (12) (1.19 g, 92%) as a colorless oil, Rf=0.57 (30% EtOAc in hexanes), [α]25 D -5.7° (c 1.09, CHC13) ; 1H-NMR (500 MHz, CDC13) δ: 3.59-3.65 (m, 2H) , 4.56 (q, 2H, J=12.2, 14.65 Hz), 5.25 (d, IH, J=10.6 Hz), 5.33 (d, IH, J=16.8 Hz), 5.57 (m, IH) , 5.84-5.91 (m, 2H) , 6.17 (dd, IH, J=6.9, 10.4 Hz), 6.44 (dd, IH, J=1.3, 16.2 Hz), 7.27-7.36 (m, 5H) ; 13C-NMR (125.8 MHz, CDC13) δ: 71 . 62 , 73 . 58 , 73 . 77 , 118 . 49 , 128 . 05 , 128 . 85 , 131 . 52 , 133 . 62 , 138 . 31 , 165 . 79 .
(5S) -5- (Benzyloxymethyl) -5H-furan-2-one (13): To a solution of acrylate (12) (1.87 g, 8.05 mmol) in CH2C12 (700 mL) was added second generation Grubbs’ catalyst (4 mol %, 170 mg, 0.322 mmol). The reaction mixture was refluxed for 5 hours, and the solvent then was removed under reduced pressure. Column chromatography on silica gel (30% EtOAc in hexanes as the eluent) yielded the furanone (13)
(1.62 g, 98%) as a brown oil, Rf=0.15 (30% EtOAc in hexanes), [α]25 D -81.3° (c 1.09, MeOH); αH-NMR (500 MHz, CDC13) δ: 3.66 (dd, IH, J=5.0, 5.5 Hz), 3.71 (dd, IH, J=5.0, 5.2 Hz), 4.57 (s, 2H) , 5.17 (m, IH) , 6.16 (dd, IH, J=1.9, 3.8 Hz), 7.29-7.37 (m, 5H) , 7.48 (dd, IH, J=1.4, 4.3 Hz); 13C-NMR (125.8 MHz, CDC13) δ: a 69.86, 74.18, 82.61, 123.03, 128.42, 128.95, 137.69, 154.32, 173.19.
(4S ,5S) -5- (Benzyloxymethyl) -4- [1 , 3] di- oxolan-2-yldihydrofuran-2-one (14) : A solution of furanone (13) (1.2 g, 5.88 mmols) and benzophenone
(108 mg, 0.588 mmols) in [1, 3] -dioxolane (108 mg) was degassed for 40 min in a stream of argon. The mixture then was irradiated using one 450 watt ACE glass medium pressure mercury lamp, from a distance of 15 cm, for 9 hours. Progress of this reaction was observed via 1H-NMR. As the reaction mixture was degassed, and throughout all of the irradiation time, the reaction flask was held in a water cooled cooling mantel. The temperature of the cooling water was constantly maintained near 0°C. Upon completion of the reaction, solvent was removed under reduced pressure, followed by column chromatography on silica gel (35% EtOAc in hexanes as the eluent), yielding the title compound (1.34 g, 82%) as a clear oil, Rf=0.14 (30% EtOAc in hexanes), [α]25 D 16.5° (c 1.2, CHC13) ; 1H-NMR (500 MHz, CDC13) δ: 2.50 (dd, IH, J=3.9, 12.9 Hz), 2.70-2.79 (m, 2H) , 3.58 (dd, IH, J=3.5, 7.2 Hz), 3.75 (dd, IH, J=2.8, 7.9 Hz), 3.87-3.92 (m, 2H) , 3.97-4.00 ( , 2H) , 4.51 (d, IH, J=11.9 Hz), 4.57-4.61 (m, 2H) , 4.88 (d, IH, J=3.6 Hz), 7.26-7.36 (m, 5H) ; 13C-NMR (125.8 MHz, CDC13) δ: 30.39, 40.53, 65.77, 71.74, 73.99, 79.52, 104.14, 128.00, 128.89, 138.07, 176.79.
(4S,5S) -4-[l,3]Dioxolan-2-yl-5-hydroxy- methyldihydrofuran-2-one (15) : To a solution of dihydrofuranone (14) (0.5 g, 1.79 mmol) in MeOH (30 mL) was added Pd/C (25 mg) . The mixture was stirred at room temperature under an H2 balloon for 24 hours, and then filtered over Celite. Removal of solvent under reduced pressure, followed by column chromatography on silica gel (35% EtOAc in hexanes as the eluent) yielded the compound (15) (301 mg, 89%) as a white solid, Rf=0.28 (50% EtOAc in hexanes), [ ]25 D 22° (c 1.32, CHC13) ; XH-NMR (500 MHz, CDC13) δ: 2.54 (dd, IH, J=6.0, 11.4 Hz), 2.68-2.81 (m, 2H) , 3.66
(dd, IH, J=3.9-8.5 Hz), 3.88-3.95 (m, 3H) , 3.97-4.02 (m, 2H) , 4.53 (m, IH) , 4.91 (d, IH, J=3.9 Hz); 13C- NMR (125.8 MHz, CDC13) δ: 30.68, 40.12, 64.36, 65.77, 81.07, 103.94, 176.83. (3S , 3aS , 6aR) -3-Hydroxyhexahydrofuro [2 , 3- b] furan (5) : To a solution of lithium aluminum hydride (76 mg, 1.98 mmols) in THF (10 ml) at 0°C was added dihydrofuranone 15 (275 mg , 1.46 mmol) in THF (30 mL ) dropwise. Upon completion of the reduction after 4 hours, the reaction was quenched with a saturated aqueous sodium sulfate solution at 0°C. The solvent then was decanted and the remaining residue was washed with THF (3x) , EtOAc (3x) , and CHC13 (3x) . The organic extracts were combined and the solvent was removed under reduced pressure, yielding a crude (2S, 3S) -3- [1, 3] dioxolan-2- ylpentane-1, 2, 5-triol, which was immediately used in the next reaction.
The crude triol was dissolved in a mixture of THF:H20 (8ml) in the ratio of a 5:1. This solu- tion then was acidified at room temperature to pH 2- 3 with 1 N hydrochloric acid, and was stirred for 40 hours. Removal of solvent with the aid of benzene under reduced pressure, followed by column chromatography purification on silica gel (5% MeOH in CHCI3 as the eluent) yielded the compound (5) (145 mg,
77%) as a white solid, Rf=0.40 (15% MeOH in CHC13) , [α]25 D -25.1° (c 1.05, CHC13) ; XH-NMR (500 MHz, CDCI3) δ: 1.67 ( , IH) , 2.13 (m, IH) , 2.31 (bs, IH) , 2.79 (m, IH) , 3.80-3.88 (m, 3H) , 3.95 (dd, IH, J=3.2, 7.1 Hz), 4.20 (d, IH, J=3.1 Hz), 5.86 (d, IH, J=4.9 Hz). Preparation of bis-THF derivative (I) (by Mitsunobu inversion of compound (5) ) : To a stirred solution of alcohol (5) (400 mg, 3.07 mmol), tri- phenylphosphine (1.6 g, 61.4 mmol), and p-nitroben- zoic acid (770 mg, 4.61 mmol) in dry benzene (30 mL) at 23 °C was added diisoproylazodicarboxylate (DIAD, 1.2 L, 6.14 mmol) dropwise. After 1.5 hours, the mixture was concentrated in vacuo, and the crude ester was dissolved in a (4:3:1) mixture of MeOH:Et3N:H20 (24 mL) , then treated with LiOH (450 mg, 10.7 mmol) . The solution was stirred at room temperature for 2 h. The mixture then was concentrated under reduced pressure and the residue was chromatographed over silica gel to provide the bis-
THF (I) (326 mg, 82%); [ ]25 D -12.4 (c 1.16 , MeOH)
In particular, the following synthetic scheme (1) illustrates the present commercial method of synthesizing compound (I). This synthesis is disclosed in detail in A.K. Ghosh et al., Tetra. hedron Letters, 36 (4) , pp. 505-508 (1995), incorporated herein by reference. Also see, A.K. Ghosh et al., J. Med. Chem . , 39, pp. 3278-3290 (1996) for the synthesis of compound (I) and a related compound of structural formula (II) (i.e., (3S, 3aR, 7aS) -3-hydroxyhexahydrofuro [2, 3-b] pyran).


The present method of synthesizing bis-THF is summarized as follows:


The synthesis of bis-THF (compound (I) ) is summarized below:

previously.


- Ghosh, Arun K.; Journal of Organic Chemistry 2004, 69(23), pg 7822-7829
- Kulkarni, Mukund G.; Tetrahedron: Asymmetry 2010, 21(19), pg 2394-2398
- Quaedflieg, Peter J. L. M.; Organic Letters 2005, 7(26), pg 5917-5920
- Black, David M.; Tetrahedron: Asymmetry 2008, 19(17), pg 2015-2019
- Ghosh, Arun K.; Synthesis 2006, (18),pg 3015-3018
- Canoy, Will L.; Organic Letters 2008, 10(6), pg 1103-1106
- Ghosh, Arun K.; ACS Medicinal Chemistry Letters 2011, 2(4), pg 298-302
- Kesteleyn, Bart Rudolf Romanie; WO 2003022853 A1 2003
- Yu, Richard H.; Organic Process Research & Development 2007, 11(6), pg 972-980
- Khmelnitsky, Yuri L.; Organic Process Research & Development 2011, 15(1), pg 279-283
//////////(3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-ol, furofuranol, DARUNAVIR
O[C@H]1CO[C@H]2OCC[C@@H]12


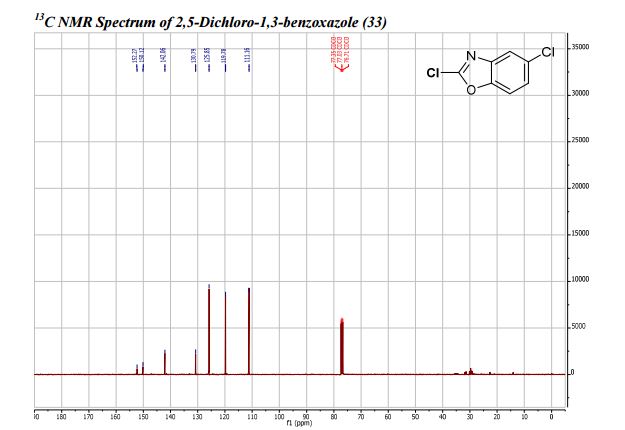
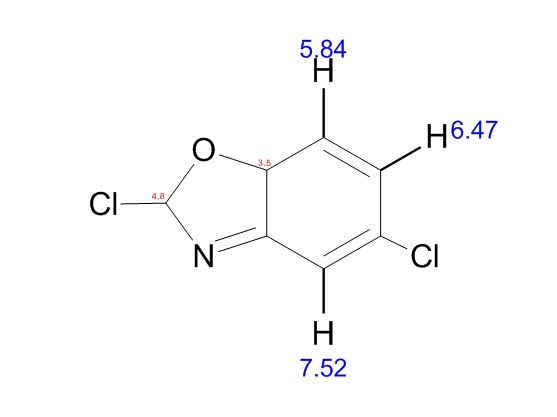
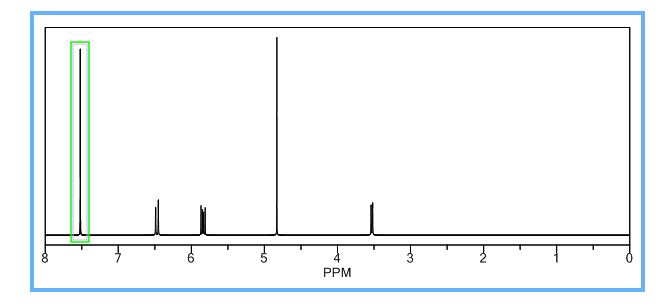
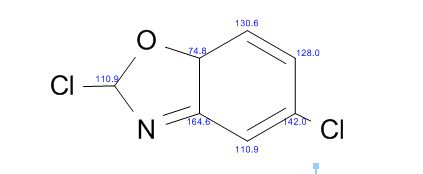

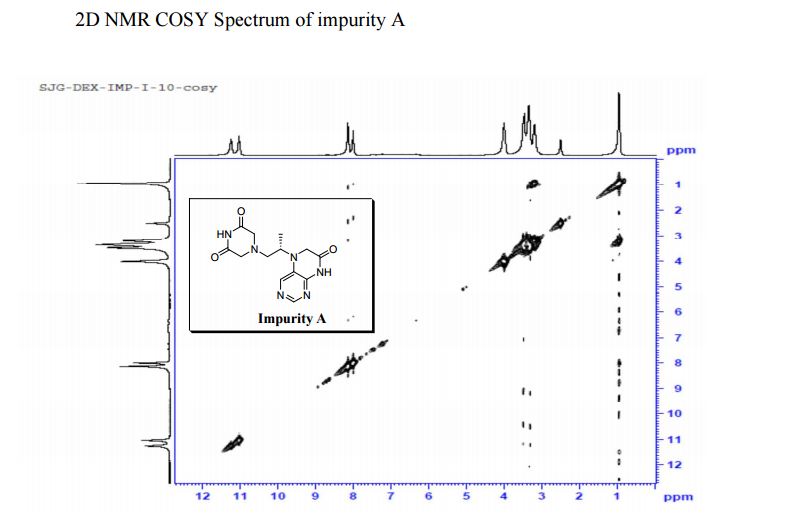

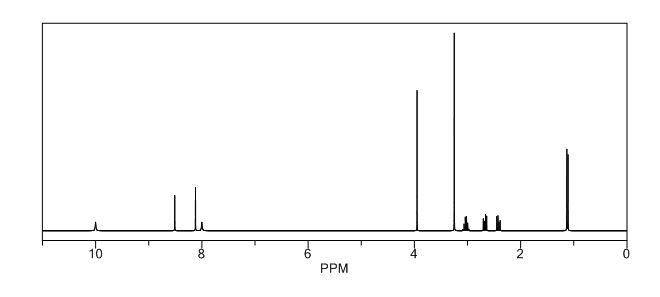
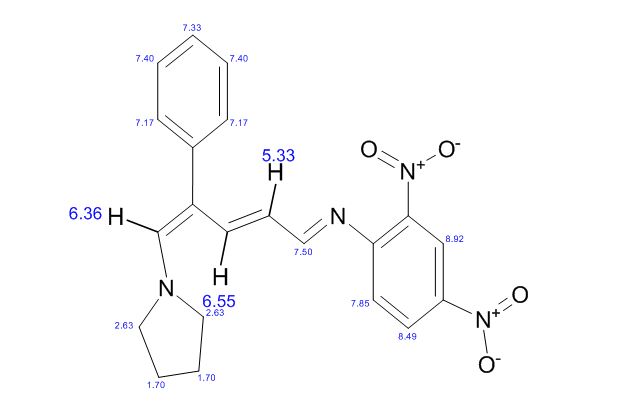
 COSY PREDICT
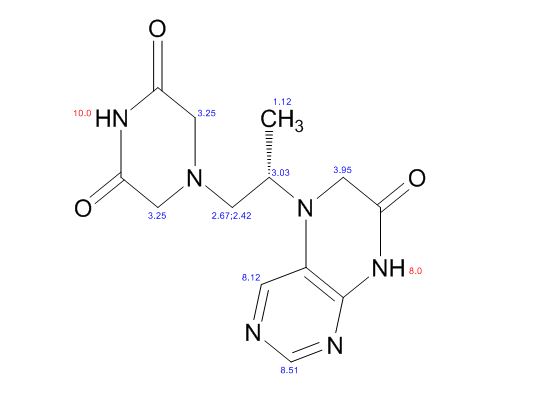
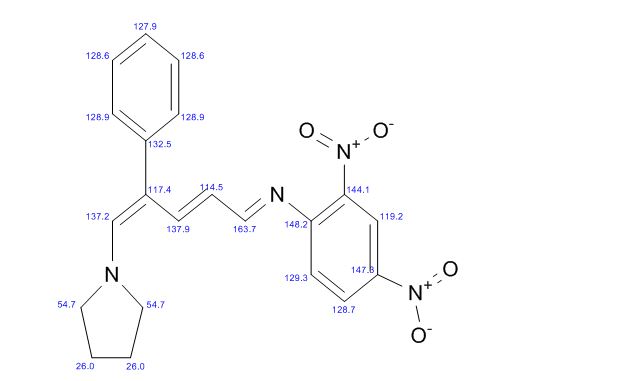
COSY PREDICT

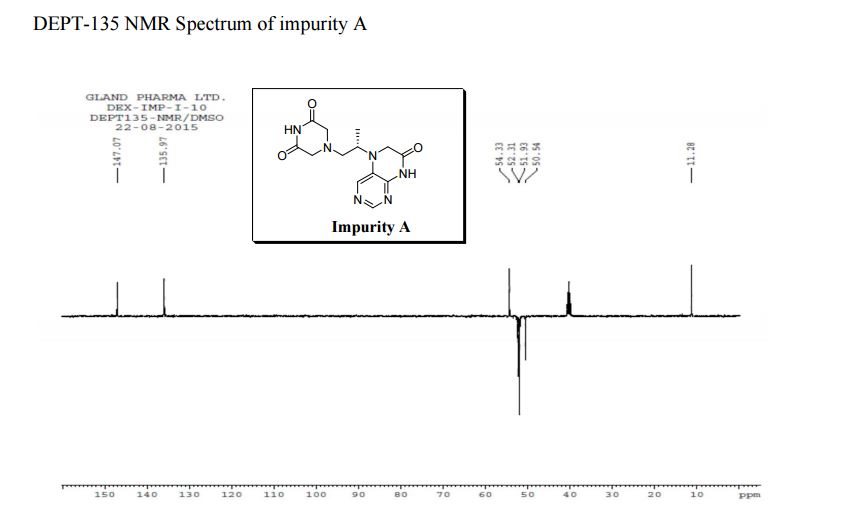
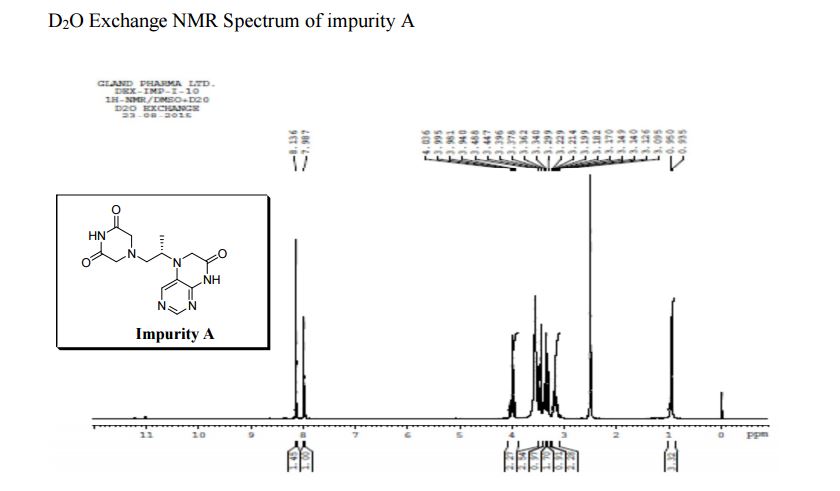
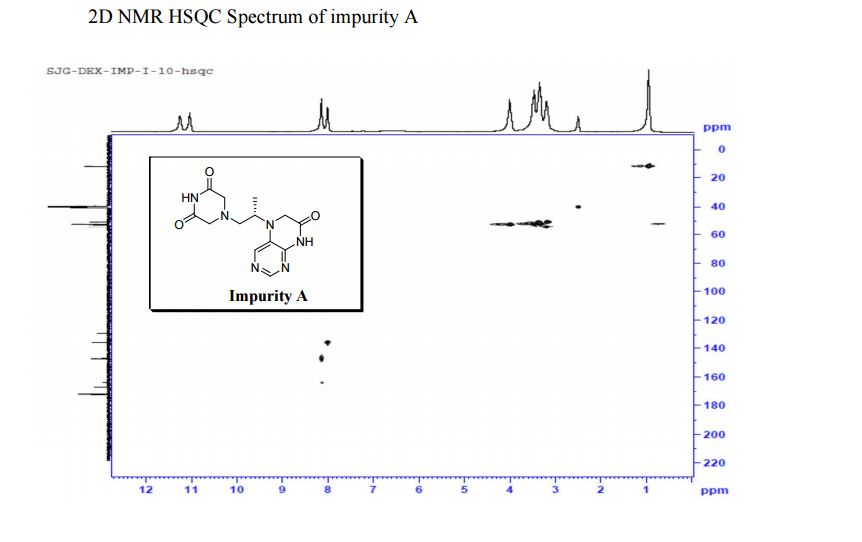
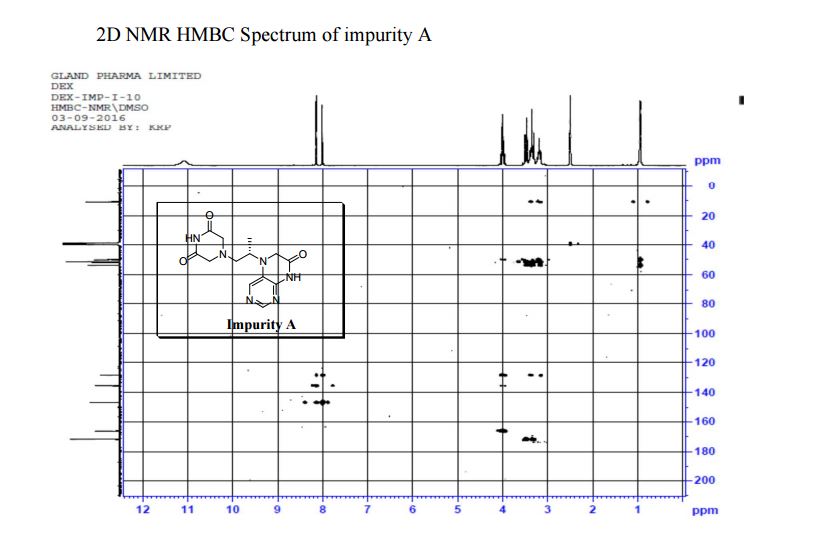



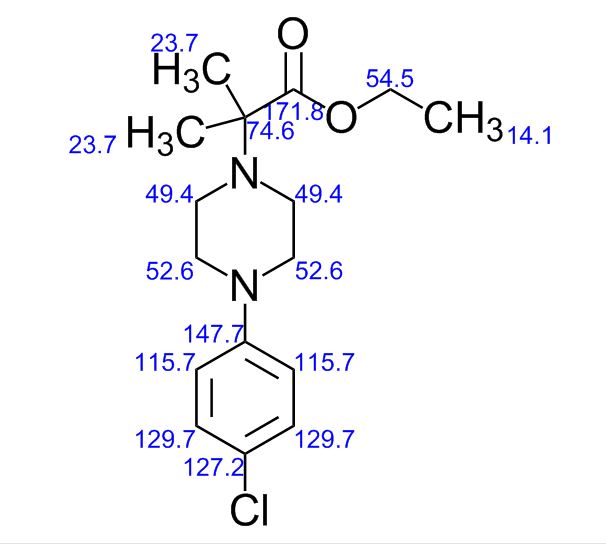
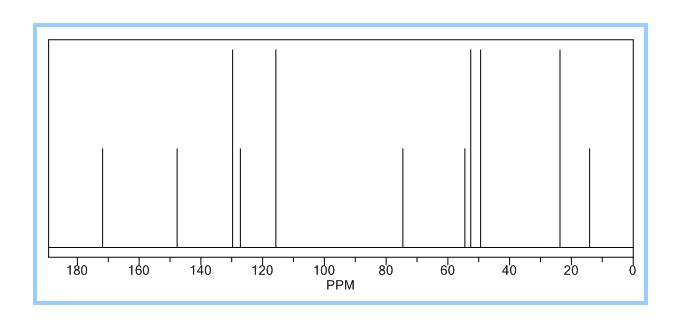
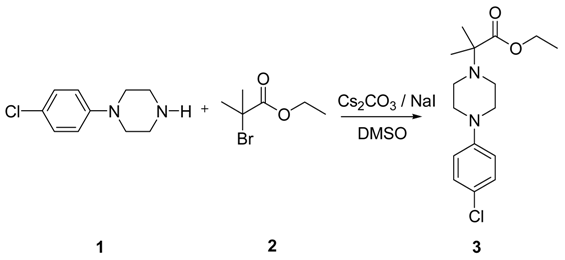
























 LinkedIn
LinkedIn Facebook
Facebook Twitter
Twitter GooglePlus
GooglePlus